
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-immolative polymers as novel pH-responsive gate keepers for drug delivery†
M. Gisbert-Garzaran ab,
D. Lozanoab,
M. Vallet-Regí*ab and
M. Manzano*ab
ab,
D. Lozanoab,
M. Vallet-Regí*ab and
M. Manzano*ab
aDepartamento de Química Inorgánica y Bioinorgánica, Facultad de Farmacia. Universidad Complutense de Madrid, Instituto de Investigación Sanitaria Hospital 12 de Octubre i + 12, Plaza de Ramón y Cajal s/n, E-28040 Madrid, Spain. E-mail: vallet@ucm.es; mmanzano@ucm.es
bNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Madrid, Spain. Fax: +34 913941786; Tel: +34 913941861
First published on 7th December 2016
Abstract
A novel pH-sensitive nanocarrier based on mesoporous silica nanoparticles with self-immolative polymers blocking the pore openings is presented. Triggered release by acid pH is demonstrated, together with their in vitro biocompatibility and effective cell internalisation, which makes this new material a promising candidate for future applications in cancer treatment.
Nanotechnology, which has the potential to impact nearly every area of modern society, is transforming medicine thanks to the development of new smart drug delivery systems.1–3 Those nanocarriers are expected to bring breakthroughs in terms of detecting, diagnosing and treating different forms of cancer.4 Recently, Mesoporous Silica Nanoparticles (MSNs) have drawn much attention for many biomedical applications, especially as drug delivery nanocarriers.5–10 The reason for this relies on their unique properties, such as a straightforward synthesis, a network of cavities ready available to be filled with different payloads, tuneable pore diameters and morphologies, facile surface functionalisation and hemo- and biocompatibility.11,12
The textural properties of MSNs, such as large surface areas (ca. 1000 m2 g−1) and large pore volume (ca. 1 cm3 g−1), are responsible for their high loading capacities. In fact, conventional polymeric nanoparticles suffer from the drawback of low drug capacity, usually less than 5% of total weight, while MSNs allow much greater values.13,14
However, MSNs present an open structure of the pores, which means that is simple to introduce molecules into their network of cavities, but it is also easy for them to diffuse out. This is the main reason to place gatekeepers on the entrance of the pores, so they can be closed once the cargo is loaded, and then opened to allow the drug release. The former process can be carried out during the production of the nanocarrier, but the later event, opening the gates of the pores, must be done on-demand inside the body. This approach leads to stimuli-responsive nanocarriers,15 in which the release of the cargo is triggered by a stimulus than can be exogenous (temperature, light, magnetic fields or ultrasounds) or endogenous (pH, redox potential or specific enzymes or analytes). Those systems allow tailoring the release profiles with temporal and dosage control.
Among the available stimuli, pH variations offer the possibility of controlling the delivery of drugs to different regions of the body such as the gastrointestinal tract, the endosomes or lysosomes, or the tumour microenvironment. In particular, the slightly acidic lysosomal pH value (4.5–5)16 has been used for the design of pH-targeted nanocarriers that release their cargo once inside the cell.17,18 Then, an efficient pH-responsive system should respond to this subtle change of pH within this organelle, triggering the release of the cargo only inside the cells (Scheme 1).
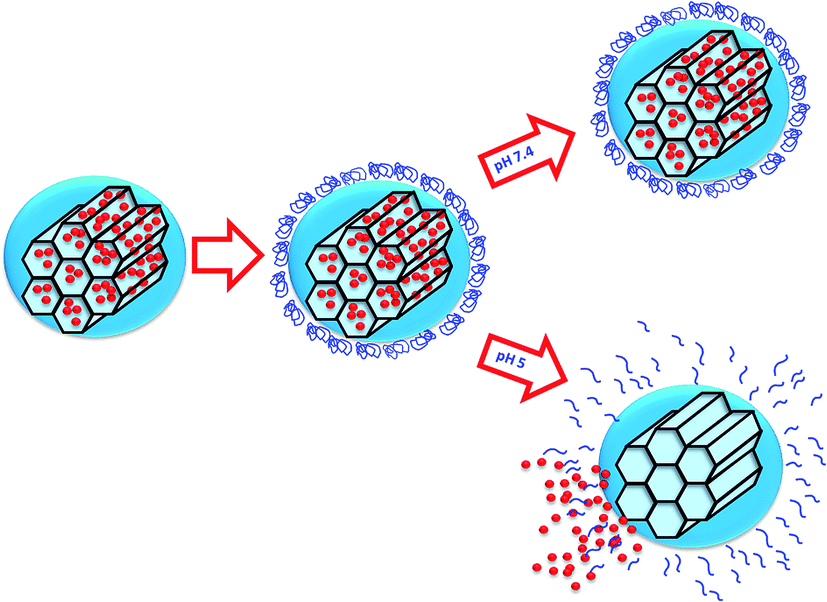 | ||
| Scheme 1 Representation of the operating principle of our pH sensitive nanocarrier. A change in the pH initiates the disassembling of the self-immolative polymer allowing cargo molecules to diffuse out of the MSNs. | ||
There are many polymeric systems with acid-sensitive bonds that can be exploited for designing responsive systems. In this sense, there are a new class of polymers, the so-called Self-Immolative Polymers (SIPs), that disassemble from head to tail completely when a specific functional group is cleaved from the polymer in response to certain stimuli.19,20 One potential advantage is that it is possible to incorporate prodrugs into the polymer chain as pendant groups. These groups would turn into actual drugs as the self-immolation takes place.21 It is also possible to modify the rate at which the chain disassembles to achieve a more controlled and sustained drug release.22 There are many triggers that can initiate the fragmentation of the polymer into its building blocks, depending on the molecule placed at the end of the polymer. Thus, it is possible to design a linear polymer based on a polyurethane backbone with a pH sensitive capping molecule as trigger. Dropping the pH would cleavage the carbamate linkage of the trigger, which would start the sequential 1–6 elimination and decarboxylation reactions yielding CO2 and the starting monomer.
Combining both materials, MSNs and SIPs, in a single functional responsive nanocarrier would provide access to the benefits of both materials, i.e., the high loading capacity of a variety of cargoes into MSNs, and the responsiveness of pH-sensitive SIPs to control the release. Decoration of the MSNs pore entrances with pH-responsive SIP gatekeepers that avoid premature release of the cargo and unblock the pores upon pH drops, represent a new and promising concept that is explored here for the first time (Scheme 2).
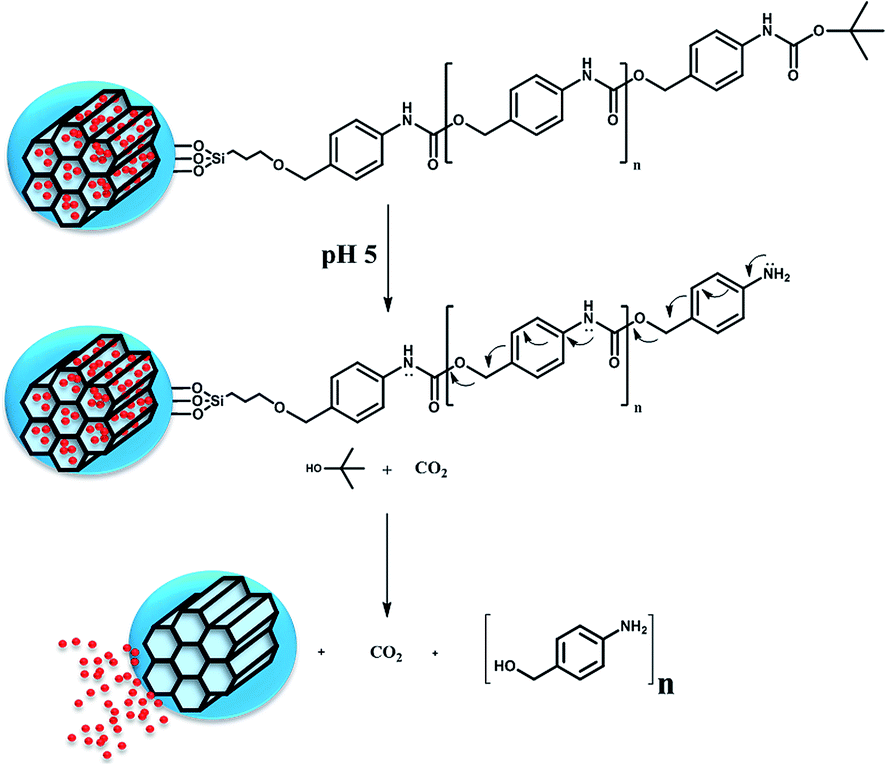 | ||
| Scheme 2 Schematic representation of the nanocarrier response to acid pH. Cleavage of the protecting group generates an intermediate that undergoes sequential 1,6-elimination and decarboxylation reactions to form the initial monomer, disassembling the immolative polymer and allowing the cargo release. | ||
In this work we have developed a smart nanocarrier able to respond to changes in the pH for potential applications in cancer treatment. For this purpose, we have selected MSNs as carriers and a SIP with a trigger sensitive to acid pH to block the pore entrances and prevent premature release. The pH sensitivity of the nanosystem was evaluated in vial showing that cargo release took place in acidic conditions, while at physiological pH there was almost no release. The biocompatibility of the system was evaluated with different cells, and internalisation of the nanoparticles was also demonstrated.
MSNs were synthesised via sol–gel method under basic conditions using hexadecyltrimethylammonium bromide (CTAB) as structure directing agent following a previously published method.23 The sol–gel method allows a fine control on the final composition of the nanoparticles under mild conditions.24,25 The spherical morphology of the as-produced MSNs was confirmed through Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM), as can be observed in Fig. 1a and b, with average sizes of ca. 150 nm (see ESI, Fig. S3† with Dynamic Light Scattering, DLS, studies).
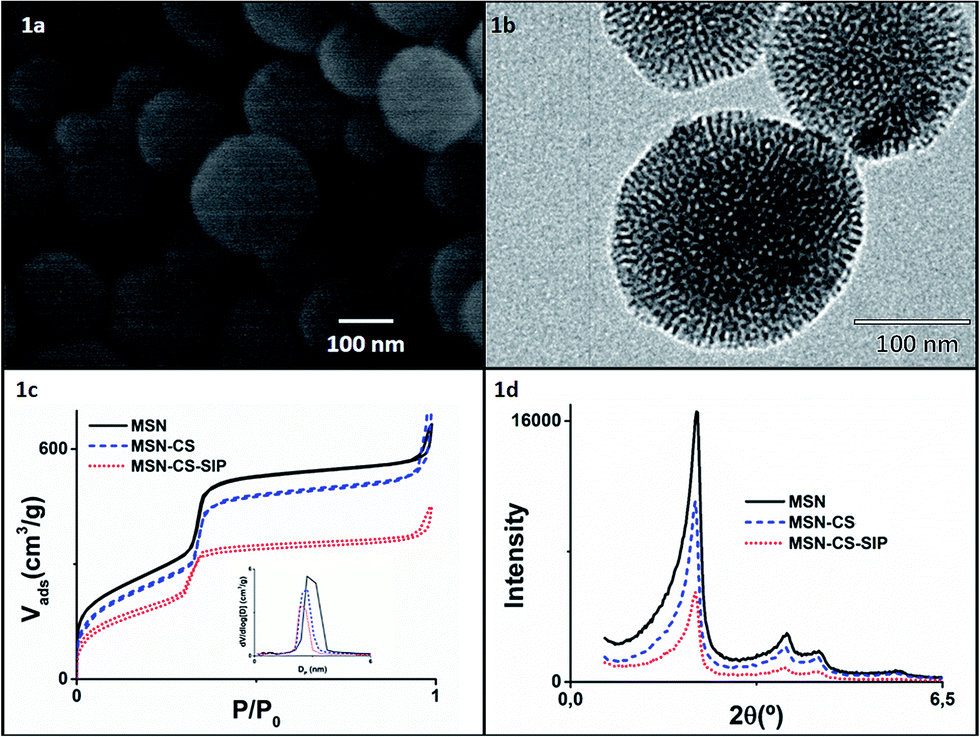 | ||
| Fig. 1 (a) SEM images of MSNs; (b) TEM images of MSNs; (c) nitrogen adsorption and desorption isotherms of MSN (black solid line), MSN–CS (blue dashed line) and MSN–CS–SIP (red dotted line) (inset: pore size distribution determined by the BJH method from the desorption branch of the isotherm); (d) XRD patterns of MSN (black solid line), MSN–CS (blue dashed line) and MSN–CS–SIP (red dotted line). | ||
Typical adsorption–desorption isotherms of MCM-41-like materials with cylindrical pores were observed (Fig. 1c), with pore diameters centred at ca. 2.7 nm. The characteristic hexagonal arrangement of the pores was confirmed through low angle X-Ray Diffraction (XRD) analyses (Fig. 1d), where typical well resolved maxima from well-ordered 2D-hexagonal structure with p6mm space group were observed.
A linear self-immolative polymer was produced to cover the pore entrances and avoid premature release of the cargo. We employed a polyurethane backbone with a BOC protecting group on the terminal amine that acted as a trigger. A pH drop would cleave the protecting group and initiate the disassembly of the polymer. The SIP was synthesised by a modification of a previously published method (Fig. 2a),19 producing the monomer 1 from 4-aminobenzyl alcohol and phenyl chloroformate. Then, the polymerisation process using DBTL as catalyst and 2 as end-group leaded to the polymeric chain 3 with an average of about 20 monomers (determined by NMR, through the ratio benzylic hydrogens at the molecule tail vs. those in the polymeric backbone).
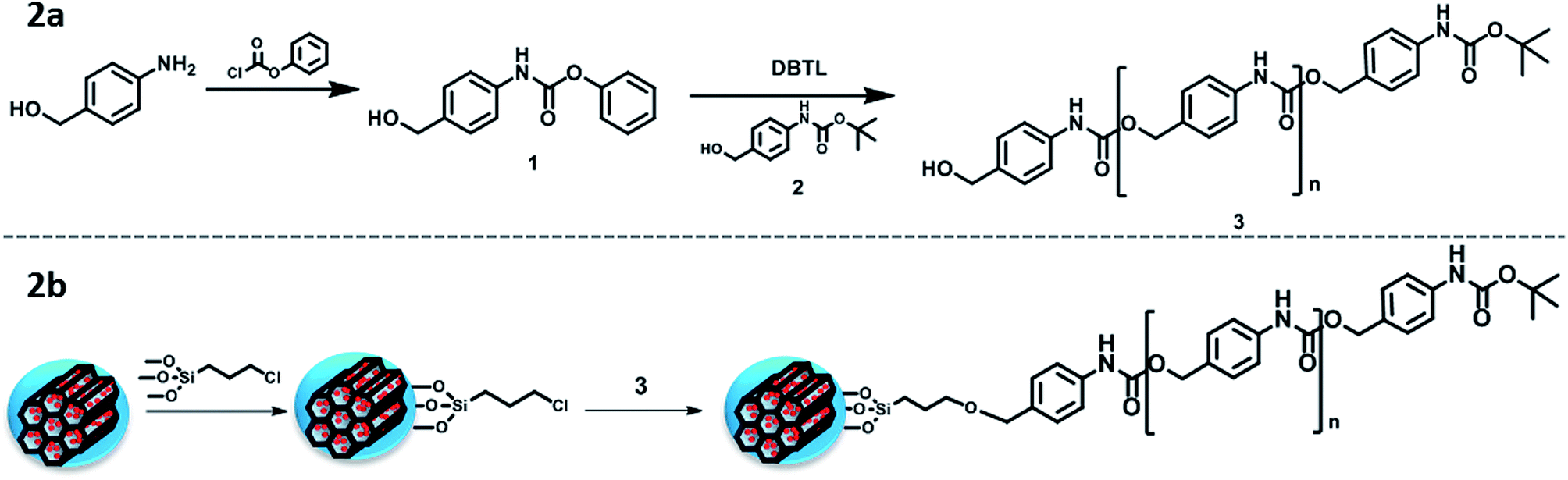 | ||
| Fig. 2 (a) Schematic representation of the SIP synthesis; (b) schematic representation of the SIP grafting to the MSNs. | ||
The self-immolative behaviour of the SIP was confirmed by treating the polymer with trifluoroacetic acid in CH2Cl2, which are the standard conditions for removing BOC protecting groups. After the acid treatment, all characteristic NMR peaks of SIP had disappeared (Fig. S8†), as expected.
The successful attachment of the SIP to the MSNs was achieved through a previous modification of the nanoparticles with an alkoxysilane bearing chloride groups at the other end (denoted CS) (Fig. 2b). Those modified-particles (MSN–CS) were characterised through thermogravimetric (TG) analysis (Fig. S1†), where the increase of weight loss confirmed the presence of organic matter, together with elemental analysis (Table 1), where the increment of C% verifies the linker grafting. The C–H stretching bands from Fourier transform infrared spectroscopy (FTIR, Fig. S2†) also confirmed the presence of the linker.
| Material | MSN | MSN–CS | MSN–CS–SIP |
|---|---|---|---|
| Organic matter by TG (%) | 5.3 | 14.4 | 20 |
| Elemental analysis (%) | C: 3.95 | C: 9.34 | C: 10.76 |
| H: 2.09 | H: 2.34 | H: 2.46 | |
| N: 0.06 | N: 0.11 | N: 0.53 | |
| Surface area (m2 g−1) | 1053 | 914 | 671 |
| Pore volume (cm3 g−1) | 1.05 | 1.02 | 0.60 |
| Pore width (nm) | 2.74 | 2.66 | 2.19 |
| Size (nm); PDI | 190; 0.467 | 190; 0.467 | 190; 0.358 |
| Zeta potential (mV) | −35.3 | −29.0 | −23.8 |
Addition of polymer 3, previously activated with DIPEA, to the MSN–CS leaded to SIP coating (MSN–CS–SIP). XRD analyses after functionalisation (Fig. 1d) showed that the ordered mesostructure survived the process, although the progressive reduction of the intensity might be ascribed to the partial filling of the pores by some polymer chains, as it was previously observed when functionalising MSNs with large dendrimers.26 The successful SIP coating onto MSNs was confirmed through different characterisation techniques. The great weight loss observed in TG analyses (Fig. S1†) is indicative of the great amount of SIP present. FTIR spectrum of MSN–CS–SIP (Fig. S2†) exhibits additional bands to those typical from silica materials at ca. 1650 cm−1, typical from C–O stretching vibrations from carbamates, verifying the presence of the polymer. This is in agreement with the data obtained from N2 analyses (Table 1), in which the reduction of the surface area and pore volume is indicative of the presence of the polymer blocking the pore entrances. Finally, the increase of N% in the elemental analysis in MSN–CS–SIP confirms the presence of the polymer.
Finally, the changes in the zeta potential to less negative values confirm the correct functionalisation. However, due to the small molecular weight of SIP the size was not affected by the functionalisation, either at pH 7.4 or pH 5.
After confirmation of the polymer grafting on the surface of MSN, drug delivery capabilities of the system were evaluated. Fluorescent cargo, tris(2,2′-bipyridine)dichloro ruthenium(II) denoted as Ru, was loaded into the network of cavities of the mesoporous nanoparticles overnight before the blocking the pore entrances with the SIP polymer (for further details see ESI†). The successful Ru loading was confirmed by the decrease in the pore volume measured through N2 adsorption (data not shown).
The responsiveness to pH of the system here developed was evaluated through an in vial Ru release from sample MSN–CS–SIP at 37 °C using Transwells at different pHs, i.e. 7.4 and 5. The data from the release kinetics, Fig. 3, showed that there was almost no cargo release at physiological pH, which means that the nanocarrier would travel through the blood stream without premature release of the payload. However, when the pH of the solution dropped to pH 5, there was a fast release of the loaded Ru, as it can be observed in Fig. 3. This behaviour confirmed that the polymer was disassembling as a consequence of the acid pH, opening the pore entrances and favouring the release of their cargo only at acid environments.
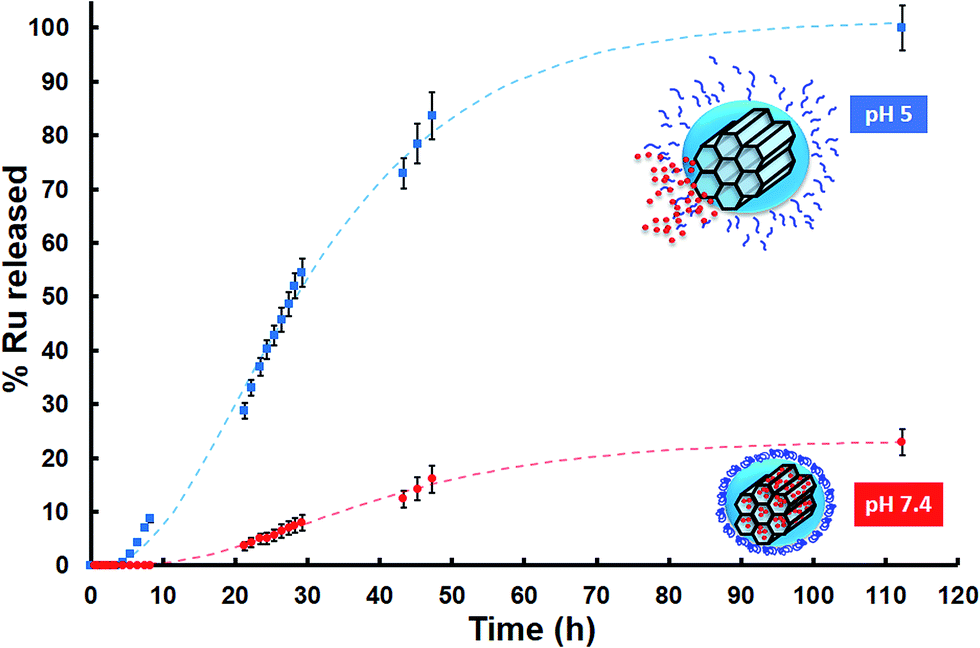 | ||
| Fig. 3 In vial cumulative Ru release (in percentage) vs. time from MSN–CS–SIP at physiological and acid pHs (repeated 3 times each measurement; Standard Error of the Mean (SEM)). | ||
Moreover, after being exposed to acid pH, the zeta potential of MSN–CS–SIP increased from −23.8 mV to −10.7 mV. That would confirm that the self-immolation actually took place, as its residue is a molecule of 4-aminobenzyl alcohol.
Once the MSNs covered with the SIPs nanocarrier were fully characterised and their pH-sensitive release capabilities demonstrated in vial, the next step was analysing their behaviour with cells. The in vitro cytotoxicity study was determined by the exposition of MC3T3-E1 pre-osteoblastic cells to different amounts of nanoparticles (50, 100, 200 and 300 μg mL−1). Fig. 4 shows that none of the studied concentrations, except 300 μg mL−1, induced significant cytotoxicity measured by an Alamar Blue assay (cell viability was >99% that of the control). Similar results were obtained in LNCaP tumoral cells (Fig. S9†).
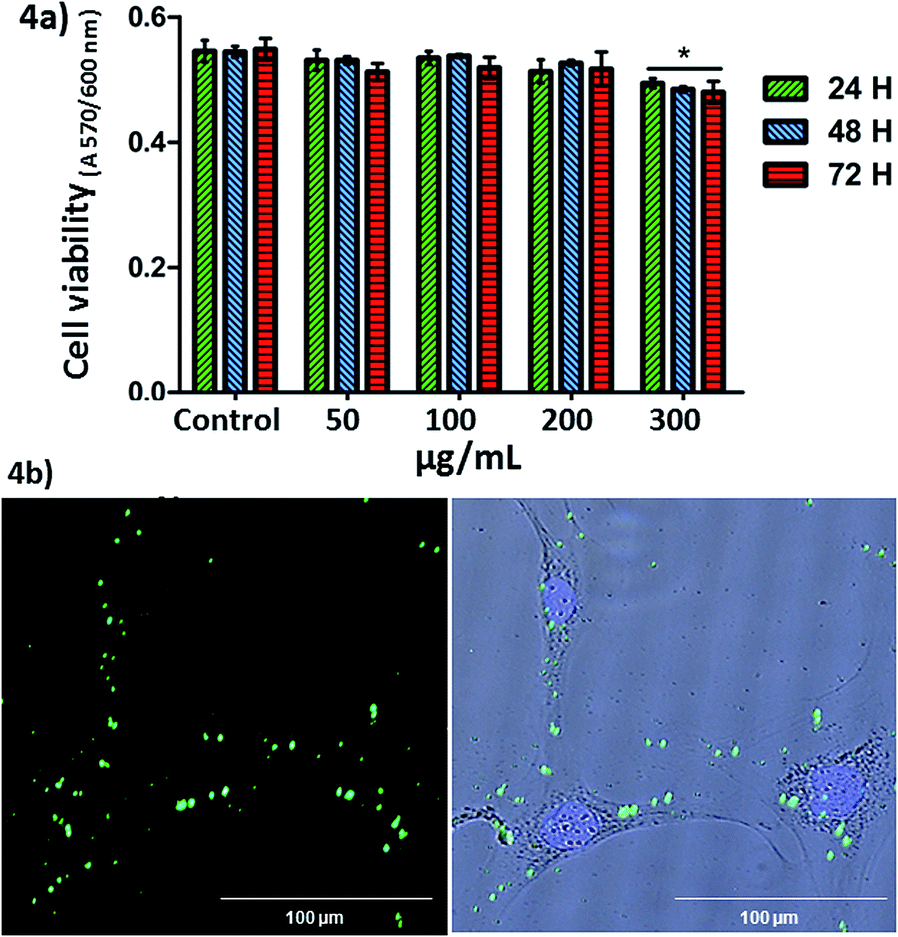 | ||
| Fig. 4 (a) MC3T3-E1 cell viability in contact with different concentrations of nanoparticles at 24, 48 and 72 h of cell culture. Similar proliferation results were obtained in LNCaP cells. *p < 0.05 vs. control without nanoparticles (student's t-test). (b) Fluorescence microscopy images of LNCaP cells incubated with nanoparticles at 2 hours of cell culture. From left to right: green fluorescence (nanoparticles with fluorescein) and overlay image with blue fluorescence (nuclei). | ||
Cellular internalisation of the nanoparticles was observable by fluorescence microscopy and analysed by flow cytometry in LNCaP cells in contact with the nanoparticles (100 μg mL−1) for 2 hours. No fluorescence was observed in the end slices corresponding to the external cellular surfaces, suggesting that the nanoparticles did not adsorb on the cell membranes. Therefore, we observed that the nanoparticles at 100 μg mL−1 were internalised by LNCaP cells. The cellular uptake observed by fluorescence microscopy was also confirmed by flow cytometry (19.25 ± 1.5% of nanoparticles incorporation) in the same cells, at 2 hours. The low cellular internalisation would be in agreement with the fact that positively charged27 or targeted28 nanocarriers show better cellular internalization than those negative, as in the case of MSN–CS–SIP.
Conclusions
In conclusion, we have developed for the first time mesoporous silica nanoparticles capped with acid responsive self-immolative polymers. The cargo release can be triggered by acid pH, typical from tumours microenvironments. The biocompatibility of this new delivery nanoplatform has been demonstrated with different cell types, and the effective nanoparticle internalization has been achieved in tumoral cells. We envision that the synergy from MSNs carriers and SIPs could be of a great interest for future applications in nanomedicine to fight against cancer.Acknowledgements
The authors thank funding from the EU H2020-NMP-PILOTS-2015 program through the grant no. 685872 (MOZART) and the European Research Council (Advanced Grant VERDI; ERC-2015-AdG Proposal no. 694160).Notes and references
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed
.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed
- R. Misra, S. Acharya and S. K. Sahoo, Drug Discovery Today, 2010, 15, 842–850 CrossRef CAS PubMed
- A. Baeza, M. Manzano, M. Colilla and M. Vallet-Regí, Biomater. Sci., 2016, 4, 803–813 RSC
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS PubMed
- I. I. Slowing, J. L. Vivero-Escoto, B. G. Trewyn and V. S.-Y. Lin, J. Mater. Chem., 2010, 20, 7924–7937 RSC
- M. Manzano and M. Vallet-Regí, J. Mater. Chem., 2010, 20, 5593–5604 RSC
- B. G. Trewyn, I. I. Slowing, S. Giri, H. Chen and V. S. Lin, Acc. Chem. Res., 2007, 40, 846–853 CrossRef CAS PubMed
- J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S.-Y. Lin, Small, 2010, 6, 1952–1967 CrossRef CAS PubMed
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS
- Y. Zhang, Z. Zhi, T. Jiang, J. Zhang, Z. Wang and S. Wang, J. Controlled Release, 2010, 145, 257–263 CrossRef CAS PubMed
- A. Baeza, M. Colilla and M. Vallet-Regí, Expert Opin. Drug Delivery, 2015, 12, 319–337 CrossRef CAS PubMed
- T. Kolter and K. Sandhoff, FEBS Lett., 2010, 584, 1700–1712 CrossRef CAS PubMed
- Z. Li, H. Li, L. Liu, X. You, C. Zhang and Y. Wang, RSC Adv., 2015, 5, 77097–77105 RSC
- Y. Yan, J. Fu, X. Liu, T. Wang and X. Lu, RSC Adv., 2015, 5, 30640–30646 RSC
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, J. Am. Chem. Soc., 2008, 8, 5434–5435 CrossRef PubMed
- W. Wang and C. Alexander, Angew. Chem., Int. Ed., 2008, 47, 7804–7806 CrossRef CAS PubMed
- S. Gnaim and D. Shabat, Acc. Chem. Res., 2014, 47, 2970–2984 CrossRef CAS PubMed
- R. A. McBride and E. R. Gillies, Macromolecules, 2013, 46, 5157–5166 CrossRef CAS
- A. Baeza, E. Guisasola, A. Torres-Pardo, J. M. González-Calbet, G. J. Melen, M. Ramirez and M. Vallet-Regí, Adv. Funct. Mater., 2014, 24, 4625–4633 CrossRef CAS
- A. S. Timin, A. R. Muslimov, K. V. Lepik, N. N. Saprykina, V. S. Sergeev, B. V. Afanasyev, A. D. Vilesov and G. B. Sukhorukov, J. Mater. Chem. B, 2016, 18, 33–37 Search PubMed
- A. S. Timin, H. Gao, D. V. Voronin, D. A. Gorin and G. B. Sukhorukov, Adv. Mater. Interfaces, 2016 DOI:10.1002/admi.201600338
- B. Gonzalez, M. Colilla, C. L. de Laorden and M. Vallet-Regi, J. Mater. Chem., 2009, 19, 9012–9024 RSC
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed
- K. Ulbrich, K. Holá, V. Šubr, A. Bakandritsos, J. Tuček and R. Zbořil, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26771h |
| This journal is © The Royal Society of Chemistry 2017 |