
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A conceptual model to predict uranium removal from aqueous solutions in water–rock systems associated with low- and intermediate-level radioactive waste disposal†
Janice P. L. Kenney *a,
Matthew E. Kirbya,
Javier Cuadrosb and
Dominik J. Weissa
*a,
Matthew E. Kirbya,
Javier Cuadrosb and
Dominik J. Weissa
aDepartment of Earth Science and Engineering, Imperial College London, London, UK
bDepartment of Mineralogy, Natural History Museum, London, UK
First published on 23rd January 2017
Abstract
Global stores of radioactive waste are housed in surface stores where actinides are susceptible to environmental release. It is imperative that waste disposal facilities are built to safely contain this waste. However, to do this we must ensure that the engineered and natural barriers are sufficient to prevent the buried materials from migrating through to the surface. Solutions migrating from repositories (ILW and LLW) will have a wide range of chemical compositions and conceptual models constraining the key mineral-water interactions with realistic lithologies are urgently needed. To this end, we conducted experiments to study U removal from solution via mineral-surface interactions with quartz, sandstone, and volcanic rock over a pH range of 2–12, with varying concentrations of U (10 ppb, 0.1 ppm, 1 ppm, and 10 ppm) and with and without bicarbonate added (2 mM) with 0.1 M NaCl electrolyte. We observed that the U concentration in solution had little effect on the extent of U removal from solution as a function of pH or bicarbonate concentration with quartz and sandstone but was important for volcanic rocks, where removal of U, due to adsorption, decreased with increasing U concentration between pH 4 and 8. When bicarbonate was added to solution then the quartz, sandstone, and volcanic rock geomaterials acted similarly in their abilities to immobilize uranium, with an adsorption envelope from pH 4–8 followed by an increase in U removal, likely via precipitation, at high pH. When bicarbonate was not added, the removal of U from solution was more controlled by the geomaterial. Bicarbonate addition at pH 6–10 lowered adsorption. However, the addition of bicarbonate in experiments with 10 ppm U at pH 10–12 allowed for precipitation of U at the rock surface, making bicarbonate an immobilizing factor. Therefore, our conceptual model shows that U is immobilised from radioactive waste-like solutions in a bimodal distribution, both at low (6) and high (11) pH.
1 Introduction
A successful radioactive waste facility prevents the escape of aqueous actinides and other hazardous and non-hazardous pollutants from the waste and migration into the biosphere. Intermediate- and low-level radioactive waste (ILW and LLW, respectively) will be encapsulated in steel containers within a cementitious grout. While a definitive repository site waits to be determined in many countries around the world, the canisters with the waste remaining in temporary repositories at the surface or just below the surface. Once buried, the steel canisters, whether LLW emplaced near the surface or ILW in the deep subsurface, will eventually fail (predicted life of 1k years in best case scenario) and groundwater will enter the canisters. The groundwater passing through the cemented ILW and LLW systems will likely have a high ionic strength and bicarbonate content.1,2 The cement will evolve in 5 stages.3 In stage 1 sodium and potassium oxides will be leached leading to high pH of 13.5. In stage 2 calcium hydroxide will dissolve and the pH will be buffer at 12.5. Then in stages 3–4 dissolution of calcium silicate hydrate (CSH) gel will take place that will eventually buffer the pH at 10.5. The 5th stage is a result of the dissolution of the remaining cement minerals such as calcite that will cause no pH buffering to the system and buffering from the local host rock would prevail. How long each stage lasts will depend greatly on the rate of groundwater flow through the cement system, but it is assumed that in the pore waters surrounding the canisters, the pH would remain above 10.5 for several hundred thousand years.4 LLW could also be stored in trenches without cementitious grout, where more acidic conditions would result due to the degradation of cellulosic waste and corrosion of the metal canisters.5 Therefore it is important to understand how both alkaline and acidic conditions can affect U mobility.If the waste is stored near the surface in crystalline rock, oxic groundwaters will enter the site. Since crystalline rocks are likely fractured, if the fracture network connects with the surface, buried canisters may see an influx of young and potentially oxic groundwaters. These waters react with U and produce mobile U(VI). It is, therefore, imperative to understand how a U-rich, high pH fluid at different stages of maturation (between pH 12.5 and 10) or low pH from cellulosic degradation interacts with the rocks in and around the repository. The pH of the actinide-containing solution will equilibrate with the groundwater of the aquifer and shift to circumneutral values.
In order to better understand how uranium, one of the most prevalent radionuclides in the radioactive waste, will migrate in the subsurface once the integrity of the container is compromised, we must understand how it will interact with the potentially fractured rocks around the repository and with fracture sealing minerals. The adsorption of U to quartz and silica gel is well studied in the pH range between 3 and 9, with the result that the majority of adsorption occurs at neutral pH with the specific adsorption edges/envelopes changing with surface area, U concentration, and ionic strength.6–9 As a specific example having similar experimental conditions as our study, Huber and Lützenkirchen9 equilibrated U solutions with 10–30 ppb U(VI) with seven different quartz samples and found that nearly all U(VI) was adsorbed at pH above 5 via surface silanol groups. Other geomaterials have been studied for their potential to immobilize soluble U(VI), such as granite, iron oxides, clays, aquifer sediments, and volcanic rock.10–13 However, most of these studies only examine the sorption of U(VI) under circumneutral pH values. Bots et al.14 noticed that at pH 13, solutions with 10 and 50 ppm U contained nano-sized precipitates, likely clarkeite ((Na, Ca, Pb)(UO2)O(OH)(H2O)0−1). This may be the same mineral that formed in the granite batch experiments conducted by Fan et al.15 at pH 9–12. Little is known about how mobile those precipitates can be in the environment, and if they change as a function of water chemistry.
The chemical composition of the aqueous solution affects the mobility of radioactive elements.8,15–18 Wazne et al.18 demonstrated that the addition of carbonate to an aqueous system decreased the sorption of U to ferrihydrite. Since carbonate tends to form strong complexes with U,19–21 this is a crucial groundwater component that must be considered when studying U mobility in fractured rocks. It is still unclear what effect carbonate species can have on the precipitation of U or how carbonate could affect the mobility of precipitates.
In this study we develop a conceptual model for U mobility in fractured rocks using a set of designed batch experiments. We examine sorption of U(VI) as a function of pH and U concentration onto quartz, a common rock-forming and fracture-sealing mineral,22 low permeability volcanic rocks from the Borrowdale Volcanic Group (BVG) and an iron-coated sandstone from the St. Bees Sandstone (SS). These two rock types are from Cumbria in the UK. The volcanic rock represents rocks which may be host to a repository and the sandstone may be a potential aquifer rock in which a contaminant plume may traverse. Since bicarbonate will be produced via cement degradation in many groundwaters,1 sodium bicarbonate is used to better represent the system and understand how varying groundwater chemistry affects the retention of U by the geomaterials.
2 Experimental details
2.1 Materials
The geomaterials used in this study are quartz, purchased from Sigma Aldrich, St. Bees sandstone (iron oxide coated sandstone) and volcanic rock from the Borrowdale Volcanic group collected from Cumbria, UK.23 All geomaterials were crushed and sieved to between 128 and 700 μm. The geomaterials were washed according to the procedure developed by Yee and Fein24 in order to remove any loosely bound surface contaminants, using a 1 M NaOH wash, then rinsed with 18 MΩ water, then washed again with 1 M HNO3, then rinsing the acid from the minerals with 18 MΩ water. Following the washing procedure, the quartz was dried at 60 °C for 24 hours and the sandstone and volcanic rock were dried at 45 °C for 24 hours. The specific surface area (BET) was measured, after being washed, with a TriStar 3000 instrument, using the adsorption of N2 at the liquid nitrogen temperature. The BET surface area of the quartz, sandstone, and volcanic rock were 0.0325, 1.3496, and 0.7549 m2 g−1, respectively. Aliquots of the geomaterials were then crushed to a fine powder in order to carry out X-ray diffraction (XRD), at the Natural History Museum of London. The XRD analyses were carried out using an Enraf-Nonius FR590 PDS120 system, with an Inel position sensitive detector covering 120° 2θ. Cobalt radiation was used, at 40 kV and 40 mA, with a primary Ge monochromator (allowing only CoKα1 radiation). Two perpendicular slits of 0.14 and 5 mm focused the incident beam at an angle of ∼4° with the surface of the sample. In this system, the diffracted radiation is collected at the same time in the entire 120° range. The time of analysis varied between 10 and 17 min. The diffraction patterns were calibrated with Ag-behenate and Si standards.Solutions were prepared using a U(VI) stock solution obtained from VWR as a 1000 ppm standard solution. This solution was diluted with 0.1 M NaCl to buffer the ionic strength, mimic groundwater salinity, and to achieve the desired U concentrations in this study. Sodium bicarbonate stock solutions were prepared by adding 0.8 g of powder (Sigma Aldrich) to 100 mL of DI water. This stock solution was then used to spike the desired solutions and suspensions to a final NaHCO3 concentration of 2 mM. Due to the open nature of the system there was fluctuation between the total alkalinity as a function of pH since high pH systems have higher alkalinity. However even at pH 12 the system with bicarbonate added consistently had 2 mM higher alkalinity than the system where bicarbonate was not added. The alkalinity of the system at high pH was measured with potentiometric titrations, data not shown.
2.2 Batch uranium(VI) experiments
Batch adsorption experiments were conducted using suspensions of quartz, sandstone, or volcanic rock with initial U concentrations of 10 ppb, 0.1 ppm, 1 ppm, and 10 ppm. To investigate how sodium bicarbonate affects the sorption of U to all geomaterials, these experiments were reproduced with an addition of sodium bicarbonate solution to a final concentration of 2 mM. This concentration was chosen to be similar to the carbonate solutions present in a groundwater system previously investigated as a potential repository host.1 Adsorption experiments were conducted in polypropylene test tubes in a 0.1 M NaCl electrolyte matrix. An aliquot of the prepared solution was taken in order to determine the precise starting concentration of U in solution, called hereafter the parent solution. Weighed sorbent was added to each test tube so that the final suspension of geomaterials contained 5 g L−1. The pH of each suspension was readjusted to targeted pH values ranging between 2 and 12 using aliquots of 1 M HCl or NaOH as needed. Experiments were mixed via end over end rotation for 3 days. Following the experiments the solutions were centrifuged at 4000 rpm to separate them from the geomaterials and the solutions were HNO3–acidified in preparation for analysis. These measurements provided the concentration of U as well as information about the dissolution of the rock materials. The elements analysed for rock dissolution were Na, Mg, Al, P, S, K, Ca, and Fe. Huber and Lützenkirchen9 found that as pH increased so did the concentration of Si in solution. They postulated that the increase in Si would complex U and possibly affect binding of U to quartz. Fox et al.,8 in contrast, studied differences in U sorption to ferrihydrite and quartz, and found that there was no change in the removal of U(VI) from solution at high pH values associated with Si dissolution, but instead found sorption decreased at lower pH values. Therefore if there is only dissolution of quartz at high pH and the presence of Si only affects U adsorption at low pH, then Si would not be an issue in our study. Experiments were conducted at least twice to ensure reproducibility; therefore all datasets represent at least two experiments. Samples from experiments conducted with 10 ppm U were also analysed with and without filtration with a 0.45 μm nylon filter to determine if there was a filterable precipitate forming that was remaining suspended after the experiments.Additional experiments were conducted to determine if a precipitate formed and determine how mobile it is (i.e., remaining suspended in solution or dropping our of solution) at pH values where U was expected to precipitate without a geomaterial present. These were done in the exact same was as the batch experiments but with no geomaterials added to ensure that any removal of U(VI) from solution was directly related to the precipitate. The experimental systems were shaken via end-over-end rotation and allowed to equilibrate for 3 days. After the final pH was measured, the supernatant was collected and half of it was filtered through a 0.45 μm nylon filter. Both the filtered and unfiltered supernatants were then diluted and acidified using HNO3 for ICP-MS measurements. At all U concentrations studied there was virtually no U removed from solution via the vessel walls. Samples were taken from the experiments without geomaterials (pH 10) to image any precipitates formed using Transmission Electron Microscopy (TEM) along with Energy Dispersive X-ray Spectroscopy (EDS) to analyse their elemental composition (details below).
The parent solutions and the supernatants from the adsorption experiments were measured using ICP-MS. Samples were analysed together with matrix-matched multi-element standards, with concentrations ranging from 0 to 10 ppm. The standards were periodically analysed during the measurements to verify that there was no machine drift or systematic analytical error. In addition, Bi was monitored as an internal standard.
2.3 Modelling uranium(VI) speciation
The aqueous speciation of U(VI) of the aqueous solutions were constrained using the sit.dat database in PHREEQC using the experimental conditions of pH range 2–12.5, U concentrations of 10 ppb, 0.1 ppm, 1 ppm, and 10 ppm, in 0.1 M NaCl, with and without the addition of 2 mM NaHCO3.2.4 Transmission electron microscopy
The TEM study was conducted on precipitates from geomaterial-free experiments using a JEOL 2100-plus TEM. Samples were prepared by evaporating a droplet of 10 ppm U solution at pH 10 onto a forvard film Cu-TEM grid. Energy dispersive spectroscopy (EDS) was used to assess the identity of the precipitate. The analyses were carried out with an operating voltage of 200 kV, and the detection limit on the EDS for each element was 1 wt%.3 Results and discussion
3.1 Characterisation of geomaterials
The quartz standard was confirmed to consist of quartz, possibly with traces of plagioclase or feldspar (very low intensity peak at ∼3.2 Å that cannot be observed in Fig. 1A). The sandstone (Fig. 1B) was composed of quartz with subordinate microcline and mica (probably muscovite, peaks at 10 and 4.5 Å which cannot be observed in Fig. 1B). The colour of the surface of the sandstone was red, indicating a thin mineral coating. This was too small for identification with XRD but is likely hematite, as this was determined to be the coating on samples from the St. Bees Sandstone observed in other studies.24,25 The volcanic rock was the mineralogically most complex of the geomaterials.26 It consisted of approximately 50 wt% quartz, with chlorite, mica (probably muscovite; Fig. 1C), plagioclase, and calcite (apparently with some Mg content because the main peak is displaced from 3.03 to 3.024 Å; Fig. 1C). This mineralogy is consistent with previous observations on the volcanic rocks of the BVG.11,27,28 | ||
| Fig. 1 XRD diagrams of the three geomaterials: quartz (A), sandstone (B), and volcanic rock (C). The figures in the plot are d-spacing values in Å. The sandstone contains quartz (Q) and microcline (M). The volcanic rock consists of quartz, chlorite (Ch; specifically clinochlore), mica (Mc; most probably muscovite), plagioclase (P), and calcite (Ca; with some Mg as indicated by the peak position at 3.024 Å). The 3.34 Å peak of quartz is truncated in (A). | ||
The extent of element release into solution via dissolution of the rock was studied in the sandstone and volcanic rock experiments. In the sandstone experiments 2–8 ppm of Fe, Na, Al, and S were released into solution, with increasing release as pH increases (Fig. 2A). In the volcanic rock experiments, between 0.5 and 8 ppm of K, S, Fe, P, and Ca were released between pH 2 and 12, with more Ca, P and Fe being released below pH 3.5 (Fig. 2B).
 | ||
| Fig. 2 Concentration of major elements (in ppb) in solution after the U adsorption experiments with (A) sandstone and (B) volcanic rock. Samples were measured using ICP-MS. | ||
3.2 Theoretical uranium(VI) speciation
Theoretical concentrations of U in solution can be seen in Fig. 3. When 2 mM of sodium bicarbonate was added to solution the species that dominate at pH values of 2–12.5 include UO22+, UO2CO3o, UO2(CO3)22−, UO2(CO3)34−, UO2(OH)3−. The relative percentage of each species in solution does not change as a function of U concentration if bicarbonate is present. If no sodium bicarbonate is added, there are significant changes in the evolution of the system with pH depending on U concentration. UO22+, and UO2(OH)3− are the dominant species at low and high pH values, respectively, in all cases. At lower U concentrations UO2OH+ and UO2(OH)2o are important species between pH 5 and 10. However, as U concentration increases the dominant species between pH 5 and 10 shift to (UO2)3(OH)5+ and (UO2)3(OH)7−. Therefore an important variable affecting U removal from solution with changing U concentration should be the different electric charge of the U-species between pH 6 and 8. This will be examined below. | ||
| Fig. 3 Speciation diagram showing the relative concentrations of the important U species in solution at 10 ppb and 10 ppm (the end-member U concentrations in this study) as a function of pH and the presence/absence of sodium bicarbonate. The species fractions do not add up to 1 above pH 12 because the species that predominates at pH 13 reached 0.5 at pH 12.5, and was removed for clarity from the graph. | ||
Speciation modelling predicts the supersaturation of U minerals at high pH, however, the specific pH at which these minerals are predicted to precipitate change as a function of U concentration and bicarbonate addition (Fig. 4). Overall, the range of pH at which some species are expected to precipitate is wider when bicarbonate is added. In the absence of bicarbonate species, precipitation is mainly expected at pH 10–12. The compounds are all sodium uranate species and their chemical formulae are sodium diuranate (Na2U2O7), sodium clarkeite ((Na)(UO2)O(OH)(H2O)0−1), schoepite (UO2)8O2(OH)12(H2O)12, and sodium compreignacite (Na2(UO2)6O4(OH)6(H2O)7).
 | ||
| Fig. 4 Speciation diagram showing the saturation index of each U-mineral predicted to precipitate in the experimental conditions with no geomaterials, with and without bicarbonate added and 10 ppb and 10 ppm U. | ||
While model calculations predict U-minerals to precipitate under our experimental conditions, Lefèvre et al.29 performed turbidimetric measurements of U(VI) solutions across a range of pH and U concentrations in line with our experimental conditions and found no evidence of U precipitation. Equally, Sylwester et al.30 and Bargar et al.31 found no precipitates in supersaturated 10 ppm uranyl samples at circumneutral pH values, with precipitation of schoepite only occurring at 17 ppm,31 observed using EXAFS. Bots et al.,14 observed precipitation of nanoparticulate U colloids with a clarkeite-type crystallographic structure at pH > 13, which was one of the predicted minerals in our thermodynamic calculations. The lack of detection of precipitated minerals in some of the above studies may be due to their very small size fraction and mobility.
3.3 Precipitation of U-minerals in high pH solutions
To investigate whether the precipitation of the U minerals, predicted in our thermodynamic calculations, occurred in solution, without the presence of minerals as a possible nucleation point, we conducted experiments as a function of pH and U concentration (10 ppb, 0.1 ppm, 1 ppm, and 10 ppm; Fig. 5). With and without added bicarbonate, less than 20% U was removed from solutions with 10 ppb, 0.1 ppm or 1 ppm solution above pH 8 using a 0.45 μm filter. Below pH 8 there was a decreasing percentage of U removed from solution as the total initial concentration of U increased. Since there was no U removed from solution via the vessel walls, this removal must be due to adsorption to the filter papers, therefore. There was up to 100% precipitation in the 10 ppm experiment at pH 10.5 where bicarbonate was not added. When bicarbonate was added 0–30% of the U was collected on the filters. In all cases it is possible that precipitates with size < 0.45 μm passed through the filter. | ||
| Fig. 5 Control experiments showing the percentage of U filtered out of the solutions as a function of pH, U concentration, and the addition of sodium bicarbonate. Open symbols denote the percentage of U removed from solution after filtration with a 0.45 μm nylon filter. Closed symbols denote the percentage of U removed from solution without filtration. Since most precipitation was expected above pH 7, these experiments were confined to areas of U supersaturation. At lower U concentrations (10 ppb and 0.1 ppm) there was interaction of the U with the filters (see text). | ||
Fig. 6 shows representative TEM images of the precipitates forming in the geomaterial free experiments without added bicarbonate. Two morphologies were observed, a gel-like morphology and another in which individual particles were more apparent. EDS analysis indicates these different morphologies may be two distinct stoichiometries coherent with sodium uranates (Fig. 6C). Chemical analysis of the gel-like area revealed higher Na/U ratios compared to areas with more distinct particles. The gel-like areas produced a Na/U ratio of 1 (A in Fig. 6A), which is consistent with sodium diuranate and clarkeite, whereas the flaky particles (B and C in Fig. 6A) had ratios that more closely resembled sodium compreignacite (Na/U 0.33). Since calculations predict the precipitation of the same phases, it is likely that these sodium uranate minerals precipitate in our systems. Similar morphologies and Na/U ratios were observed in the minerals formed in the systems with bicarbonate added.
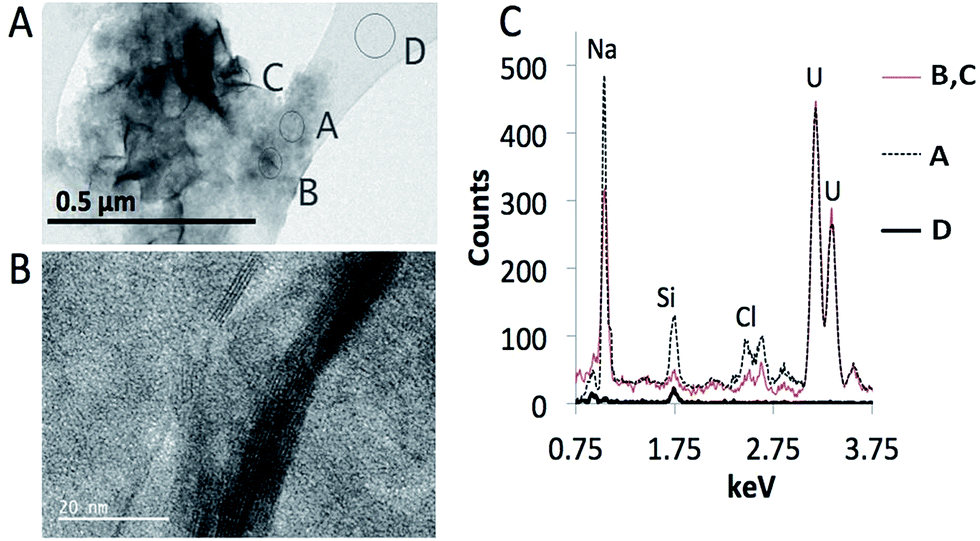 | ||
| Fig. 6 Representative TEM images of the sodium uranate minerals precipitated in a geomaterial-free 10 ppm solution of U at pH 10 without the addition of sodium bicarbonate. (A) Shows the low-magnification image of particles with two morphologies, flaky and gel-like. (B) Shows a high-resolution image of a few flaky particles, with layered structure. (C) Shows spectra of the corresponding spots on the TEM image. Spectra from spots A and B, C are significantly different. Spectrum D is from the carbon film in the Cu grid. | ||
3.4 Batch uranium(VI) experiments
Batch experiments were conducted as a function of pH, U concentration, bicarbonate concentration, and type of geomaterial (Fig. 7). Fig. S1 and S2† show different configurations of the data to more easily understand the effects of bicarbonate content and geomaterial, respectively, on U removal. Across all experimental conditions, we observe an adsorption envelope of U onto the geomaterials between pH 4 and 7–10 and in some cases (quartz and sandstone with bicarbonate, and in all BVG experiments) a second “envelope” between pH 10 and 12 is seen. Therefore, we identify two regions of U removal for discussion. Region 1 is adsorption, occurring between pH 4 and 7 (or 10 in the case of sandstone without bicarbonate addition). Below pH 4 there is little adsorption in all experiments. This most likely relates to the positively charged minerals repelling the positively charged UO22+ species. Region 2 is between pH 9 and 12 and may either be adsorption or precipitation. | ||
| Fig. 7 Batch adsorption data showing the percentage of U(VI) removed from solution in contact with quartz (Qtz), sandstone (SS), and volcanic rock (BVG) as a function of pH, U concentration, and addition of sodium bicarbonate (denoted with HCO3− in the figure). | ||
The point of zero charge (pzc) of quartz is 2.5–3.5,32 therefore as pH increases above the pKa values the charge of the quartz surface becomes increasingly negatively changed. Since the sandstone is coated with hematite, the pzc of hematite is approximately 8.5, therefore the hematite coating on the sandstone would allow the surface of the sandstone to be positively charged until over pH 8.5, where the surface would begin to be more negatively charged. The pzc of the minerals predominating in the volcanic rock are below 3.5, similar to the quartz only system,32 and therefore the surface charge would act similarly to that of quartz. Therefore, the adsorption in region 1 follows the electrostatic attraction of U in solution to the surface of the geomaterials, whereby as the surface and the U become negatively charged at the same time, they repel each other, limiting adsorption. Wainipee et al.33 saw As adsorbed to clays at pH 8 via adsorption to Na+ at the negatively charged clay mineral surface. Therefore, since U would act as a similar oxyanion as As, region 2 in our study may result in adsorption via bridging Na+ or K+ cations (K+ cations in the sandstone or volcanic rock experiments). Bridging adsorption occurs when cations sorb to the negatively charged mineral surface, which produces a positively charged bridge onto which the negatively charged U species may sorb. This would also explain the results in the experiments with bicarbonate, since the adsorption in region 1 decreases when the UO2(CO3)34− species becomes prevalent due to its high negative charge. As UO2(OH)3− become prevalent above pH 10.5 the negative charge is significantly lessened and this would allow the bridging adsorption.
Saturation index calculations (Fig. 4), previous studies,14,31 and TEM-EDS analysis of experiments with no geomaterial (Fig. 6), indicate that the removal in region 2 is due to precipitation rather than adsorption onto the geomaterials surface. This is corroborated by the fact that removal of U in region 2 is often only observed when the U concentration is 10 ppm, thereby indicating a threshold concentration for precipitation. If bridging adsorption were taking place it would occur at all concentrations. It is possible that the precipitation is controlled by sodium compreignacite when no bicarbonate is added since the peak between pH 10–12 matches the saturation index of the sodium compreignacite (Fig. 4B). However this may not be the case with bicarbonate added since there is a much wider pH range for the precipitation of sodium compreignacite in those experiments (Fig. 4C). Therefore it is possible that precipitation and adsorption occur, depending on the geomaterials studied, as discussed below.
The removal of U from solution in the volcanic rock experiments at pH values greater than 10 (region 2) may be a result of adsorption via bridging Na+ or K+ or via precipitation at the surface of the volcanic rock as one of the sodium uranate-type minerals, which are predicted to be supersaturated at high pH for all U concentrations studied (Fig. 4). There is the same percentage of U removed from solution in the pH 10–12 region for the volcanic rock for the 10 ppb, 0.1 ppm, and 1 ppm U experiments, but a significant increase in the 10 ppm experiment. Therefore, it is assumed that bridging adsorption is responsible for significant U removal in both regions 1 and 2 at the lower U concentrations and that either precipitation or a combination of adsorption and precipitation is responsible for removal in region 2 of the volcanic rock experiments.
All the geomaterials remove similar percentages of U from solution at pH 7–10 when bicarbonate was added (Fig. 7, S2B†). This is mostly controlled by the negative charge of UO2(CO3)34− at this pH range (Fig. 3) that impede adsorption.
All geomaterials studied consist of at least 50% quartz, therefore surface silanol groups will account for a significant proportion sites for U adsorption. Deprotonated silanol groups readily adsorb metal cations and positively charged U species would be adsorbed. These results have been echoed by studies looking at U sorption to silica gels and quartz,6,8,9 where U sorption increased with increasing pH from no adsorption at pH 2 to nearly 100% adsorption by pH 7.
Fox et al.8 examined the sorption of U onto quartz and ferrihydrite in equilibrium with air and noted that as pH increased above pH 7.75 significant less U sorbed and there was no sorption above pH 8.75. This is similar to what was observed in our quartz and volcanic rock experiments. However, the sandstone experiments had substantially more sorption above pH 7. Lefèvre et al.29 found that the U sorption envelope on hematite was shifted to higher pH values, with respect to that on ferrihydrite. The wider sorption envelope observed in sandstone in our study is likely a combination of the adsorption on hematite above pH 6 and the sorption to silanol groups between pH 4 and 7. Contribution to the removal of U from solution by hematite requires adsorption of the neutral and negatively charged U species (Fig. 3) to hematite on the sandstone. The iron oxide component in the St. Bees sandstone becomes negatively charged above pH 8.5.34 Sorption of U above 8.5 decreases as the concentration of negatively charged U species increases. The removal of U between pH 8.5 and 10 is possibly due to cation bridging as shown to occur with As,33 likely Na+ or K+ in the case of sandstone (Fig. 2). In addition, a significant amount of iron (up to 6 ppm) is released into solution, especially at high pH (Fig. 2A). Uranium tends to co-precipitate with Fe,35,36 and therefore there may also be an effect of Fe–U co-precipitation at the surface of the sandstone. However thermodynamic modelling including the concentrations of elements released into solution does not predict the precipitation of any Fe–U minerals. Because Fe release increases above pH 9 (Fig. 2A) adsorption on hematite is likely the main cause of U retention envelope below pH 10 as compared with quartz and volcanic rock.
In the experiments with volcanic rock, the sorption envelope of U below pH 8 (region 1) is similar to that of quartz. Of these two, U removal is higher in the volcanic rock between pH 8 and 12. A similar bimodal adsorption pattern has been reported for adsorption of U between pH values of 3 and 11 onto granite.18 Wazne et al.18 assigned this second region of U removal to precipitation of sodium diuranate on the surface of the granite. We see a combination of what we believe is at least two different sodium uranate minerals, according to TEM-EDS analysis, precipitating in our system at high pH. This increase in removal of U from solution onto the volcanic rock at high pH would significantly reduce the mobility of U at high pH values associated with near surface radioactive waste disposal.
Between pH 10.5 and 11.5, at low U concentrations the presence of sandstone leads to more removal of U from bicarbonate-rich solutions than the other rock types (Fig. 7). In contrast, at high U concentrations, volcanic rock and quartz immobilise more U from solution between pH 10.5 and 11.5 (Fig. 7). Regardless of the addition of bicarbonate or type of geomaterial, no U is removed from solution at pH > 12.
To test if U precipitates were remaining dispersed in solution, i.e. were highly mobile, we filtered the solution through a 0.45 μm filter after the 10 ppm batch adsorption experiments. These were chosen as the most likely to produce precipitation. When bicarbonate was added to the system little U was removed from solution as a precipitate larger than 0.45 μm (Fig. 8B). However, when no bicarbonate was added in the range of pH 9–11.5 precipitates contained from nearly 100% to a few % of U, depending on the geomaterial (Fig. 8A). Therefore when bicarbonate was not added to the system there was a significant percentage of highly mobile U precipitates remaining in suspension between pH 8 and 12.
 | ||
| Fig. 8 Percentage of U that was removed from solution by filtration through a 0.45 μm nylon filter after the 10 ppm U batch adsorption experiments with quartz (Qtz), sandstone (SS), and welded tuff (BVG). Sodium bicarbonate not added (A) and added (B). | ||
3.5 Uranium mobility in the near surface environment
Uranium exhibits a bimodal immobilization as a function of pH; with adsorption accounting for most of the immobilization at pH 4–7 and a mixture of adsorption and precipitation between pH 10.5 and 12. While U is predicted to precipitate in high pH environments, it is important to understand the potential mobility of the precipitates. Bots et al.14 saw the precipitation of nano-sized U minerals. These nanomaterials may remain suspended in solution and if so they would be highly mobile in the environment. In our experiments, we observed the precipitation of similar sodium uranate-type minerals, whose aggregation may be pH dependent, with virtually no precipitation observed in our system at pH 12. Modelling of the solutions indicate that precipitation decreases steeply above pH 12 (Fig. 4). If there are any precipitates at pH 12 they may be small enough to pass through our 0.45 μm filters. In the case of sandstone and quartz when no bicarbonate is added, the precipitates remained in water as highly mobile colloids between pH 9 and 12 (Fig. 8). The precipitates in the sandstone experiments may also be co-precipitating as (Na, K) compreignacite, as noted above since there is K released into solution during the batch experiments (Fig. 2). In the case of the volcanic rock there are only minor precipitates collected on the filters between pH 8 and 11, regardless of the addition of bicarbonate to the system. Additionally, phosphate is released into solution at low pH from the volcanic rock, and therefore there may also the precipitation uranyl phosphates. However we see no removal of U from solution in this region, meaning that if a precipitate is formed it is too small to be filtered and remains in solution.Fig. 9 shows a conceptual model of U immobilisation onto quartz, sandstone, and volcanic rock with and without the addition of bicarbonate, as a function of pH. Our model shows that in the immediacy of the repository after the waste material is leached, volcanic rock would perform the best at immobilising U at the expected high pH caused by leaching of the cement. This immobilisation would occur also in the presence of bicarbonate if concentrations of U are ≥10 ppm, despite predictions that U would be more mobile as a carbonate species.8,15,21 In the case of LLW disposal in trenches, where pH is buffered below pH 6 due to cellulose decomposition, the U would adsorb to all geomaterials regardless of U concentration or bicarbonate addition and would therefore be immobile.
 | ||
| Fig. 9 Conceptual diagram showing U immobilised for each geomaterial (quartz, Qtz; sandstone, SS; and volcanic rock, BVG) as a function of pH and bicarbonate addition. The lines on the diagram represent when U is immobilised, with dashed lines representing when U immobilised is concentration dependent. | ||
As the fluids travel away from the repository, the pH would approach the typical pH values of aquifers, which in the case of sandstone and volcanic rocks range between pH 6 and 8.37,38 In this pH range sandstone and volcanic rock would successfully immobilise U from solution, and volcanic rock less so, regardless of the concentration of U. However, if bicarbonate was present in values similar to that in brines (2 mM) there would only be a more limited U immobilisation, high only close to pH 6. At high pH, however there would be U precipitates suspended in solution and it is necessary to assess if these precipitates would be mobiles in the subsurface. Most pores will be larger than 0.45 μm and, in principle, it should be expected that U precipitates are mobile. It is possible, though, that connectivity between pores is low which would reduce some mobility. When bicarbonate is present and the concentration of U is ≥10 ppm, U is more likely to precipitate at the mineral surface further impeding mobility.
4 Conclusions
In our study we examined how variations in pH, concentration, type of geomaterial, and bicarbonate content affect U removal from solution. Quartz, sandstone, and volcanic rock adsorb U onto their surfaces between pH 4 and 8 in sufficient concentrations to be efficient at retaining radioactive U after the failure of a LLW or ILW repository. They are also efficient at retaining U at pH 10–12 where U concentrations are ≥10 ppm, mainly through U precipitation as Na (and sometimes K) precipitates. The presence of bicarbonate, a common ion in groundwaters reduces U retention only in the pH range 8–10, but enhances it at pH 10–12 when U ≥ 10 ppm. These results are important to the understanding of how U may interact and be immobilised in areas where U may escape into environments with similar lithology, and important to predicting the fate of this contaminant.Acknowledgements
We acknowledge the Natural Environment Research Council, Radioactive Waste Management Limited, and Environment Agency for the funding received for this project through the Radioactivity and the Environment (RATE) programme (NE/L000660/1). We thank Dr Mahmoud Ardakani at the Department of Materials at Imperial College London for his help using the TEM. We thank Jennifer Peng for her help with alkalinity determinations and Dr Simon Norris, Alan Hooper, Simon Norris and Rebecca Beard for their insights in writing this manuscript.Notes and references
- A. Bath, H. Richards, R. Metcalfe, R. McCartney, P. Degnan and A. Littleboy, J. Geochem. Explor., 2006, 90, 24–44 CrossRef CAS.
- A. Stockale and N. D. Bryan, Earth-Sci. Rev., 2013, 121, 1–17 CrossRef.
- A. J. Francis, R. Cather and I. G. Crossland, Nirex Science Report, 1997, S/97/014 Search PubMed.
- K. M. Krupa and R. J. Serne, U.S. Nuclear Regulatory Commission, NUREG/CR-6377, 1998, PNNL-11408 Search PubMed.
- R. Cummings and D. Raaz, LLW Repository Ltd Environmental Safety Case LLWR/ESC/R(11)10021, 2011 Search PubMed.
- P. Michard, E. Guibal, T. Vincent and P. Le Cloirec, Microporous Mater., 1996, 5, 309–324 CrossRef CAS.
- J. D. Prikryl, A. Jain, D. R. Turner and R. T. Pabalan, J. Contam. Hydrol., 2001, 47, 241–253 CrossRef CAS PubMed.
- P. M. Fox, J. A. Davis and J. M. Zachara, Geochim. Cosmochim. Acta, 2006, 70, 1379–1387 CrossRef CAS.
- F. Huber and J. Lützenkirchen, Aquat. Geochem., 2009, 15, 443–456 CrossRef CAS.
- K. V. Ticknor, Radiochim. Acta, 1994, 64, 229–236 CrossRef CAS.
- J. A. Berry, A. J. Baker, K. A. Bond, M. M. Cowper, N. L. Jefferies and C. M. Linklater, Geol. Soc. Spec. Publ., 1999, 157, 101–116 CrossRef CAS.
- D. L. Stoliker, D. B. Kent and J. M. Zachara, Environ. Sci. Technol., 2011, 45, 8733–8740 CrossRef CAS PubMed.
- Y. Gao, Z. Shao and Z. Xiao, J. Radioanal. Nucl. Chem., 2015, 303, 867–876 CrossRef CAS.
- P. Bots, K. Morris, R. Hibberd, G. T. W. Law, J. F. W. Mosselmans, A. P. Brown, J. Doutch, A. J. Smith and S. Shaw, Langmuir, 2014, 30, 14396–14405 CrossRef CAS PubMed.
- Q. H. Fan, L. M. Hao, C. L. Wang, Z. Zheng, C. L. Liu and W. S. Wu, Environ. Sci.: Processes Impacts, 2014, 16, 534–541 CAS.
- D. McCubbin and K. S. Leonard, Radiochim. Acta, 1995, 69, 97–102 CrossRef CAS.
- M. Kohler, G. P. Curtis, D. B. Kent and J. A. Davis, Water Resour. Res., 1996, 32, 3539–3551 CrossRef CAS.
- M. Wazne, G. P. Korfiatis and X. Meng, Environ. Sci. Technol., 2003, 37, 3619–3624 CrossRef CAS PubMed.
- G. Meinrath and T. Kimura, J. Alloys Compd., 1993, 202, 89–93 CrossRef CAS.
- S. D. Kellya, K. M. Kemnera and S. C. Brooks, Geochim. Cosmochim. Acta, 2007, 71, 821–834 CrossRef.
- B. D. Stewart, M. A. Mayes and S. Fendorf, Environ. Sci. Technol., 2010, 44, 928–934 CrossRef CAS PubMed.
- S. E. Laubach, R. H. Lander, L. M. Bonnell, J. E. Olson and R. M. Reed, Geol. Soc. Spec. Publ., 2004, 231, 1–9 CrossRef.
- J. Quirke, C. M. B. Henderson, R. A. D. Pattrick, K. M. Rosso, A. Dent, J. W. Sharples and C. I. Pearce, Mineral. Mag., 2015, 1353–1367 CrossRef CAS.
- N. Yee and J. B. Fein, Chem. Geol., 2002, 185, 303–319 CrossRef CAS.
- G. E. Strong, A. E. Milodowski, J. M. Pearce, S. J. Kemp, S. V. Prior and A. C. Morton, Proc. Yorks. Geol. Soc., 1994, 50, 77–89 CrossRef.
- A. E. Milodowski, M. R. Gillespie, J. Naden, N. J. Fortey, T. J. Shepherd, J. M. Pearce and R. Metcalfe, Proc. Yorks. Geol. Soc., 1998, 52, 215–241 CrossRef.
- J. A. Berry, H. E. Bishop, M. M. Cowper, P. R. Fozard, J. W. McMillan and S. A. Mountfort, Analyst, 1993, 118, 1241–1246 RSC.
- D. C. Entwisle, P. R. N. Hobbs, L. D. Jones, D. Gunn and M. G. Raines, Geotechnical and Geological Engineering, 2005, 23, 793–809 CrossRef.
- G. Lefèvre, S. Noinville and M. Fédoroff, J. Colloid Interface Sci., 2006, 296, 608–613 CrossRef PubMed.
- E. R. Sylwester, E. A. Hudson and P. G. Allen, Geochim. Cosmochim. Acta, 2000, 64, 2431–2438 CrossRef CAS.
- J. R. Bargar, R. Reitmeyer, J. J. Lenhart and J. A. Davis, Geochim. Cosmochim. Acta, 2000, 64, 2737–2749 CrossRef CAS.
- H. Churchill, H. Teng and R. M. Hazen, Am. Mineral., 2004, 89, 1048–1055 CrossRef CAS.
- W. Wainipee, J. Cuadros, M. A. Sephton, C. Unsworth, M. G. Gill, S. Strekopytov and D. J. Weiss, Geochim. Cosmochim. Acta, 2013, 121, 487–502 CrossRef CAS.
- M. Kosmulski, Adv. Colloid Interface Sci., 2009, 152, 14–25 CrossRef CAS PubMed.
- M. C. Duff, J. U. Coughlin and D. B. Hunter, Geochim. Cosmochim. Acta, 2002, 66, 3533–3547 CrossRef CAS.
- Z. Niu, Q. Fan, W. Wang, J. Xu, L. Chen and W. Wu, Appl. Radiat. Isot., 2009, 67, 1582–1590 CrossRef CAS PubMed.
- A. H. Bath, R. A. McCartney, H. G. Richards, R. Metcalfe and M. B. Crawford, Q. J. Eng. Geol. Hydrogeol., 1996, 29, S39–S57 CrossRef.
- M. Z. Jaafar, J. Vinogradov and M. D. Jackson, Geophys. Res. Lett., 2009, 36, 1–6 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26773d |
| This journal is © The Royal Society of Chemistry 2017 |