
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring opening polymerization of lactides and lactones by multimetallic alkyl zinc complexes derived from the acids Ph2C(X)CO2H (X = OH, NH2)†
Yahya F. Al-Khafajia,
Mark R. J. Elsegood b,
Josef W. A. Freseb and
Carl Redshaw*a
b,
Josef W. A. Freseb and
Carl Redshaw*a
aSchool of Mathematics and Physical Sciences, The University of Hull, Cottingham Rd, Hull, HU6 7RX, UK. E-mail: C.Redshaw@hull.ac.uk
bChemistry Department, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK
First published on 16th January 2017
Abstract
The reaction of the dialkylzinc reagents R2Zn with the acids 2,2-Ph2C(X)(CO2H), where X = NH2, OH, i.e. 2,2′-diphenylglycine (dpgH) or benzilic acid (benzH2), in toluene at reflux temperature afforded the tetra-nuclear ring complexes [RZn(dpg)]4, where R = Me (1), Et (2), 2-CF3C6H4 (3), and 2,4,6-F3C6H2 (4); complex 2 has been previously reported. The crystal structures of 1·(2MeCN), 3 and 4·(4(C7H8)·1.59(H2O)) are reported, along with that of the intermediate compound (2-CF3C6H4)3B·MeCN and the known compound [ZnCl2(NCMe)2]. Complexes 1–4, together with the known complex [(ZnEt)3(ZnL)3(benz)3] (5; L = MeCN), have been screened, in the absence of benzyl alcohol, for their potential to act as catalysts for the ring opening polymerization (ROP) of ε-caprolactone (ε-CL), δ-valerolactone (δ-VL) and rac-lactide (rac-LA); the co-polymerization of ε-CL with rac-LA was also studied. Complexes 3 and 4 bearing fluorinated aryls at zinc were found to afford the highest activities.
Introduction
The last decade has seen a continued interest in the development of catalytic systems for the production of biodegradable polymers, typically via the ring opening polymerization (ROP) of cyclic esters.1 This interest stems from the use of polycaprolactone (PCL) and polylactic acid (PLA) in the packaging and medical (as implants) industries.2 Furthermore, given the differing properties that can be associated with both PCL and PLA, there is interest in ε-Cl/LA copolymers of varying compositions; low molecular weight ε-Cl/LA copolymers have been employed as bio-adhesives.3In terms of control, as well as judicious choice of metal centre, it has become clear that the ligand set can also play a pivotal role in determining the catalytic behaviour of the ROP system. Chelating N,O- or O,O-ligand sets have shown great potential.1 With this in mind, we have been investigating the use of acids containing the motif Ph2C(X), where X = OH, NH2, i.e. benzilic acid (X = OH) and diphenylglycine (X = NH2), which is known to impart crystallinity.4 A search of the CSD revealed 33 hits for the Ph2C(N)(CO2) moiety, the majority of which were organic in nature; a chart of the non-organic structures is given in the ESI (Chart S1†).5 We have recently screened a number of these systems for the ROP of cyclic esters, in particular alkylaluminium species,6 and a number of complexes comprising LixOy rings and ladders7 have revealed reasonable activity. Given that zinc complexes have also shown promise over the years as ROP pre-catalysts,8 we have now turned our attention to alkylzinc complexes incorporating the Ph2C(X) motif. We have previously published a number of intriguing molecular structures derived from these acids and alkylzinc and aluminium reagents including tetra-, hexa- and octanuclear ring systems,9 however the ROP capability of such systems was not examined. Herein, we extend the family of complexes available by employing zinc reagents where R = Me or a fluorinated aryl group (see Scheme 1), and assess their ability as catalysts towards the ROP of ε-caprolactone (ε-CL), δ-valerolactone (δ-VL) and rac-lactide (rac-LA); the co-polymerization of ε-CL with rac-LA has also been studied.
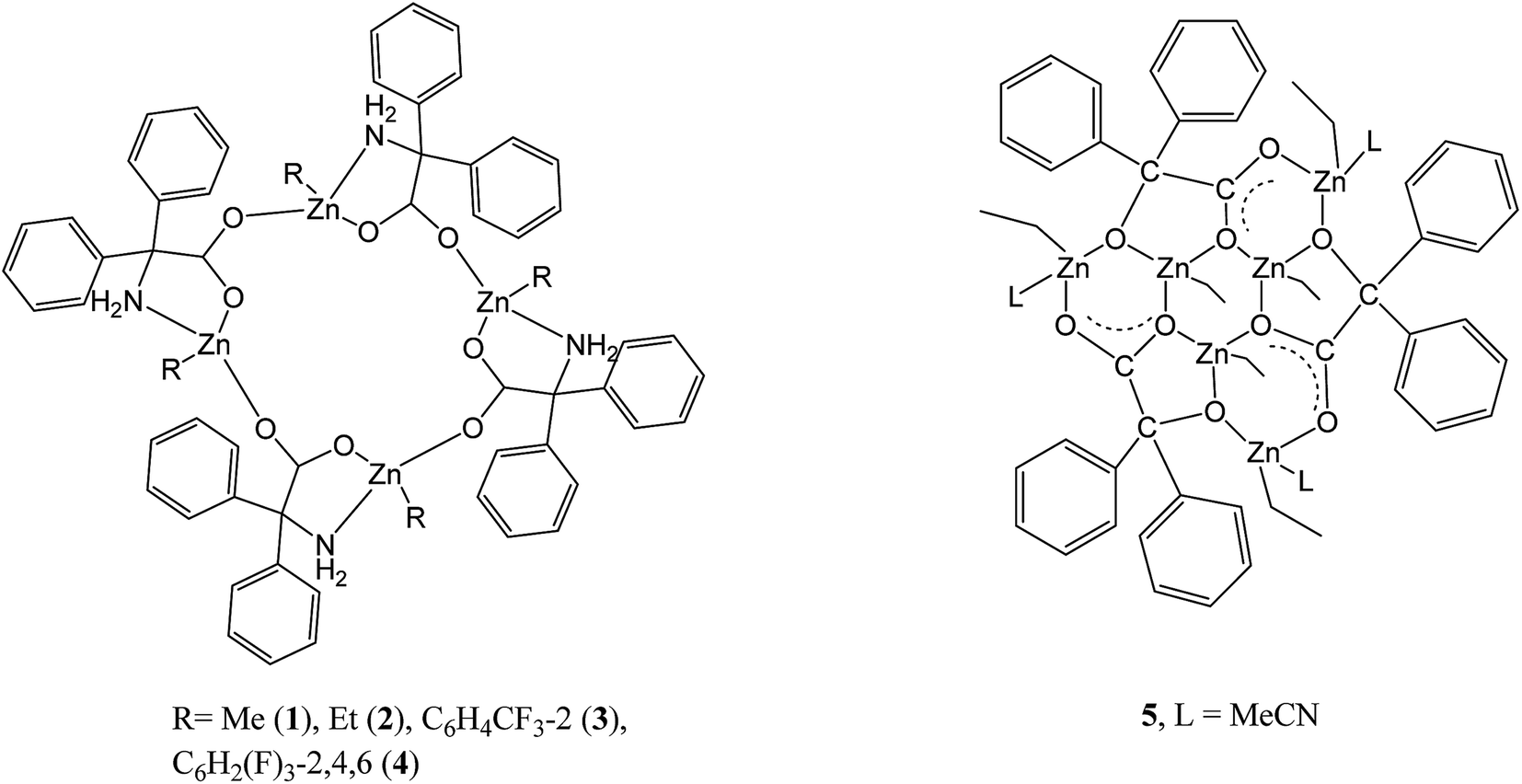 | ||
| Scheme 1 Organozinc complexes 1–5 studied herein. | ||
Results and discussion
Use of 2,2′-diphenylglycine (dpgH)
Using our previously published procedure,9b we here reacted Me2Zn (2.1 equiv.) with dgpH and following work-up, the resulting white solid was characterized via elemental analysis, IR spectroscopy with νNH stretches at 3345 and 3233 cm−1, and by 1H NMR spectroscopy with doublets at 3.02 and 5.79 ppm assigned to exo and endo NH2 protons respectively. Single crystals suitable for X-ray diffraction were grown from a saturated solution of acetonitrile. The molecular structure of 1·2MeCN is shown in Fig. 1 (for an alternative view of the core of the molecule see Fig. S1, ESI†), with selected bond lengths and angles given in the caption. As for the ethyl derivative [EtZn(dpg)]4 (2), each zinc is four coordinate with a Ph2C(NH2)-CO2 ligand binding in N,O fashion, whilst the other oxygen of this ligand binds to an adjacent zinc. The result is a 16-membered ring adopting an up-down–up-down conformation, in which the carboxylates all bind in anti/syn fashion. One of the NH2 groups is involved in an intramolecular H-bond to a neighbouring dpg carboxylate oxygen, whilst another forms an H-bond to an acetonitrile. In terms of packing, there are two channels containing acetonitriles that run parallel to b and c (see Fig. S2, ESI†).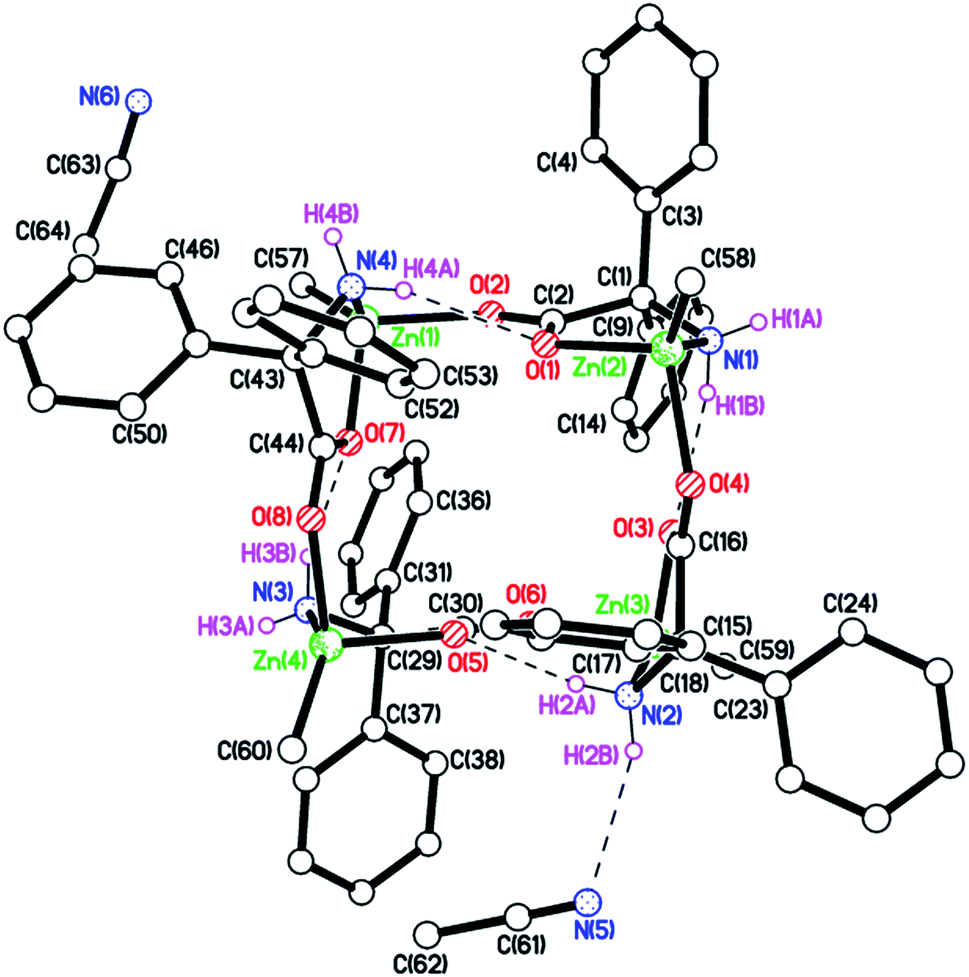 | ||
| Fig. 1 Molecular structure of 1. Selected bond lengths (Å) and angles (°): Zn(1)–O(2) 2.017(2), Zn(1)–O(7) 2.056(3), Zn(1)–N(4) 2.090(3), Zn(1)–C(57) 1.958(4), Zn(2)–O(1) 2.090(3), Zn(2)–O(4) 2.029(3), Zn(2)–N(1) 2.083(3), Zn(2)–C(58) 1.937(5); Zn(1)–O(2)–C(2) 132.0(2), Zn(1)–O(7)–C(44) 117.1(2), Zn(1)–N(4)–C(43) 109.2(2), Zn(2)–O(1)–C(2) 115.7(2), Zn(2)–O(4)–C(16) 130.5(2), Zn(2)–N(1)–C(1) 110.4(2). | ||
| Run | Cat | Monomer | [Monomer]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cat:BnOH :Cat:BnOH |
Time | Conva (%) | Mn,GPCb | Mn,Calc | PDId |
|---|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy.b Mn,GPC values corrected considering Mark–Houwink factors (0.56 poly(ε-caprolactone), 0.58 (polylactide)) from polystyrene standards in THF.c Calculated from ([monomer]0/[cat]0) × conv (%) × monomer molecular weight.d From GPC. | ||||||||
| 1 | 1 | ε-CL | 150:1:0 |
1 | 74 | 9580 | 12670 |
1.37 |
| 2 | 2 | ε-CL | 150:1:0 |
1 | 82 | 10600 |
14040 |
1.31 |
| 3 | 3 | ε-CL | 150:1:0 |
1 | 90 | 12840 |
15400 |
1.40 |
| 4 | 4 | ε-CL | 150:1:0 |
1 | 92 | 12900 |
15750 |
1.31 |
| 5 | 5 | ε-CL | 150:1:0 |
1 | 84 | 10800 |
14380 |
1.10 |
| 6 | 1 | rac-LA | 100:1:0 |
12 | 55 | 4550 | 7930 | 1.88 |
| 7 | 2 | rac-LA | 100:1:0 |
12 | 57 | 5010 | 8220 | 1.25 |
| 8 | 3 | rac-LA | 150:1:0 |
12 | 66 | 7030 | 14270 |
1.51 |
| 9 | 4 | rac-LA | 100:1:0 |
12 | 67 | 6150 | 9660 | 1.23 |
| 10 | 5 | rac-LA | 100:1:0 |
12 | 61 | 5970 | 8790 | 1.27 |
| 11 | 1 | δ-VL | 100:1:0 |
24 | 42 | 1300 | 4210 | 2.12 |
| 12 | 2 | δ-VL | 100:1:0 |
24 | 50 | 1810 | 5010 | 2.18 |
| 13 | 3 | δ-VL | 100:1:0 |
24 | 54 | 2810 | 5410 | 1.14 |
| 14 | 4 | δ-VL | 100:1:0 |
24 | 58 | 2670 | 5810 | 1.13 |
| 15 | 5 | δ-VL | 100:1:0 |
24 | 51 | 1740 | 5110 | 1.73 |
When the reagent [EtZnCl]10 was reacted with dpgH, the only crystalline material isolated following work-up was the known compound [ZnCl2(NCMe)2]. Although the structure has been previously reported, both of the previous data collections were conducted at room temperature and neither refinement included H atoms.11 We have thus presented our improved, low temperature structure of [ZnCl2(NCMe)2] in the ESI (Fig. S3 and S4†), which reveals a layer structure.
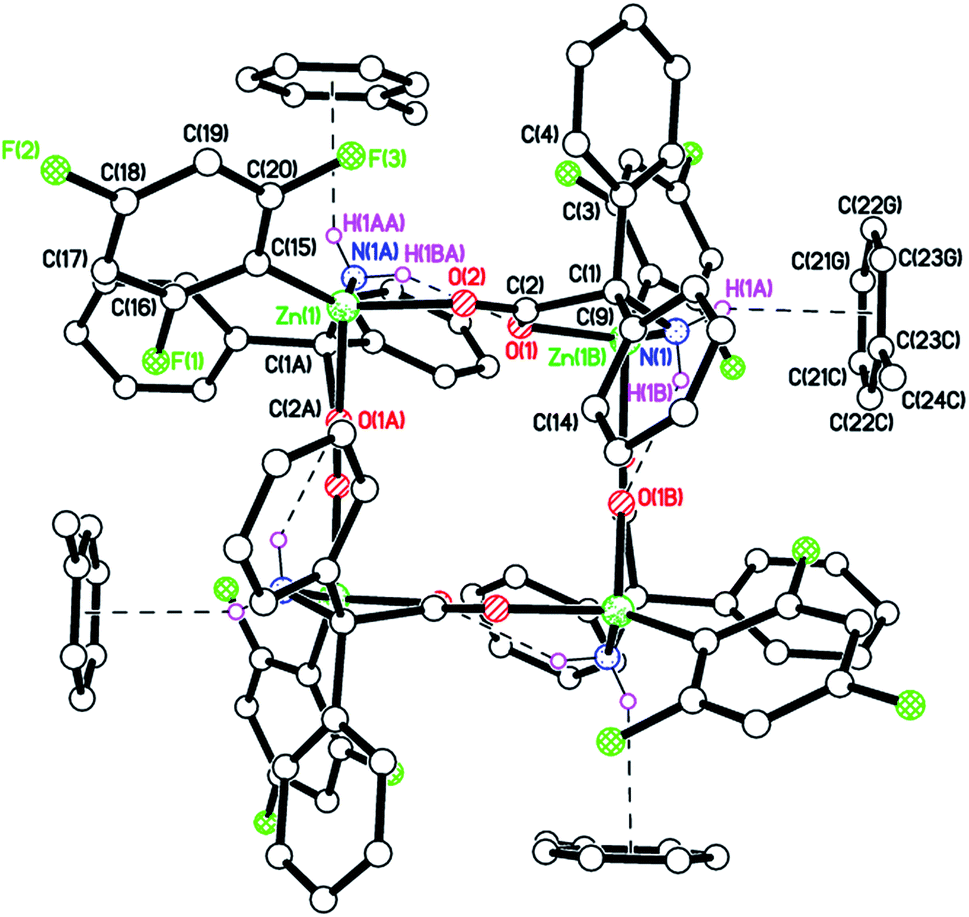
Similar use of (2-CF3C6H4)2Zn led to the isolation of [(2-CF3C6H4)Zn(dpg)]4 (3) in good yield (68%). The product was characterized by IR spectroscopy with νNH stretches at 3306, 3242 and 3172 cm−1, and by 1H NMR spectroscopy with doublets at 3.02 and 5.79 ppm assigned to exo and endo NH2 protons respectively. Crystals suitable for X-ray diffraction were grown from a saturated solution of acetonitrile at ambient temperature. The structure, shown in Fig. 2 (for an alternative view see Fig. S5, ESI†), was crystallographically challenging in that it was both merohedrally twinned via the twin law 010 100 00−1 (major component 54(2)%), and racemically twinned (Flack parameter 0.472(2)). A structure solution was only possible after temporary ‘de-twinning’ of the reflection data. The full data and the twin law were then used for the structure refinement.
 | ||
| Fig. 2 Molecular structure of 3 with dpg Ph groups omitted for clarity. Selected bond lengths (Å) and angles (°): Zn(1)–O(2) 1.993(7), Zn(1)–O(1A) 2.049(6), Zn(1)–N(1A) 2.091(8), Zn(1)–C(15) 1.966(10), Zn(1B)–O(1) 2.048(6), Zn(1B)–N(1) 2.091(8) Zn(1)–C(15)–C(16) 125.9(9), Zn(1)–C(15)–C(20) 118.5(8), Zn(1B)–O(1)–C(2) 116.6(6), Zn(1B)–N(1)–C(1) 109.7(5). Symmetry codes: A = y + 1/2, −x + 1/2, −z + 1/2; B = −y + 1/2, x − 1/2, −z + 1/2. | ||
One quarter of the Zn4 complex is unique, and the zinc centres are arranged up-down-up-down with ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) symmetry in the macrocycle. There is no solvent of crystallization; the packing is shown in Fig. S6, ESI.†
symmetry in the macrocycle. There is no solvent of crystallization; the packing is shown in Fig. S6, ESI.†
Extending the methodology to the use of (2,4,6-F3-C6H2)2Zn led to the isolation of the complex [(2,4,6-F3-C6H2)Zn(dpg)]4 (4) in moderate yield (ca. 40%), for which the IR spectrum contained νNH stretches at 3318 and 3172 cm−1, and the 1H NMR spectrum doublets at 3.43 and 5.38 ppm assigned to exo and endo NH2 protons respectively. Single crystals were grown from a saturated solution of toluene at ambient temperature. The molecular structure is shown in Fig. 3 (for an alternative view see Fig. S7, ESI†), with selected bond lengths and angles given in the caption. The asymmetric unit comprises one quarter of [(2,4,6-F3-C6H2)Zn(dpg)]4·4(C7H8)·1.59(H2O) given the symmetry. One of the NH protons H-bonds to the Zn-bound O(1) carboxylate atom, while the other forms a C–H⋯π interaction to the toluene of crystallisation with the distance from the ring centroid to the H atom being 2.549 Å. The core structure is very similar to that in described in 1 above. In the packing of 4 (see Fig. S8, ESI†), there are some weak intermolecular C–H⋯F interactions between 2,4,6-F3-C6H2 groups and the toluene molecule, with the toluene bridging two 2,4,6-F3-C6H2 groups.
 | ||
| Fig. 3 Molecular structure of 4. Selected bond lengths (Å) and angles (°): Zn(1)–O(2) 2.008(2), Zn(1)–O(1A) 2.0304(19), Zn(1)–N(1A) 2.065(2), Zn(1)–C(15) 1.965(3); Zn(1)–O(2)–C(2) 124.62(18), Zn(1B)–N(1)–C(1) 111.26(16). Symmetry codes: A = −y + 1, x, −z + 1; B = y, −x + 1, −z + 1. | ||
Use of benzilic acid
For comparative studies, the previously reported complex [(ZnEt)3(ZnL)3(benz)3] (5; L = MeCN) has also been prepared.9bMolecular structure of tris(boron) intermediate
During the preparation of (2-CF3C6H4)2Zn, we isolated and structurally characterized the acetonitrile solvate of (2-CF3C6H4)3B(NCMe). This intermediate was prepared using a modification of the synthesis of B(C6F5)3 reported by Lancaster;12 subsequent treatment with Et2Zn affords (C6F5)2Zn, i.e. C6F5 transfer. However, herein we limited the scale of this preparation (<5 g) and conducted it behind a safety shield given the precedent for explosions when using halogeno(trifluoromethyl)benzenes for Grignard synthesis.13 We note that Ashley and coworkers have employed i-PrMgCl for metal halogen exchange with 1-bromo-3,5-bis(trifluoromethyl)benzene, which was subsequently reacted with BF3·OEt2 to afford tris[3,5-bis(trifluoromethyl)phenyl]borane.14The molecular structure of (2-CF3C6H4)3B(NCMe) is shown in Fig. S9,† with selected bond lengths and angles given in the caption. The three phenyl groups are twisted by 43.5, 45.6 and 36.4°, forming a propeller-like orientation. The acetonitrile-free complex has previously been characterized, which possessed near C3 symmetry and twist angles of 45.6, 49.3 and 52.9°, reflecting the increased space available around the 3-coordinate B centre.15
Ring opening polymerization
:[4] of 150:1 for ε-CL or 100:1 for rac-LA and δ-VL at a temperature of 110 °C over 1 h for ε-CL or 12 h or 24 h for rac-LA and δ-VL, respectively. Under these conditions, each of 1–5 was screened for the ROP of ε-CL, rac-LA and δ-VL; the results are presented in Table 1.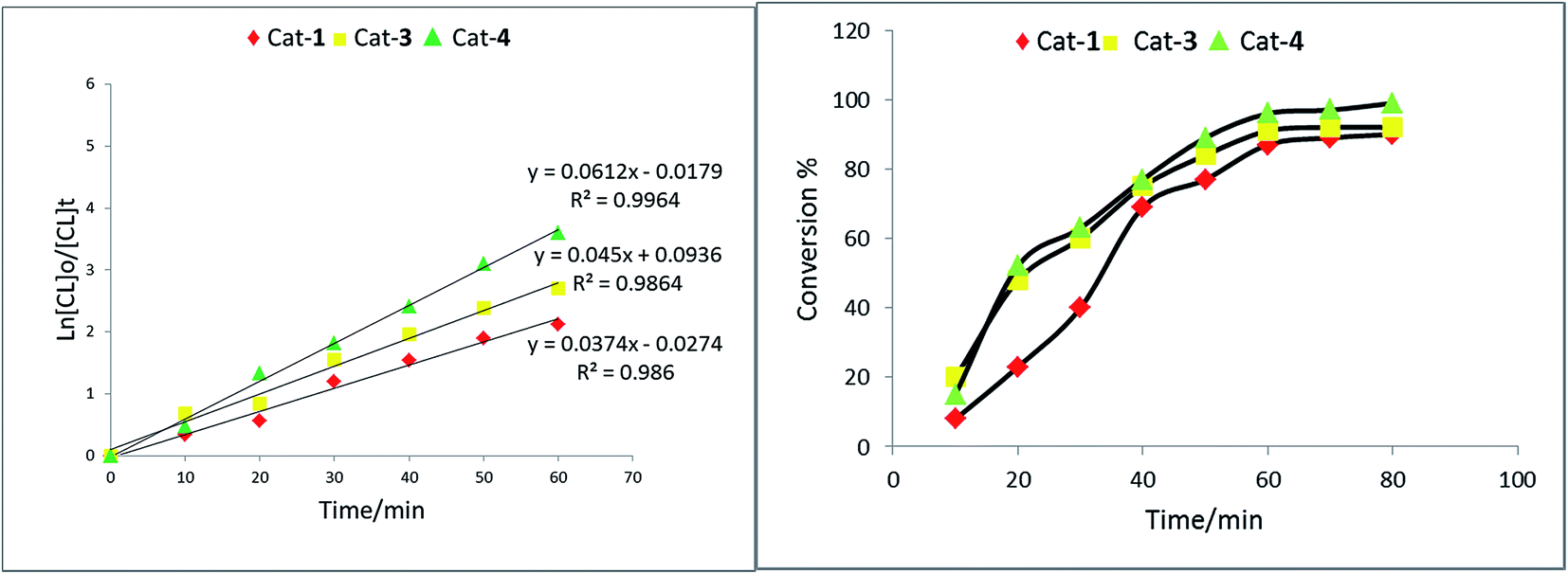 | ||
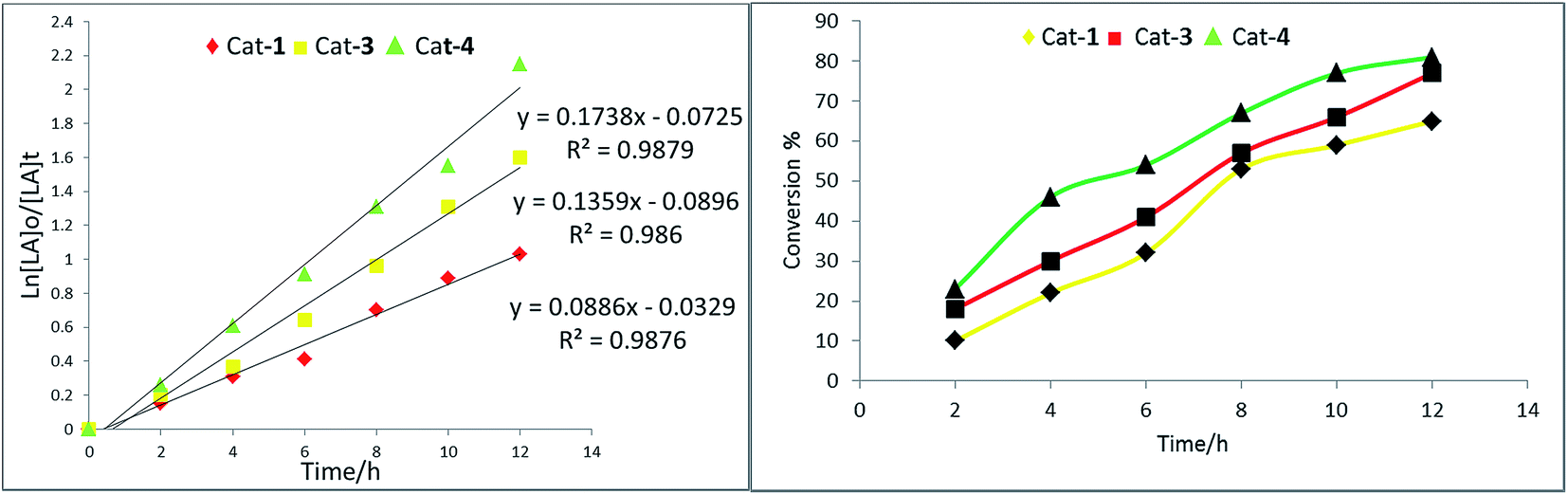
| Fig. 4 Left: Plot of ln[CL]0/[CL]t vs. time using complex 1, 3 and 4; right: relationship between conversion and time for the polymerization of CL. | ||
 | ||
| Fig. 5 Left: Plot of ln[rac-LA]0/[rac-LA]t vs. time using 1, 3 and 4; right: relationship between conversion and time of polymerization. | ||
The plot of [δ-VL]/[4](Fig. S22†) is near linear. From the 1H NMR spectra of the PVL (Fig. S23†), the peak at δ 3.62 revealed the presence of a CH2OH end group. In general, it was evident that the ROP of δ-VL required more robust conditions than were required for ε-CL, which is consistent with the thermodynamic parameters for these lactones.18
| Runa | Cat | CL:LAb |
Yield | Mnc | PDId |
|---|---|---|---|---|---|
| a Optimum conditions: 1 h ε-CL 110 °C/12 h rac-LA 110 °C, (150 [ε-CL]:150 [rac-LA]:[cat]).b Ratio of CL to LA observed in the co-polymer by 1H NMR spectroscopy.c Mn values were determined by GPC in THF vs. PS standards and were corrected with a Mark–Houwink factor (Mn,GPC × 0.56 × % PCL + Mn,GPC × 0.58 × % Pr-LA).d PDI were determined by GPC.e Reverse addition (i.e. rac-LA added first). |
|||||
| 1 | 1 | 62:38 |
65 | 5310 | 1.09 |
| 2 | 2 | 60:40 |
72 | 2900 | 1.12 |
| 3 | 3 | 53:47 |
77 | 5160 | 1.19 |
| 4 | 4 | 55:45 |
58 | 2210 | 1.25 |
| 5 | 5 | 56:44 |
69 | 3590 | 1.08 |
| 6e | 4 | — | — | — | — |
In conclusion, we have synthesised a series of alkylzinc complexes of formula [RZn(dpg)]4, where R = Me (1), Et (2), 2-CF3C6H4 (3), 2,4,6-F3C6H2 (4) and dpg is derived from Ph2C(X)CO2H (dpgH). In each complex, the core structure is very similar, with the zinc macrocycle adopting an up-down-up-down conformation. Complexes 1–4 and the known benzilic acid (benzH2)-derived complex [(ZnEt)3(ZnL)3(benz)3] (5) were found to be active for the ROP of ε-Cl, rac-LA and δ-VL, without benzyl alcohol present. Complexes 3 and 4, bearing fluorinated aryls at zinc, were found to afford the highest activities. All complexes were also capable of affording copolymers via sequential addition of the monomers ε-Cl and rac-LA.
Experimental
All manipulations were carried out under an atmosphere of nitrogen using standard Schlenk line and cannula techniques or a conventional N2 filled glove box. Solvents were refluxed over the appropriate drying agents, and distilled and degassed prior to use. Elemental analyses were performed at London Metropolitan University and the University of Hull. NMR spectra were recorded on a Varian VXR 400 S spectrometer at 400 MHz, a Gemini at 300 MHz or a Bruker DPX300 spectrometer at 300 MHz (1H) and 75.5 MHz (13C) at 298 K; chemical shifts are referenced to the residual protio impurity of the deuterated solvent. IR spectra (KBr discs) were recorded on a Perkin-Elmer 577 or 457 grating spectrophotometer. DSC analyses of polymer samples were performed on a TA Instruments DSC Q 1000. Matrix Assisted Laser Desorption/Ionization-Time Of Flight (MALDI-TOF) mass spectrometry was performed on Bruker autoflex III smart beam in linear mode. MALDI-TOF mass spectra were acquired by averaging at least 100 laser shots. 2,5-Dihydroxylbenzoic acid was used as matrix and tetrahydrofuran as solvent. Sodium chloride was dissolved in methanol and used as the ionizing agent. Samples were prepared by mixing 20 μL of polymer solution in tetrahydrofuran (2 mg mL−1) with 20 μL of matrix solution (10 mg mL−1) and 1 μL of a solution of ionizing agent (1 mg mL−1). Then 1 mL of these mixtures was deposited on a target plate and allowed to dry in air at room temperature. The dialkylzinc complexes were prepared either via the Chisholm method involving addition of lithium salts to ZnCl2 or via the Bochmann method whereby trisaryl(boron) reagents are used as aryl transfer agents with either Me2Zn or Et2Zn.12,20 [EtZn(dpg)]4 (2) was prepared by our previously reported procedure.9b All other chemicals were obtained commercially and were used as received unless stated otherwise.Synthesis of [MeZn(dpg)]4·2MeCN (1·2MeCN)
A solution of dimethylzinc (9.25 mL, 9.25 mmol, 1 M in toluene) was added at room temperature to a solution of diphenylglycine (1.00 g, 4.40 mmol) in toluene (30 mL). The resulting solution was heated under reflux for 12 h, volatiles were removed in vacuo, and the residue was extracted into warm acetonitrile (30 mL). Prolonged standing at 0 °C afforded colourless crystals of 1. Yield 0.98 g, 68.1%. Elemental analysis calculated for C60H60N4O8Zn4·2(C2H3N): C, 58.68; H, 5.04; N, 6.44%. Found: C, 58.67; H, 4.91; N, 6.58%. IR (nujol null, KBr, cm−1): 3345 (m), 3233 (bw), 2955 (s), 2923 (s), 2853 (s), 2358 (w), 1659 (w), 1644 (m), 1602 (s), 1491 (w), 1460 (s), 1378 (s), 1260 (s), 1211 (w), 1152 (w), 1135 (w), 1108 (w), 1079 (m), 1042 (m), 1022 (s), 945 (m), 808 (s), 769 (m), 730 (m), 701 (s), 730 (m), 671 (m), 636 (w), 624 (w), 615 (m), 571 (w), 535 (w), 500 (w), 473 (w). MS (ES, positive mode): m/z 1036 [M–Ph2C(NH2)CO2H–3Me], 971 [M–Ph2C(NH2)CO2H–ZnMe–2Me], 744 [M–2Ph2C(NH2)CO2H–ZnMe–2Me], 517 [M–3Ph2C(NH2)CO2H–ZnMe–2Me]. 1H NMR (CDCl3, 400 MHz) δ: 7.02–7.60 (3 × m, 40H, arylH), 5.79 (d, 4H, 2JHH = 10.8, endo-NH2), 3.02 (d, 4H, 2JHH = 10.8, exo-NH2), 2.35 (s, 6H, MeCN), 0.22 (s, 6H, Zn–CH3), 0.07 (s, 6H, Zn–CH3).Synthesis of [(2-CF3C6H4)Zn(dpg)]4 (3)
As for 1, but using (2-CF3C6H4)2Zn (3.28 g, 9.24 mmol) and diphenylglycine (1.00 g, 4.40 mmol). Prolonged standing at 0 °C afforded colourless crystals of 3. Yield 1.14 g, 59.0%. Elemental analysis calculated for C84H56F12N4O8Zn4·0.2(C2H3N): C, 57.70; H, 3.22; N, 3.35%. Found: C, 57.91; H, 3.28; N, 3.31%. IR (nujol null, KBr, cm−1): 3306 (w), 3242 (bw), 3175 (w), 1661 (s), 1644 (s), 1608 (w), 1493 (w), 1343 (s), 1262 (s), 1195 (w), 1160 (w), 1131 (m), 1090 (m), 1079 (s), 1051 (m), 1040 (m), 1026 (s), 803 (s), 773 (w), 760 (w), 744 (w), 731 (w), 694 (s), 673 (w), 615 (s). MS (ES, positive mode): m/z 1519 [M–Ph2C(NH2)CO2H], 1373 [M–Ph2C(NH2)CO2H–Ar-CF3]. 1H NMR (CDCl3, 400 MHz) δ: 7.57–7.02 (4 × m, 56H, arylH), 5.79 (d, 4H, 2JHH = 10.7 Hz, endo-NH2), 3.02 (d, 4H, 2JHH = 10.7 Hz, exo-NH2), 2.35 (s, H, MeCN). 19F NMR (dmsod6) δ: −61.09. 19F NMR (dmsod6) δ: −61.09.Synthesis of [(2,4,6-F3C6H2)Zn(dpg)]4·4(C7H8)·(1.59H2O) (4·4(C7H8)·(1.59H2O))
As for 1, but using (2,4,6-FC6H2)2Zn (3.03 g 9.24 mmol) and diphenylglycine (1.00 g 4.40 mmol). Prolonged standing of a saturated toluene solution at 0 °C afforded colourless 4. Yield 1.14 g, 54.0%. Sample dried in vacuo for number of hours; – 3.58C7H8·1.59H2O. Elemental analysis calculated for C66H40F12N4O8Zn4·0.42(C7H8): C, 53.53; H, 2.80; N, 3.62%. Found: C, 52.91; H, 2.79; N, 3.62%. IR (nujol null, KBr, cm−1): 3318 (s), 3172 (w), 2725 (w), 1660 (m), 1605 (s), 1587 (m), 1460 (s), 1377 (s), 1305 (w), 1260 (m), 1148 (m), 1101 (m), 1042(w), 1022 (m), 990 (m), 839 (w), 802 (m), 774 (m), 743 (m), 728 (m), 698 (m), 631 (w), 609 (w), 512 (w), 472 (w). MS (ES, positive mode): m/z 1147 [M–0.42 toluene–Ph2C(NH2)CO2H–aryl-F3], 920 [M–0.42 toluene–Ph2C(NH2)CO2H–aryl-F3–Ph2C(NH2)CO2H]; 1H NMR (CDCl3, 400 MHz) δ: 7.46–7.12 (4 × m, 48H, arylH + 0.42 C6H5CH3), 5.38 (d, 4H, 2JHH = 11.2 Hz, endo-NH2), 3.43 (d, 4H, 2JHH = 11.2 Hz, exo-NH2), 2.35 (s, 1.2H, 0.42C6H5CH3). 19F NMR (dmsod6) δ: −83.91 (s, 4F, o-F), −84.37 (s, 4F, o-F), −107.29 (s, 1F, p-F), −112.81 (s, 2F, p-F), −114.78 (s, 1F, p-F).Synthesis of (2-CF3C6H4)3B·MeCN
2-Bromobenzotrifluoride (5.60 mL, 0.041 mol) was slowly added to Mg turnings (1.00 g, 0.041 mol) in diethyl ether (150 mL) at 0 °C and the system was stirred for 2 h. The Grignard solution was then added to BF3·Et2O (1.60 mL, 0.013 mol) in toluene (100 mL) at 0 °C, and the system was slowly allowed to warm to ambient temperature. The diethyl ether was then removed under vacuo, and the system was heated to 100 °C on a water bath for 1 h. Following removal of volatiles, the residue was extracted into hexane (150 mL). Removal of the hexane and crystallization from acetonitrile (80 mL) afforded (2-CF3C6H4)3B·MeCN. Yield 4.60 g, 79.8%. IR (nujol null, KBr, cm−1): 2922 (bw), 2345 (w), 2341 (w), 1954 (w), 1661 (m), 1596 (m), 1462 (s), 1376 (s), 1313 (s), 1258 (m), 1164 (m), 1142 (m), 1060 (w), 1032 (m), 999 (m), 899 (s), 801 (m), 771 (m), 669 (m), 655 (w), 645 (w), 616 (w), 581 (w), 497 (w), 441 (w). MS (ES, positive mode): m/z 446 [M–MeCN], 300 [M–MeCN–Ar-CF3]. 1H NMR (CDCl3, 400 MHz) δ: 7.72–7.22 (4 × m, 12H, arylH), 1.99 (s, 3H, MeCN). 11B NMR (CDCl3, 400 MHz) δ: 70.13 (bs).Ring opening polymerization procedure
Typical polymerization procedures are as follows. A mixture of monomer (3.44 mmol of ε-caprolactone), 2.29 mmol rac-lactide or (2.29 mmol δ-valerolactone) and catalyst 1 (0.02 mmol) were added into a Schlenk tube at room temperature under nitrogen protection and toluene (5 mL) was added to the reaction mixture. The reaction mixture was then placed into an oil bath pre-heated to the required temperature, and the solution was stirred for the prescribed time. The polymerization mixture was then quenched by addition of an excess of glacial acetic acid (0.2 mL) into the solution, and the resultant solution was then poured into methanol (200 mL). The resultant polymer was then collected on filter paper and was dried in vacuo.Copolymerization procedure
Co-polymerizations were conducted by adding ε-CL (3.44 mmol) to the catalyst (0.02 mmol) in toluene (10 mL) pre-heated to 110 °C and stirring for 1 h, and then adding rac-lactide (3.44 mmol) and stirring for a further 12 h. The reaction mixture was quenched by addition (200 mL) of methanol. The precipitate was collected and dried in vacuo.X-ray crystallography
Diffraction data were collected on a Bruker APEX 2 CCD diffractometer and were corrected for absorption and Lp effects.21 Further details are provided in Table 3. Structures were solved by direct methods {charge flipping for 4·4(C7H8)·1.59(H2O)} and refined by full matrix least squares methods.22 N–H distances were restrained for structures 1·2MeCN and 4·4(C7H8)·1.59(H2O). The structure 3 was refined as merohedrally twinned via the twin law 010 100 00−1 (major component 54(2)%), and racemically twinned (Flack parameter 0.472(2)). A structure solution was only possible after temporary ‘de-twinning’ of the reflection data. The full data and the twin law were then used for the structure refinement. For 4·4(C7H8)·1.59(H2O) the toluene lies on a symmetry element and the methyl group was 50/50 disordered over two positions as a result. There was also a significant residual electron density peak close to an aromatic ring H atom. This was modelled as a partial water molecule with 39.7(9)% occupancy with H atoms not located.| Compound | 1·2MeCN | 3 |
| Formula | C60H60N4O8Zn4·2(C2H3N) | C84H56F12N4O8Zn4 |
| Formula weight | 1308.70 | 1746.87 |
| Crystal system | Monoclinic | Tetragonal |
| Space group | P21/n | I |
| Unit cell dimensions | ||
| a (Å) | 15.021(2) | 18.401(5) |
| b (Å) | 16.504(3) | |
| c (Å) | 25.151(4) | 11.347(3) |
| β (°) | 96.420(2) | 90 |
| V (Å3) | 6196.0(17) | 3842(2) |
| Z | 4 | 2 |
| Temperature (K) | 150(2) | 150(2) |
| Wavelength (Å) | 0.71073 | 0.71073 |
| Calculated density (g cm−3) | 1.403 | 1.510 |
| Absorption coefficient (mm−1) | 1.59 | 1.32 |
| Transmission factors (min./max.) | 0.535 and 0.763 | 0.504 and 0.890 |
| Crystal size (mm3) | 0.45 × 0.43 × 0.18 | 0.60 × 0.11 × 0.09 |
| θ(max.) (°) | 27.5 | 26.5 |
| Reflections measured | 56708 |
16703 |
| Unique reflections | 14215 |
3988 |
| Rint | 0.062 | 0.064 |
| Reflections with F2 > 2σ(F2) | 10644 |
3856 |
| Number of parameters | 769 | 256 |
| R1 [F2 > 2σ(F2)] | 0.053 | 0.055 |
| wR2 (all data) | 0.138 | 0.133 |
| GOOF, S | 1.07 | 1.11 |
| Largest difference peak and hole (e Å−3) | 1.08 and −0.89 | 1.54 and −1.13 |
| Compound | 4 | BAr3 | ZnCl2(NCMe)2 |
| Formula | C66H40F12N4O8Zn4·4(C7H8)·1.59H2O | C23H15BF9N·C2H3N | C4H6Cl2N2Zn |
| Formula weight | 1903.68 | 528.22 | 218.38 |
| Crystal system | Tetragonal | Monoclinic | Orthorhombic |
| Space group | I |
P21/c | Pnma |
| Unit cell dimensions | |||
| a (Å) | 13.5944(5) | 9.3027(5) | 12.7113(11) |
| b (Å) | 18.1466(10) | 9.9296(8) | |
| c (Å) | 22.3469(9) | 14.1566(8) | 6.5812(6) |
| β (°) | 94.8366(10) | ||
| V (Å3) | 4129.9(3) | 2381.3(2) | 830.67(12) |
| Z | 2 | 4 | 4 |
| Temperature (K) | 150(2) | 150(2) | 150(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| Calculated density (g cm−3) | 1.531 | 1.473 | 1.746 |
| Absorption coefficient (mm−1) | 1.24 | 0.14 | 3.52 |
| Transmission factors (min./max.) | 0.637 and 0.808 | 0.911 and 0.966 | 0.154 and 0.401 |
| Crystal size (mm3) | 0.40 × 0.36 × 0.18 | 0.70 × 0.45 × 0.26 | 0.75 × 0.36 × 0.26 |
| θ(max.) (°) | 30.6 | 30.0 | 31.7 |
| Reflections measured | 24843 |
27132 |
9419 |
| Unique reflections | 6188 | 6939 | 1440 |
| Rint | 0.026 | 0.040 | 0.027 |
| Reflections with F2 > 2σ(F2) | 5685 | 4851 | 1284 |
| Number of parameters | 291 | 336 | 48 |
| R1 [F2 > 2σ(F2)] | 0.032 | 0.043 | 0.020 |
| wR2 (all data) | 0.082 | 0.105 | 0.055 |
| GOOF, S | 1.04 | 1.05 | 1.04 |
| Largest difference peak and hole (e Å−3) | 0.60 and −0.39 | 0.33 and −0.21 | 0.36 and −0.30 |
Acknowledgements
The National Mass Spectrometry Service at Swansea and Dr Kevin Welham (University of Hull) are both thanked for mass spectrometry data. We wish to acknowledge the use of the EPSRC's Chemical Database Service hosted by the RSC. The higher committee for education development in Iraq and the EPSRC are thanked for financial support.References
- For recent reviews see: (a) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed; (b) C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1 CrossRef CAS; (c) E. S. Place, J. H. George, C. K. Williams and M. H. Stevens, Chem. Soc. Rev., 2009, 38, 1139 RSC; (d) M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484 RSC; (e) A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801 RSC; (f) X. Rong and C. Chunxia, Progress in Chemistry, 2012, 24, 1519 Search PubMed.
- (a) R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS; (b) M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS; (c) A. L. Sisson, D. Ekinci and A. Lendlein, Polymer, 2013, 54, 4333–4350 CrossRef CAS.
- S. Sriputtirat, W. Boonkong, S. Pengprecha, A. Petsom and N. Thongchul, Adv. Chem. Eng. Sci., 2012, 2, 15 CrossRef CAS.
- M. Braun, Angew. Chem., 1996, 108, 565 (Angew. Chem. Int. Ed., 1996, 35, 519) CrossRef.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- X. Wang, K.-Q. Zhao, S. Mo, Y. F. Al-Khafaji, T. J. Prior, M. R. J. Elsegood and C. Redshaw, unpublished results.
- Y. F. Al-Khafaji, T. J. Prior, L. Horsburgh, M. R. J. Elsegood and C. Redshaw, unpublished results.
- See for example, C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed and references therein.
- (a) C. Redshaw, M. R. J. Elsegood and K. E. Holmes, Angew. Chem., Int. Ed., 2005, 44, 1884–1887 CrossRef; (b) C. Redshaw and M. R. J. Elsegood, Angew. Chem., Int. Ed., 2007, 46, 7453–7457 CrossRef CAS PubMed.
- A. Guerrero, D. L. Hughes and M. Bochmann, Organometallics, 2006, 25, 1525–1527 CrossRef CAS.
- (a) I. V. Isakov and Z. V. Zvonkova, Dokl. Akad. Nauk SSSR, 1962, 145, 808 Search PubMed; (b) V. K. Bel'sky, N. R. Stetsova, B. M. Balychev, P. A. Storozhenko, L. V. Ivankina and A. I. Gobumov, Inorg. Chim. Acta, 1989, 164, 211–220 CrossRef.
- S. J. Lancaster, ChemSpider, 2003, 215 10.1039/sp215.
- N. A. Yakelis and R. G. Bergman, Organometallics, 2005, 24, 3579–3581 CrossRef CAS PubMed.
- T. J. Herrington, A. J. W. Thom, A. J. P. White and A. E. Ashley, Dalton Trans., 2012, 41, 9019–9022 RSC.
- S. Toyota, M. Asakura, M. Ōki and F. Toda, Bull. Chem. Soc. Jpn., 2000, 73, 2357–2362 CrossRef CAS.
- N. Iwasa, S. Katao, J. Liu, M. Fujiki, Y. Furukawa and K. Nomura, Organometallics, 2009, 28, 2179–2187 CrossRef CAS.
- M. J. Walton, S. J. Lancaster and C. Redshaw, ChemCatChem, 2016, 6, 1892–1898 CrossRef and references therein.
- See Handbook of Ring Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH, 2009 Search PubMed.
- See for example: (a) P. J. A. Int Veld, E. M. Velner, P. Van de Witte, J. Hamhuis, P. J. Dijkstra and J. Feijen, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 219–226 CrossRef CAS; (b) Y. Liu, W.-S. Dong, J.-Y. Liu and Y.-S. Li, Dalton Trans., 2014, 43, 2244–2251 RSC.
- (a) M. H. Chisholm, J. C. Gallucci, H. Yin and H. Zhen, Inorg. Chem., 2005, 44, 4777–4785 CrossRef CAS PubMed; (b) D. A. Walker, T. J. Woodman, D. L. Hughes and M. Bochmann, Organometallics, 2001, 20, 3772 CrossRef CAS.
- APEX 2 & SAINT software for CCD diffractometers, Bruker AXS Inc., Madison, USA, 2006-2014 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed; (c) L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1516751–1516755. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26746g |
| This journal is © The Royal Society of Chemistry 2017 |