
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two new molybdates based on ∞[MonO3n+1]2− units (n = 11, 4) with proton conduction under ionothermal†
Y. H. Zhaoa,
X. C. Jina,
L. Chena,
Y. F. Dinga,
Z. Maoa,
H. Fu *ab and
Y. H. Ao*ab
*ab and
Y. H. Ao*ab
aSchool of Chemistry and Life Science and Advanced Institute of Materials Science, Changchun University of Technology, Changchun, Jilin, P. R. China. E-mail: Mr.fuhai@163.com; aoyuhui@mail.ccut.edu.cn
bSchool of Chemistry and Life Science, Jilin Province Key Laboratory of Carbon Fiber Development and Application Changchun University of Technology, Changchun, Jilin, P. R. China
First published on 23rd January 2017
Abstract
Two new hybrid organic–inorganic molybdates have been synthesized under ionothermal conditions, namely (H4C3N2(C6H4N)2N2C3H4)2/∞[Mo11O34]·H2O (1), and (H6C5N(CH2)3NC5H6)1/∞[Mo4O13] (2), which were based on layered and chained ∞[MonO3n+1]2− (n = 11, 4) blocks and two different sizes of organoammonium dications bis-(4-imidazol-1-yl-phenyl)-diazene and 1,3-di-(4-pyridyl)-propane (+HL1H+ and +HL2H+), respectively. The 2/∞[Mo11O34]2− and 1/∞[Mo4O13]2− units in 1 and 2 are unprecedented members of the ∞[MonO3n+1]2− family with the new n value extended to 11 and 4, whose structure is similar with the ∞[MoO3] slabs in α-MoO3. Single crystal X-ray analysis shows that the ∞[MonO3n+1]2− layers and chains in 1 and 2 are pillared in the three-dimensional (3D) networks by the organic dications, while the two connections at the organic–inorganic interface are similar. At the same time, the different ∞[MonO3n+1]2− blocks (n = 11, 4) in hybrid organic–inorganic layered and chained molybdate materials are clearly evidenced by the efficient Raman spectroscopy. Moreover, the electrochemical impedance spectroscopy (EIS) measurements of 1 show a high conductivity (2.3 × 10−4 S cm−1 at 75 °C and 90% relative humidity), with an activation energy of 0.45 eV for proton conduction. The mechanism of proton conduction for this molybdate material is proved to be Vehicular mechanism.
1. Introduction
Polyoxometalate (POM) based hybrid organic–inorganic materials are of great interest for their potential applications in a wide range of fields including fuel cells,1 medicine,2 photocatalysis.3 These hybrid materials could offer a good chance to combine the unique properties of the organic and inorganic components in one single phase. And these properties of the hybrid materials depend on the connection of POMs and organic components.4 Owing to the fact that POMs have discrete and mobile ionic structure, POMs have been regarded as attractive candidates for proton conduction.5Inspired by the application of proton conductors in fuel cells, a large amount and a high mobility of proton carriers are required to achieve high proton conductivity.6 Proton transport may occur through water molecules and hydroxonium ions, which moves in the direction of the pathway.7 In POMs based hybrid organic–inorganic materials, conductivity generally arises from guest water and coordinated water as proton donors or carriers; furthermore, it could also be caused by the characteristics of a “pseudo-liquid phase” with high proton mobility. In that context, the organic–inorganic hybrid materials as proton conductors based on POMs and organoammonium cations (OACs) are quite representative systems.8 Therefore, the synthesis of such materials is one of the most challenging issues in synthetic chemistry. Considering that, introducing highly soluble ionic liquids to organic ligands and inorganic subunits could promote the interaction between organic and inorganic components well.9 Ionothermal is now regarded as a more effective and environmentally friendly synthetic method to overcome the challenges.
The modern room temperature ionic liquids (RTILs) are, as the name suggests, liquid at room temperature while near room temperature ionic liquids (nRTILs) are often defined as being liquid below a certain temperature, often 100 °C, although this varies depending on the application envisaged for the liquids. For ionothermal synthesis, nRTILs are often defined as being liquid below about 200 °C, which traditionally used in hydrothermal synthesis. RTILs show a range of properties used as media for the preparation of inorganic and inorganic–organic hybrid materials. ILs can be used as the solvent and the template to prepare many types of solid. One of the most interesting and important classes of material that has been recently developed is that of coordination polymers. A great many of the materials prepared are relatively low dimensional solids under ionothermal synthesis, and this is clearly a very ideal method for the preparation of such materials. It is clear that in these systems changing the chemistry of the solvent to ionothermal leads to great possibilities in this area.10,11
In the planning synthesis, we introduced the efficient ionothermal reaction system to easily synthesize the rare molybdates ∞[MonO3n+1]2− (n = 11, 4) blocks, using Na2MoO4·2H2O as the molybdenum source and two sized organoammonium dications labeled as +HL1H+ and +HL2H+ (bis-(4-imidazol-1-yl-phenyl)-diazene, L1 = H14C18N6, and 1,3-di-(4-pyridyl)-propane, L2 = H14C13N2). Because the dications act as structure-directing agents for molybdate condensation in these systems, we predicted that the topology of the chained and layered molybdates should be strongly correlated to the nature of the H-bonding connected dications. Hence ∞[MonO3n+1]2− blocks with the larger value of n can be obtained by using dications with larger sizes.12 Under this consideration, we have investigated the reaction of [MoO4]2− anions toward two different kinds of dications under the ionothermal condition in order to enlarge the value of n in the family of ∞[MonO3n+1]2− blocks. In this paper, two new hybrid molybdates based organic–inorganic materials (H4C3N2(C6H4N)2N2C3H4)2/∞[Mo11O34]·H2O (1), and (H6C5N(CH2)3NC5H6)1/∞[Mo4O13] (2), have been obtained under the ionothermal condition. 1 contains an unprecedented layered molybdate block, i.e., the 2/∞[Mo11O34]2− which turns out to be a new member of the ∞[MonO3n+1]2− family with the n value extended to 11, until now it is the largest in the family.12 While, 2 is built by the well-known 1/∞[Mo4O13]2− chains. Moreover, a powerful method Raman spectroscopy is used to perfectly discriminate different ∞[MonO3n+1]2− blocks. According to the single crystal X-ray diffraction, 1 possesses layered structures with water molecules interacting with the organic ligands and the molybdates based polyanion. These structural characteristics prompted us to investigate its proton conduction properties. Then, we measured the water-assisted proton conduction in 1, which operates at a low temperature (45–75 °C). Vehicular mechanism of 1 is dominant for the proton conduction under different relative humidity (RH) of 70%, 80%, and 90%. The electrochemical impedance spectroscopy (EIS) measurements show a high conductivity (2.3 × 10−4 S cm−1 at 75 °C and 90% RH), with an activation energy of 0.45 eV for proton conduction.
2. Experimental
2.1. Materials and general methods
All reagents were purchased commercially and used without further purification. The organic ligand L1 was prepared based on the literature procedures.13 Raman spectroscopy was conducted on Horiba JY LabRAM HR Evolution Raman spectrometer. The TGA and DTA spectroscopy were conducted on a TA TGAQ500. The single-crystal X-ray crystallography was conducted on BRUKER D8 Venture Single-Crystal Diffractometer. Impedance measurement of the product was performed on PARSTAT 2273 Advanced electrochemical impedance analyzer.2.2. Preparation of (1)
A mixture of [Emim]Br (2.000 g, 10.500 mmol), Na2MoO4·2H2O (0.019 g, 0.08 mmol), L1 (0.002 g, 0.006 mmol) was stirred for about half an hour at room temperature. During the period, the final pH of the mixture was adjusted to about 4 with HCl (conc.) and NaOH (solid). Then the suspension was sealed in a 23 mL Teflon-lined stainless steel autoclave reactor, kept under autogenous pressure at 175 °C for five days. After cooling to room temperature at a rate of 10 °C h−1, it stood for one day. Then the autoclave was opened, and the obtained brown block-shaped crystals of 1 were filtered off, washed with acetone, and dried in the air.2.3. Preparation of (2)
A mixture of [Emim]Br (2.000 g, 10.500 mmol), Na2MoO4·2H2O (0.019 g, 0.08 mmol), L2 (0.003 g, 0.015 mmol) was stirred for about half an hour at room temperature. During the period, the final pH of the mixture was adjusted to about 3.5 with HCl (conc.) and NaOH (solid). Then the suspension was sealed in a 23 mL Teflon-lined stainless steel autoclave reactor, kept under autogenous pressure at 160 °C for five days. After cooling to room temperature at a rate of 10 °C h−1, it stood for one day. Then the autoclave was opened, and the obtained brown block-shaped crystals of 2 were filtered off, washed with acetone, and dried in the air.2.4. Single-crystal X-ray crystallography
Diffractive quality single crystals were mounted on a glass fiber and the crystallographic data were collected at 293 (K) for 1 and 2 on a Bruker D8 venture diffract meter using graphite monochromatic Mo-Kα radiation (λ = 0.71073 Å) and SMART CCD diffractometer. The structures of 1 and 2 were solved by the direct method and refined by the full-matrix least-squares method on F2 using the SHELXTL-97 crystallographic software package.14 All non-hydrogen atoms were refined with anisotropic thermal parameters. The H atoms on the C atoms were fixed in calculated positions. The detailed crystal data and structure refinements for 1 and 2 were summarized in Table 1. And the hydrogen bonding of 1 and 2 were summarized in Table 2.†| Compounds | 1 | 2 |
|---|---|---|
| a R1 = Σ||Fo| − |Fc||/Σ|Fo|.b wR2 = Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]1/2. | ||
| Empirical formula | C18H18Mo11N6O35 | C13H16Mo4N2O13 |
| Formula weight | 1933.72 | 792.04 |
| T/K | 153(2) | 153(2) |
| Wavelength/Å | 0.71073 | 0.71073 |
| Cryst. syst. | Monoclinic | Monoclinic |
| Space group | P2/c | P21/c |
| a/Å | 13.2348(10) | 10.929(2) |
| b/Å | 5.4270(4) | 9.7226(19) |
| c/Å | 29.837(2) | 19.633(4) |
| α/° | 90 | 90 |
| β/° | 96.396(2) | 101.82(3) |
| γ/° | 90 | 90 |
| V/Å3 | 2129.7(3) | 2041.8(7) |
| Z | 2 | 4 |
| Dcalc (mg m−3) | 3.016 | 2.577 |
| μ/mm−1 | 3.237 | 2.473 |
| F(000) | 1820 | 1520 |
| θ range (°) | 2.75 to 24.99 | 3.13 to 25.00 |
| Rint | 0.0366 | 0.0252 |
| Data/restraints/parameters | 3748/0/322 | 3588/0/289 |
| GOF on F2 | 0.948 | 0.951 |
| Final R1a, wR2b [I > 2σ(I)] | R1 = 0.0341, wR2 = 0.1367 | R1 = 0.0252, wR2 = 0.0813 |
| Final R1a, wR2b (all data) | R1 = 0.0426, wR2 = 0.1503 | R1 = 0.0276, wR2 = 0.0833 |
| X–H⋯O (X = N, C) | X–H | H⋯O | X⋯O |
|---|---|---|---|
| Compound 1 | |||
| N3–H3⋯O3 | 0.8804(86) | 2.0297(58) | 2.8344(104) |
| C2–H2A⋯O1 | 0.9505(100) | 2.2791(58) | 3.2148(117) |
| O1W–H1O⋯O2 | 0.9016(1058) | 2.0518(1133) | 2.8606(67) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Compound 2 | |||
| C18–H18a⋯O12 | 0.9498(40) | 2.4536(33) | 3.2948(55) |
| N1–H1⋯O9 | 0.8810(44) | 1.9001(29) | 2.7494(51) |
| N2–H2⋯O6 | 0.8802(38) | 1.9009(28) | 2.7795(48) |
3. Results and discussion
3.1. Structure description of 1
The single-crystal X-ray analysis of 1 reveal that 1 is built on 2/∞[Mo11O34]2− layers, +HL1H+ dications. Until now the n value of the known 2/∞[MonO3n+1]2− layers was limited to 5, 7 and 9, and thus the 2/∞[Mo11O34]2− block is the very first member of this family with the n value equal to 11. Here we present one complementary description of the overall topology of the 2/∞[Mo11O34]2− layers. As shown in Fig. 1d, the overall topology of the 2/∞[Mo11O34]2− layers can be schematically described from infinite [MonO4n+2] ribbons built upon corner-sharing [MoO6] octahedral as shown in Fig. 1a. The ribbons defined in yz planes are one octahedral thick and n octahedral broad and then developed along y-axis. Each ribbon is sandwiched via edge-sharing condensation between two identical ribbons shifted along x-axis in two opposite directions with half-overlapping (Fig. 1b). It results in stair-like layered structures in which the step widths are exactly half of those of the [MonO4n+2] ribbons (Fig. 1b). The 2/∞[Mo11O34]2− blocks are constructed on linear [MonO4n+2] segments of the 1/∞[MonO4n] chains described above (Fig. 1b). These segments condense via edge-sharing to form zigzag infinite 1/∞[MonO4n] chains. These chains are then corner-shared-connected to give rise to the 2/∞[MonO3n+1]2− layers as shown in Fig. 1c and d. As shown in Fig. 2, the 2/∞[MonO3n+1]2− layers crystallographic x-axis and they are pillared by the +HL1H+ dications. The connection between the organic and inorganic components is ensured by strong hydrogen-bonding interactions and follows the same interacting scheme as shown in Fig. 2. Namely, each ammonium head of the +HL1H+ dications is anchored at the step of the 2/∞[MonO3n+1]2− layers and develops two C–H⋯O interactions, one with a terminal O atom of a given [MonO4n+2] ribbon (O1 in 1) and the other with a bridging μ2-O (O3) that is common with the two vicinal ribbons constitutive of the 2/∞[MonO3n+1]2− layer. The complete hydrogen-bonding network in 1 also includes crystallized water molecules that develop two short hydrogen bonds (Table 2) with O2, which are the more nucleophilic oxide of the heptamolybdate blocks with two adjacent +HL2H+ cations. At first sight, the hydrogen-bonding network in 1 matches well with the H-atom-donor and -acceptor strengths considering that, as is often observed in such hybrid materials, the short O–H⋯O interactions imply the water molecule that is the strong H-atom donor and the most nucleophilic oxide of the molybdates, while longer and weaker C–H⋯O interactions are established with less nucleophilic oxides.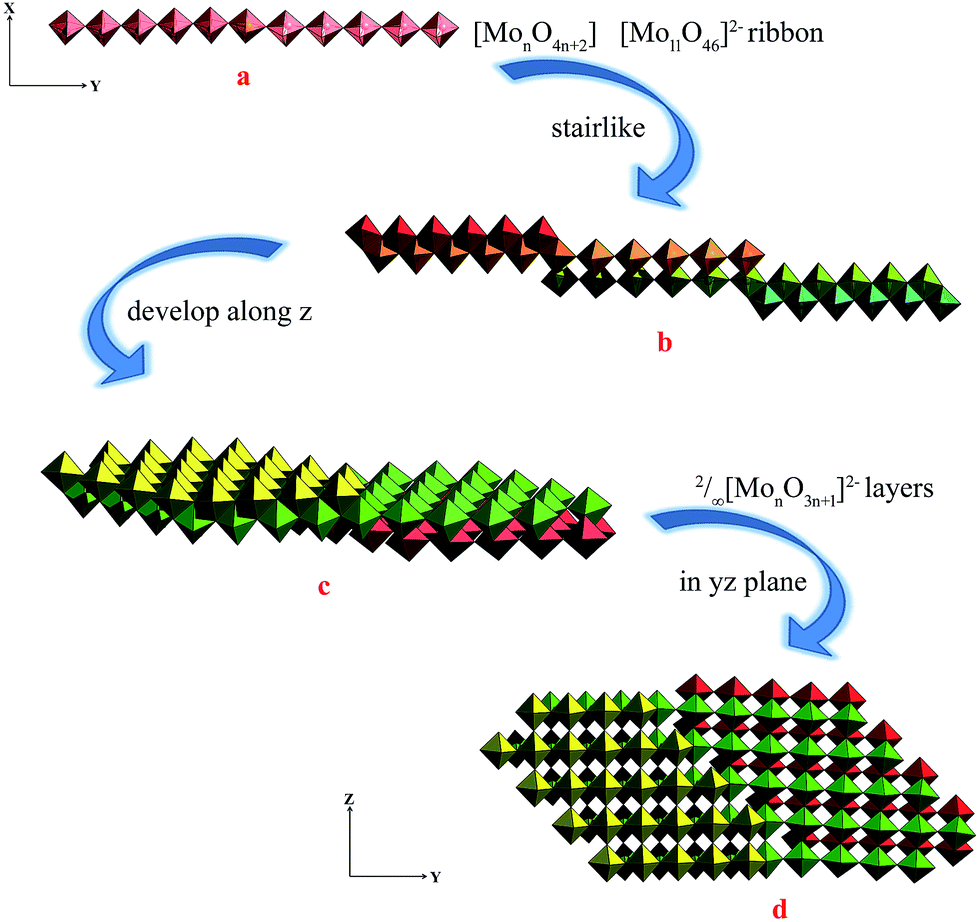 | ||
| Fig. 1 (a) The representations of the infinite 1D chain-like [Mo11O46]2− ribbon, (b) the representations of the stair-like stacking of different adjacent ribbons in xy plan, (c) the representations of the 2/∞[MonO3n+1]2− layers resulted from the stair-like stacking ribbon plane developed along z, (d) the representations of the 2/∞[MonO3n+1]2− layers in yz plane view from x. | ||
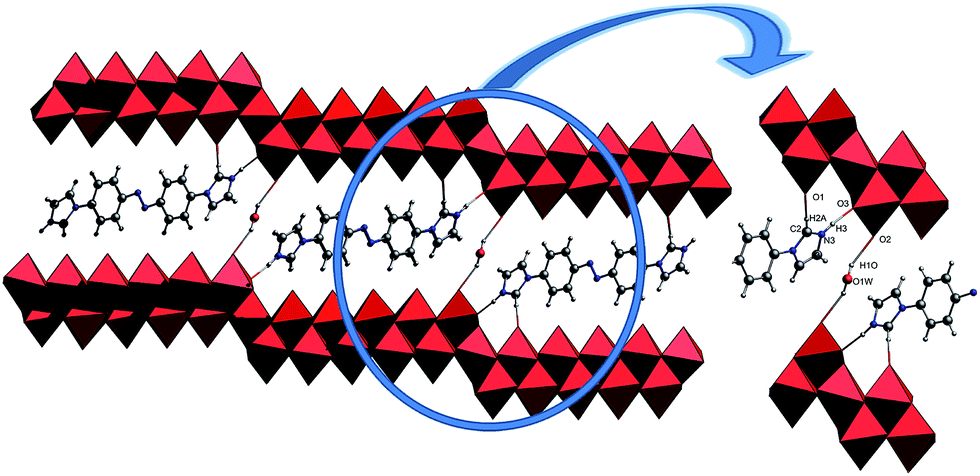 | ||
| Fig. 2 The representations of the H-bond in compound 1, C2–H2A⋯O1, N3–H3⋯O3, O1W–H1O⋯O2, N atoms blue balls, C atoms gray balls, water molecules yellow balls. | ||
3.2. Structure description of 2
The single-crystal X-ray data reveals that 2 was crystallized in the monoclinic space group P21/c. And reveal that 2 is built on 1/∞[Mo8O26]4− chains and two +HL2H+ dications. The well-known β-[Mo8O26]4− anion is a centrosymmetric block composed of eight distorted edge-sharing [MoO6] octahedral as shown in Fig. 3a. In 2, the subunit [Mo8O28] is quite similar to β-[Mo8O26]4− anion, based on eight edge-shared distorted [MoO6] octahedral as shown in Fig. 3b and S2.† And then, the [Mo8O28] subunits connected via the sharing of the two vertices to form the infinite 1/∞[Mo8O26]4− chains via Mo2–O8–Mo1 along the crystallographic y-axis. As shown in Fig. 3c, the 1/∞[Mo8O26]4− chains in 2 are stacked along the crystallographic x-axis and they are pillared by the +HL2H+ dications. The connection between the organic and inorganic components is ensured by strong hydrogen-bonding interactions and follows the same interacting scheme. Namely, each 1,3-di-(4-pyridyl)-propane head of the +HL2H+ dications is anchored at the step of the 1/∞[Mo8O26]4− chains and develops two N–H⋯O interactions, one with two terminal O atoms (O6 and O9), resulted 2D infinite layers, as shown in Fig. S3† and Table 2. These 2D infinite layers are maintained parallel via van der Waals and electrostatic interactions to give rise to a supermolecular 3D network though three terminal O atoms and one bridging μ2-O atom (O7, O10, O12 and O3) as shown in Fig. S4.†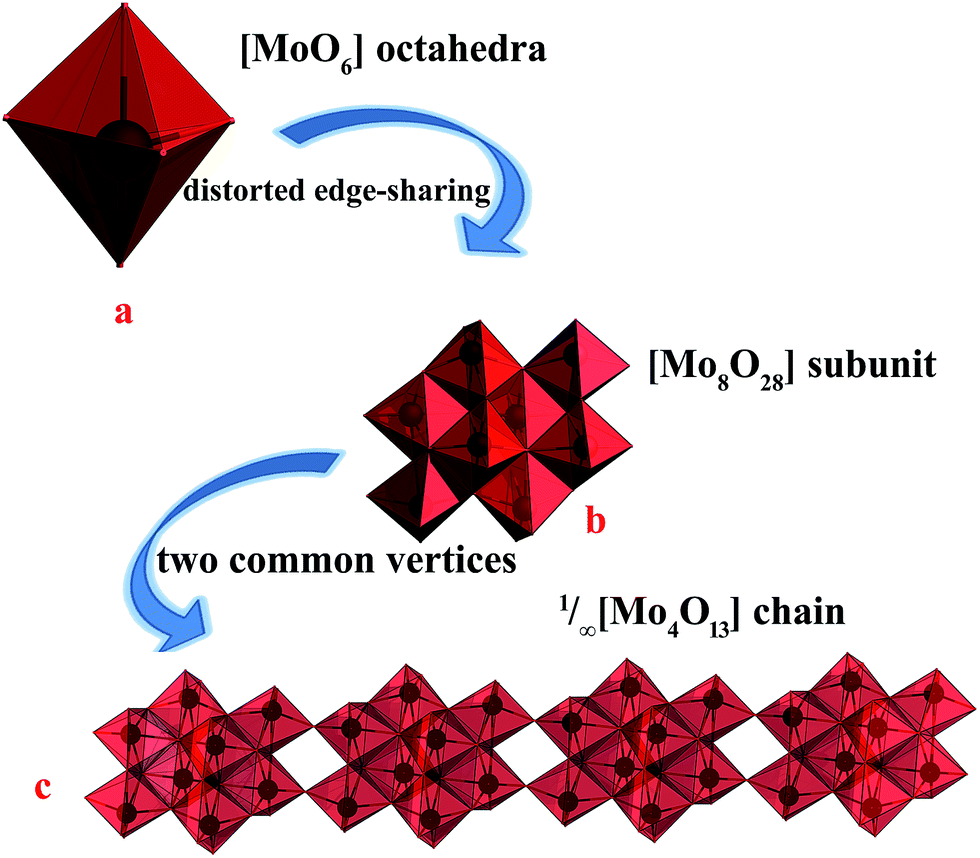 | ||
| Fig. 3 (a) The representations of the well-known [MoO6] octahedral, (b) the representations of [Mo8O28] subunit, (c) the representations of the 1D infinite chain of 1/∞[Mo8O26]4− chains. | ||
3.3. Synthesis discuss
Ionothermal synthesis is the method in which ionic liquids (ILs) act as solvents, potential templates or structural directing agents in the formation of solid materials. Compared to the solvents in traditional hydro/solvothermal synthesis, ILs, as the polar solvents, possess reasonably good solubility to most of the inorganic precursors and are environmentally friendly. And it is also ionic with a range of significant advantages, including their low vapor pressure, higher chemical and thermal stabilities, as well as their low toxicities. The advantages in the ionothermal synthesis introduced to the synthesis of the isopolymolybdate-based hybrid organic–inorganic materials are as follows: the weak coordinating ability, the good solubility and tunable acidity. In summary, all the advantages of the ionothermal synthesis may play very important parts in the synthesis of isopolymolybdate-based hybrid organic–inorganic materials.In this paper, as shown in Fig. S5,† two different kind of organic ligands were introducing into the reaction system towards the ∞[MonO3n+1]2− blocks. And the sizes of the two ligands are about 16.1548(160) Å and 8.8773(63) Å for 1 and 2, respectively. The size of the organic ligand in 1 is about twice than that in 2. According to the structures of the two compounds, the n value in 1 is larger than that in 2. The size of the ligand in 2 is about the same to the classical 1/∞[Mo8O26]4− chains. In order to enlarge the n value of the family ∞[MonO3n+1]2− blocks, the larger size organic ligand bis-(4-imidazol-1-yl-phenyl)-diazene was introduced into the ionothermal synthesis system. As a result, the n value was extended to 11 in 1. The result of the n value confirm the idea that ∞[MonO3n+1]2− blocks with the larger value of n can be obtained by using dications with larger sizes. Because the dications act as structure-directing agents for molybdate condensation in these systems, and the topology of the chained and layered molybdates should be strongly correlated to the nature of the H-bonding connected dications.12
3.4. Characterization of the Mo–POM blocks in 1 and 2 by Raman spectroscopy
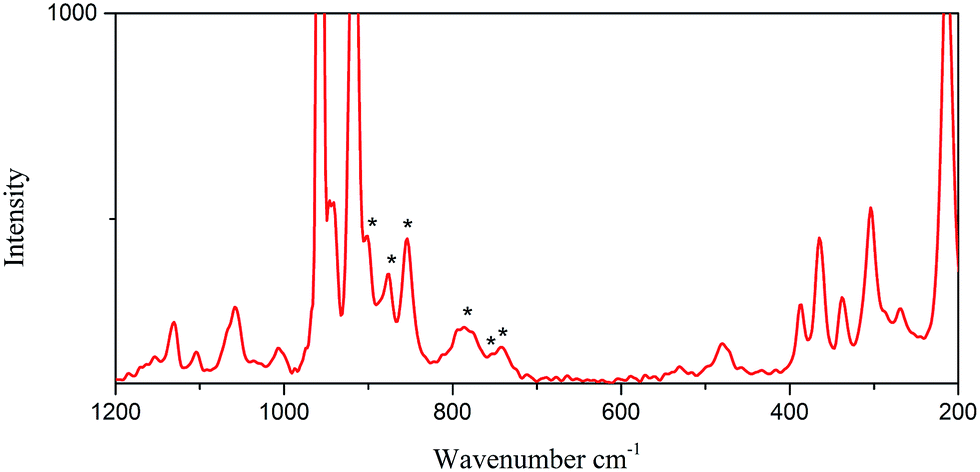
The Raman spectra were measured using a LabRam HR Evolution Raman Spectrometer (Jobin-Yvon Horiba Scientific), equipped with an air cooled frequency doubled Nd:Yag 532 nm laser for excitation (1 mW). The laser was focused through a 50 objective. The scattered light was analyzed by spectrograph with holographic grating (600 g mm−1), and opened confocal hole (100 μm). The time of acquisition of a particular spectral window was optimised for individual sample measurements (ca. 30 s). As aforementioned, the Raman spectroscopy appears as an appropriate and powerful method to identify the nature of the Mo–POM blocks, especially for materials containing different Mo–POM isomers. Fig. 4 and 5 show a comparison between the Raman spectra of 1 and 2 in the 1100–500 and 1200–200 cm−1 range, which contains the absorption bonds relative to the mineral components. Practically, this domain may be restricted to the 900–600 cm−1 range to unambiguously and pragmatically discriminate the two octamolybdate isomers in 1 and 2.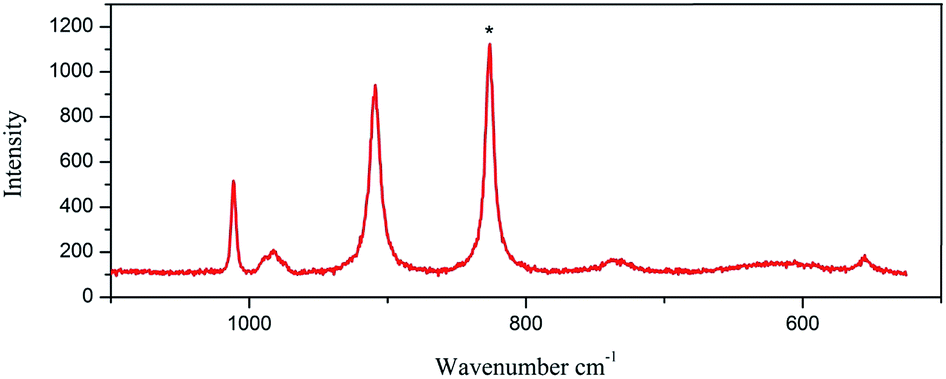 | ||
| Fig. 4 The Raman spectra of 1. | ||
 | ||
| Fig. 5 The Raman spectra of 2. | ||
As shown in Fig. 4, the Raman signatures of the 2/∞[MonO3n+1]2− blocks (n = 11) in 1 are perfectly distinguishable by comparing the position in frequency of the strongest and sharp lines located at 825 cm−1 for the 2/∞[Mo11O34]2− units. This frequency shifts at 820 cm−1 for α-MoO3 and is assigned by Seguin et al.15 to the stretching mode of the Mo–O–Mo vibration (labeled as Mo2O vibration). This specific vibration implies the O atoms that connect two 1/∞[MoO4] chains (Fig. 1a). Thus, by analogy with α-MoO3, the strong line observed in the Raman spectra of the 2/∞[MonO3n+1]2− blocks (n = 11) should be attributed to the Mo2O vibration frequency (νMo2O), implying the corner-shared O atoms in common with two [MonO4n+2] segments (Fig. 1b). In the experiment, νMo2O vibration frequency reaches its lower value for α-MoO3. This already has been interpreted by Dessapt et al.12 The Raman signatures of the 2/∞[MonO3n+1]2− blocks (n = 5, 7, 9) are perfectly distinguishable by comparing the position in frequency of the strongest and sharp lines located at 886, 872, and 855 cm−1 for the 2/∞[Mo5O16]2−, 2/∞[Mo7O22]2−, and 2/∞[Mo9O28]2− units, respectively.12 Consequently, n increases with the length of the [MonO4n+2] segments in the 2/∞[MonO3n+1]2− layers to reach its maximum value for the 1/∞[MoO4] chains of α-MoO3. The result of the Raman of 1 confirm the predictive character, which could be useful to easily identify new 2/∞[MonO3n+1]2− layers (especially with n higher than 9) in future powdered hybrid material.12
As shown in Fig. 5, the Raman signature of the octamolybdate block in 2 (n = 4) shows six absorption bands at 900, 877, 853, 782, 755 and 740 cm−1. As the above mentioned, 2 is built on 1/∞[Mo8O26]4− chains the well-known β-[Mo8O26]4− anion. The structure of 2 is an infinite 1D chain, and it is different from the 2D layered structure in 1. However, in 2, because of the lack of the O atoms that connect two infinite 1D 1/∞[MoO4] chains in 1, the stretching mode of the Mo–O–Mo vibration is different from that in 1. So the positions in frequency in 2 are not in accordance with the tendency in 1. In fact, the [Mo8O26]4− isomer is a flexible block that can adopt numerous different geometries. For example, the isolated [Mo8O26]4− cluster has been characterized in the solid state into nine isomers, namely, α, β, γ, δ, ε, ζ, ξ, η and θ. It is lack of Raman characterizations for these species.12
3.5. The proton conductivity study
According to the result of the thermogravimetric analysis and differential thermal analysis of 1 (as shown in Fig. S7 in ESI†), it is shown that 1 possesses high stability (thermal and solvent stability, especially in water). As well, 1 possesses layered structures with water molecules that interact with the organic ligands and the molybdates based polyanion. These structural characteristics prompted us to investigate its proton conduction properties. The homogeneous dry crystalline powder of 1 was obtained with a mortar and pestle. And then the powder was added to a standard 13.2 mm die, sandwiched between two stainless steel electrodes and pressed at 5000 kg for about 2 min; the pellet was 13.2 mm in diameter and 1.2 mm in thickness. Measurements were implemented by using an impedance and gain-phase analyzer (PARSTAT 2273, Ametek, USA), 1 Hz to 1 MHz, with a two-probe electrochemical cell and an applied ac voltage of 50 mV (unless otherwise stated). We measured the water-assisted proton conduction in 1, which operates at a low temperature (45–75 °C). Among reported water-mediated molybdates-based proton-conductors, highly humid conditions (commonly near to 100% RH) are usually require.16 Because most of these materials have poor hydrophilicity and water retention, water molecules easily desorb from them at low humidity resulting in disconnected proton-conducting pathways. Avoiding this desorption and retaining efficient proton transport at low humidity is of great significance for practical application.As Fig. 6 shown, the Nyquist plots obtained under different humidity were used to determine the proton conductivity of 1 using the relation, σ = L/(S × R) equation, where R is the resistance, L is the width, and S is the area of the sample plate. The electroconductibility of 1 increases with the increase of temperature under different humidity (70% RH, 80% RH, 90% RH), and the conductivity of 1 is the largest under the condition of 90% RH. The increase of the RH make water molecules be more easily up taken into the 1D hydrophilic channels, facilitating the proton transport to give larger proton conductivities. The activation energy of 1 is 0.40 eV, 0.42 eV, 0.45 eV under different humidity (90% RH, 80% RH, 70% RH), respectively, calculated by the Arrhenius equation. The electrochemical impedance spectroscopy (EIS) measurements show a high conductivity (2.3 × 10−4 S cm−1 at 75 °C and 90% RH), with an activation energy of 0.45 eV for proton conduction. However, it is near with the graphene oxide (GO) nanosheets (≈10−4 S cm−1 at 25 °C and 60% RH), which Matsumoto et al. have reported.17 And it exhibits better proton conductivities than Ni-POM-EN and Cu3Mo5P2 which were synthesized by Banerjee et al. (at 39 °C, 59 °C and 98% RH are 2.09 × 10−5, 1.69 × 10−5 S cm−1 and 2.2 × 10−5 S cm−1 at 28 °C and 98% RH. respectively).18,19 And also, the proton conductivity values of 1 at 75 °C and 90% RH are much higher than those of MIL-53-based MOFs (ca. 10−6 to ca. 10−9 S cm−1 reported by Kitagawa et al. at 25 °C under 95% RH)20 and those of a zinc-phosphonate MOF (ca. 10−5 S cm−1 reported by Shimizu et al. at 25 °C under 98% RH).21
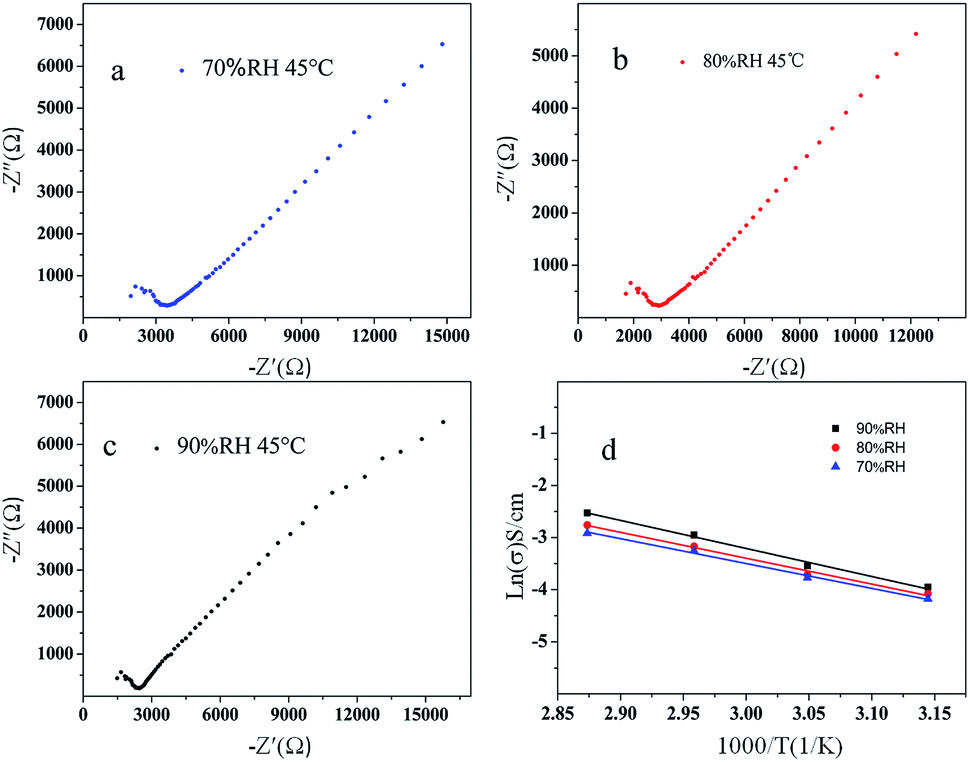 | ||
| Fig. 6 Nyquist plot of proton conduction for 1 with 45 °C condition (a) at 70% RH, (b) at 80% RH and (c) at 90% RH. (d) Arrhenius plots of proton conductivities for 1 at 70% RH, 80% RH, 90% RH, respectively. | ||
Vehicular mechanism of 1 is dominant for the proton conduction under different humidity (70% RH, 80% RH, 90% RH). The reduced activation energy and the increased proton conductivity of 1 suggest the presence of continuous H-bonding networks among the L1 organic ligands and water molecules (lattice water) in the layered structures. In an appropriate range, the number of water molecules increases which come into the layered structures, with the increasing of humidity. The conductivity of the material increases with the increasing of free protons, owing to the water molecules combination with layered structures. The Vehicular mechanism is consistent with the architectural feature that a vehicular transfer mechanism is predominant, which is reasonable because of the architectural feature and the limited number of water molecules and organic ligands in the channels acting as vehicles.22 That is, the direct diffusion of additional protons with water molecules, due to the loss of the hydrogen-bonding network from solvent water molecules. Under higher temperature and humidity, the +HL1H+ ligands can be deprotonated and the protons can be incorporated by the water molecules.23
4. Conclusions
Two new hybrid organic–inorganic molybdates based on chained and layered have been synthesized under ionothermal conditions. It is clearly evidenced that the different ∞[MonO3n+1]2− blocks (n = 11, 4) in hybrid organic–inorganic layered molybdate materials can be easily discriminated by the Raman spectroscopy. Moreover, high proton conductivity is achieved above 2.3 × 10−4 S cm−1 at 75 °C under 90% RH. The activation energy for proton transfer in 1 is 0.45 eV, and the proton conduction mechanism is typical vehicle mechanism. As a consequence, molybdates-based organic–inorganic hybrids with water chains and additional protons will probably become an important class of proton conductive materials in the future.Acknowledgements
This work was financially supported by the National Nature Science Foundation of China (No. 21301019), the Nature Science Foundation of Technology Department of Jilin Province, China (No. 20160101299JC), the National Science Foundation for Post-doctoral Scientists of China (No. 2013M530135), Technology Department of Jilin Province (No. ZDZH2011003).Notes and references
- N. Li, Z. X. Sun, R. Liu, L. Xu, K. Xu and X. M. Song, Sol. Energy Mater. Sol. Cells, 2016, 157, 853 CrossRef CAS; K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535 CrossRef PubMed; K. D. Kreue, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637 CrossRef; H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780 CrossRef PubMed.
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327 CrossRef CAS PubMed; T. J. Yamase, Mater. Chem., 2005, 15, 4773 RSC; B. Asenknopf, Front. Biosci., 2005, 10, 275 CrossRef; J. D. Compain, P. Mialane, J. Marrot, F. Secheresse, W. Zhu, E. Oldfield and A. Dolbecq, Chem.–Eur. J., 2010, 16, 13741 CrossRef PubMed.
- A. Hiskia, A. Mylonas and E. Papaconstantinou, Chem. Soc. Rev., 2001, 30, 62 RSC; Y. Guo and C. Hu, J. Mol. Catal. A: Chem., 2007, 262, 136 CrossRef CAS; Y. Yang, Y. Guo, C. Hu, Y. Wang and E. Wang, Appl. Catal., A, 2004, 273, 201 CrossRef; D. Schaming, C. Allain, R. Farha, S. Lobstein, A. Giraudeau, B. Hasenknopf and L. Ruhlmann, Langmuir, 2010, 26, 5101 CrossRef PubMed; T. Yamase, Chem. Rev., 1998, 98, 307 CrossRef PubMed.
- J. D. Compain, P. Deniard, J. Marrot and P. Mialane, Chem. Commun., 2010, 46, 7733 RSC; Y. Wang, W. Li and L. Wu, Langmuir, 2009, 25, 13194 CrossRef CAS PubMed; B. B. Gai, H. He, Y. H Zhao, Z. Mao and H. Fu, Chem. Res. Chin. Univ., 2016, 32, 527 CrossRef; T. Zhang, S. Liu, D. G. Kurth and C. F. Faul, Adv. Funct. Mater., 2009, 19, 642 CrossRef.
- S. Bureekaew, S. Horike, M. Higuchi, M. Mizuno, T. Kawamura, N. Yanai and S. Kitagawa, Nat. Mater., 2009, 8, 831 CrossRef CAS PubMed; J. A. Hurd, R. Vaidhyanathan, V. Thangadurai, C. I. Ratcliffe, I. L. Moudrakovski and G. K. H. Shimizu, Nat. Chem., 2009, 1, 705 CrossRef PubMed; E. L. Zhou, C. Qin, X. L. Wang, K. Z. Shao and Z. M. Su, Chem.–Eur. J., 2015, 21, 13058 CrossRef PubMed.
- T. Norby, Solid State Ionics, 1999, 125, 1 CrossRef CAS.
- J. Miao, Y. W. Liu, S. X. Liu and Q. Y. Wu, Dalton Trans., 2014, 43, 14749 RSC; C. Dey, T. Kundu, H. B. Aiyappa and R. Banerjee, RSC Adv., 2015, 5, 2333 RSC.
- P. Mialane, G. Zhang, Y. P. Mbomekalle, J. D. Compain, A. Dolbecq, J. Marrot, B. Keita and L. Nadjo, Chem.–Eur. J., 2010, 16, 5572 CrossRef CAS PubMed; R. Dessapt, M. Collet, V. Coue, S. Jobic, C. Lee and M. H. Whangbo, Inorg. Chem., 2009, 48, 574 CrossRef PubMed; V. Coue, R. Dessapt, M. Evain and S. Jobic, Inorg. Chem., 2007, 46, 2824 CrossRef PubMed.
- Y. P. Ren, X. J. Kong, X. Y. Hu, M. Sun, L. S. Long, R. B. Huangand and L. S. Zheng, Inorg. Chem., 2006, 45, 4016 CrossRef CAS PubMed; A. Demessence, G. Rogez, R. Welter and P. Rabu, Inorg. Chem., 2007, 46, 3423 CrossRef PubMed; X. N. Cheng, W. Xue, W. X. Zhang and X. M. Chen, Chem. Mater., 2008, 20, 5345 CrossRef; S. Ma, D. Sun, X. S. Wang and H. C. Zhou, Angew. Chem., Int. Ed., 2007, 46, 2458 CrossRef PubMed; W. X. Chen, S. T. Wu, L. S. Long, R. B. Huang and L. S. Zheng, Cryst. Growth Des., 2007, 7, 1171 Search PubMed; R. Q. Zou, H. Sakurai, S. Han, R. Q. Zhong and Q. Xu, J. Am. Chem. Soc., 2007, 129, 8402 CrossRef PubMed.
- R. E. Morris, Chem. Commun., 2009, 2990 RSC.
- H. Fu, Y. Lu, Z. L. Wang, C. Liang, Z. M. Zhang and E. B. Wang, Dalton Trans., 2012, 41, 4084 RSC.
- B. Martine, D. Remi, D. Philippe and J. Stephane, Inorg. Chem., 2012, 51, 142 CrossRef PubMed; R. Dessapt, D. Kervern, M. Bujoli-Doeuff, P. Deniard, M. Evain and S. Jobic, Inorg. Chem., 2012, 49, 11309 CrossRef PubMed.
- M. K. Sharma and P. K. Bharadwaj, Inorg. Chem., 2011, 50, 1889 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Gçttingen, Gçttingen, Germany, 1997 Search PubMed; G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, University of Gçttingen, Gçttingen, Germany, 1997 Search PubMed.
- L. Seguin, M. Figlarz, R. Cavagnat and J. C. Lassegues, Spectrochim. Acta, Part A, 1995, 51, 1323 CrossRef.
- Y. W. Liu, X. Yang, Z. Shi and S. X Liu, Chem. Commun., 2014, 50, 10023 RSC.
- K. Hatakeyama, M. R. Karim, C. Ogata, H. Tateishi, A. Funatsu, T. Taniguchi, M. Koinuma, S. Hayami and Y. Matsumoto, Angew. Chem., Int. Ed., 2014, 53, 6997 CrossRef CAS PubMed.
- C. Dey, T. Kundu, H. B. Aiyappa and R. Banerjee, RSC Adv., 2015, 5, 2333 RSC.
- C. Dey, T. Kundu and R. Banerjee, Chem. Commun., 2012, 48, 266 RSC.
- A. Shigematsu, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2011, 133, 2034 CrossRef CAS PubMed.
- J. M. Taylor, R. K. Mah, I. L. Moudrakovski, C. I. Ratcliffe, R. Vaidhyanathan and G. K. H. Shimizu, J. Am. Chem. Soc., 2010, 132, 14055 CrossRef CAS PubMed.
- K. D. Kreuer, A. Rabenau and W. Weppner, Angew. Chem., Int. Ed. Engl., 1982, 21, 208 CrossRef.
- M. L. Wei, X. X. Wang and X. Y. Duan, Chem.–Eur. J., 2013, 19, 1607 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, crystallographic data (CIF) and additional supporting data. CCDC 1510385 and 1510386. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26729g |
| This journal is © The Royal Society of Chemistry 2017 |