
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of metallo-supramolecular polymers from thiophene-based unimers bearing pybox ligands†
Sviatoslav
Hladysh
 *a,
Daniela
Václavková
b,
David
Vrbata
a,
Dmitrij
Bondarev
bc,
David
Havlíček
d,
Jan
Svoboda
e,
Jiří
Zedník
a and
Jiří
Vohlídal
a
*a,
Daniela
Václavková
b,
David
Vrbata
a,
Dmitrij
Bondarev
bc,
David
Havlíček
d,
Jan
Svoboda
e,
Jiří
Zedník
a and
Jiří
Vohlídal
a
aDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University in Prague, Hlavova 8, CZ-12840 Prague 2, Czech Republic. E-mail: sviatoslavhladysh@gmail.com
bTomas Bata University in Zlín, Faculty of Technology, Department of Polymer Engineering, Vavrečkova 275, CZ-762 72 Zlín, Czech Republic
cPolymer Institute, Slovak Academy of Sciences, Dubravska cesta 9, 845 41 Bratislava, Slovakia
dDepartment of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 8, CZ-12840 Prague 2, Czech Republic
eInstitute of Macromolecular Chemistry AS CR, v.v.i, Heyrovsky Sq. 2, 16206, Prague 6, Czech Republic
First published on 8th February 2017
Abstract
A series of novel metallo-supramolecular polymers was successfully prepared, based on 2,6-bis(2-oxazolinyl)pyridine building blocks consisting of pyridine flanked by two oxazoline rings as a tridentate binding site bridged with thiophene, bithiophene and thienothiophene as a linker, beginning from a cheap and commercially available 1,4-dihydro-4-oxo-2,6-pyridinedicarboxylic (chelidamic) acid. Metallo-supramolecular polymers were obtained and spectroscopically characterized upon treatment of the synthesized building blocks, also known as unimers, with the following metal ions: Fe2+, Zn2+, Ni2+, Cu2+. During the self-assembly process of the prepared unimers with a Cu2+ ion coupler, UV/vis investigation showed the highest shift of absorption maxima to lower energies, contrary to the Fe2+ ion couplers where the lowest value of shift was detected, compared to the free unimer. Upon the complexation of the Fe2+ ion coupler with selected unimers, the appearance was observed of new absorption bands around 600 nm ascribable to metal-to-ligand charge-transfer transitions. The luminescence study of the complexation of the synthesized unimers with Zn2+ exhibited a high fluorescence increase with an increase of metal ion concentration. Adversely, all of the other metals only showed fluorescence quenching.
Introduction
Supramolecular materials have attracted the attention of many researches during the past decades.1,2 An important subclass of these materials is represented by metallo-supramolecular polymers (MSPs).3 They are typically built from low-molar-mass (multidentate) organic molecules (denoted as unimers (U) – analogous to the monomer unit in the classic polymer or micelle building units) that are self-assembled into larger arrays or chains by non-covalent (labile) interactions typical of coordination bonds with metal ion couplers (ICs). This lability of the coordination bonds gives the resulting materials a dynamic nature and, consequently, these materials belong to the family of materials called dynamers (a term introduced by Nobel Prize laureate Jean-Marie Lehn).4 Labile bonding introduces interesting new properties and allows the possibility of precise system tailoring by the structure of unimers and character and concentration of ion couplers.5–8 The self-assembly process is achieved by introducing the ion coupler into the unimer solution, or vice versa. This process proceeds by the formation of coordination bonds (complexation) between the Us and ICs, leading to the formation of MSPs by the self-assembly process. The term complexation is also used for the formation of “model” complexes that are also presented in this contribution. Therefore, complexation is a general tool for the preparation of either MSPs by the self-assembly process or simple metal–ligand complexes prepared by coordination with standard ligand possessing only one chelating binding site. Large self-assemblies are formed in the case of multifunctional Us possessing at least two chelating binding sites. Most MSPs are typically processed in the solid state as thin films. There are two aspects of MSP stability in solution. The first is represented by the thermodynamic stability constant of the coordination sites of the U molecules and selected metal ICs. The second aspect is related to the kinetics of the labile coordination bonding, characterized by the exchange rate of the free unimer and in the supramolecular assembly. The kinetic aspect of equilibrium in solution is denoted as a constitutional dynamic in further text.9,10The application potential of MSPs is broad across various fields: MSPs with low Tg are utilized in self-healing applications11,12 as well as various responsive systems,13–16 including MSPs that contain totally conjugated Us exploited in optoelectronic devices (light-emitting diodes, photovoltaic cells)17–20 or biomedical applications.21 Despite extensive work in our laboratory with tridentate 2,2′:6′2′′-terpyridine-4-yl units,22–27 the decision to synthesize ditopic unimers with another tridentate ligand – 2,6-bis(oxazolyl)pyridine (pybox) – has been taken into account. The main reasons behind this decision were as follows: (i) oxazoline complexes have a large application potential in microelectronics and also as catalytic systems (especially Ru-based pybox derivatives);28–32 (ii) pybox based ligands allow an easy derivatization through attaching substituents at the 4-position of the pyridine rings (in this case, optical isomerism is introduced into the pybox motif, allowing further tailoring of the prepared systems); (iii) pybox derivatives exhibit much better solubility in organic solvents, which is an important parameter in MSP processing.33 Therefore, all these above-mentioned potential advantages of the bis(oxazolines), in combination with the thiophene based central blocks, bring their own benefits such as the possibility of band gap tuning, high absorption coefficients, charge transport properties, etc. The tridentate ditopic pybox ligand has been partly described by A. Duerrbeck.34 But, to the best of our knowledge, the synthesis, characterization and optical properties of α,ω-bis(2,6-bis(oxazoline-2-yl)pyridin-4-yl)thiophene (P-Th), α,ω-bis(2,6-bis(oxazoline-2-yl)pyridin-4-yl)bithiophene (P-Bt) and α,ω-bis(2,6-bis(oxazoline-2-yl)pyridin-4-yl)thieno[3,2-b]thiophene (P-Te) are new. Moreover, the process of self-assembly of the prepared unimers into MSPs by means of complexation with the selected model ion couplers, Fe2+, Zn2+, Ni2+, Cu2+, performed like a detailed properties survey, is also new. Cu2+ radioisotopes can coordinate suitable ligands and such a method opens applications in diagnostic and therapeutic areas.35 The Zn2+ complexes are used as sensitizers in solar cells.36 Both Ni2+ and Fe2+ ions could catalyze various types of chemical reactions. The Ni2+ complexes became attractive catalysts for the reduction of alkynes, olefins, ketones and aldehydes in high yields.37–39 The Fe2+ complexes are used for various organic transformations, such as olefin polymerization,40 Mukaiyama-aldol reaction,41etc. These observations of d-transition metals make the self-assembly process with synthesized unimers a highly attractive research field. The presented investigations introduce novel information concerning the coordination of unimers and the consequent corresponding MSP formation. Thiophene, bithiophene and thieno[3,2-b]thiophene linkers between pybox groups were chosen as competing units to phenyl linkers that were studied in the above mentioned work.34 The incorporation of these units can positively influence the photophysical and electrochemical properties due to a lower stiffness and a higher conjugation extent of the pybox based unimers used for MSP preparation.42
Results and discussion
Synthesis
The synthesis pathway to the unimers studied is depicted in the ESI as Scheme S1.† The precursor (7) 4-bromo-2,6-bis(4,5-dihydro-2-oxazolyl)-pyridine was synthesized from a cheap chelidamic acid in high yield (82%) by a 6-step procedure applying a simple type of chemical reaction. The compound successfully obtained (7) was further transformed through a Stille reaction43 into appropriate unimers using 2,5-bis(trimethylstannyl)thiophene, 5,5′-bis(trimethylstannyl)-2,2′-bithiophene or 2,5-bis(trimethylstannyl)-thieno[3,2-b]thiophene as the coupling reagents. As compound (7) possesses a reactive bromide in the para position of the pyridine moiety, it allows the introduction of various substituents into the molecule. Based on this fact, the Pybox-Th ligand (see Scheme 1 and ESI†) was synthesized using a Suzuki coupling reaction. The prepared unimers and ligand were characterized by common spectroscopic methods. | ||
| Scheme 1 Synthesized unimers (U) and ligands with formal group numbering for NMR. | ||
UV/vis spectroscopy analysis
Upon the IC addition to the ligand solution the absorption maxima shifted to lower energies. The metal-to-ligand charge transfer transition (MLCT) at 580 nm was observed in the case of the iron salt.44,45 Isosbestic points are present in both titrations with the mentioned ICs, indicating the presence of a coordinated (ligand–IC–ligand) symmetric form, referred to in the literature as a “butterfly dimer”,46 and free ligand in solution. The equilibrium character of the mixture and an approximate character of the described model must always be kept in mind. All the possible compounds (IC, free unimer, ligand–IC and ligand–IC–ligand) are always present in the reaction mixture, dependent upon the thermodynamic stability constant of each species.
| IC | P-Th | P-Bt | P-Te | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r | λ A nm | λ e nm | ε max | λ F nm | S cm−1 | λ A nm | λ e nm | ε max | λ F nm | S cm−1 | λ A nm | λ e nm | ε max | λ F nm | S cm−1 | |
| U | 0 | 350 | 491 | 8.9 | 404 | 3820 | 400 | 526 | 12.6 | 532 | 6210 | 373 | 516 | 15.3 | 510 | 7209 |
| Fe2+ | 1 | 371 | 698 | 7.2 | 408 | 2445 | 411 | 687 | 10.5 | 480 | 3490 | 398 | 702 | 10.9 | 440 | 2390 |
| Zn2+ | 1 | 380 | 516 | 8.7 | 434 | 3270 | 445 | 625 | 13.3 | 542 | 4020 | 422 | 566 | 14.6 | 483 | 2990 |
| Ni2+ | 1 | 375 | 523 | 10.6 | 409 | 2210 | 449 | 594 | 15.6 | 496 | 2110 | 427 | 567 | 17.9 | 465 | 1910 |
| Cu2+ | 1 | 388 | 538 | 10.2 | 421 | 2020 | 457 | 632 | 12.8 | 501 | 1920 | 433 | 583 | 14.9 | 474 | 1990 |
A set of solutions of constant unimer's concentration (2 × 10−4 M) and metal ions (2 × 10−2 M) was prepared as the chosen mixed solvent (chloroform/methanol) for the UV/vis titration study. By the method of a stepwise increase of the ion coupler to unimer ratio, MSPs systems were obtained that were subsequently studied spectroscopically. The [IC]/[U] ratio is denoted as r in further text.
The results are summarized in Fig. 1 for P-Bt, and the rest of the titration results of P-Th and P-Te with various ICs are shown in Fig. S3 and S4 in the ESI.† The optical parameters of all the unimers and metallo-supramolecular polymers are shown in Table 1.
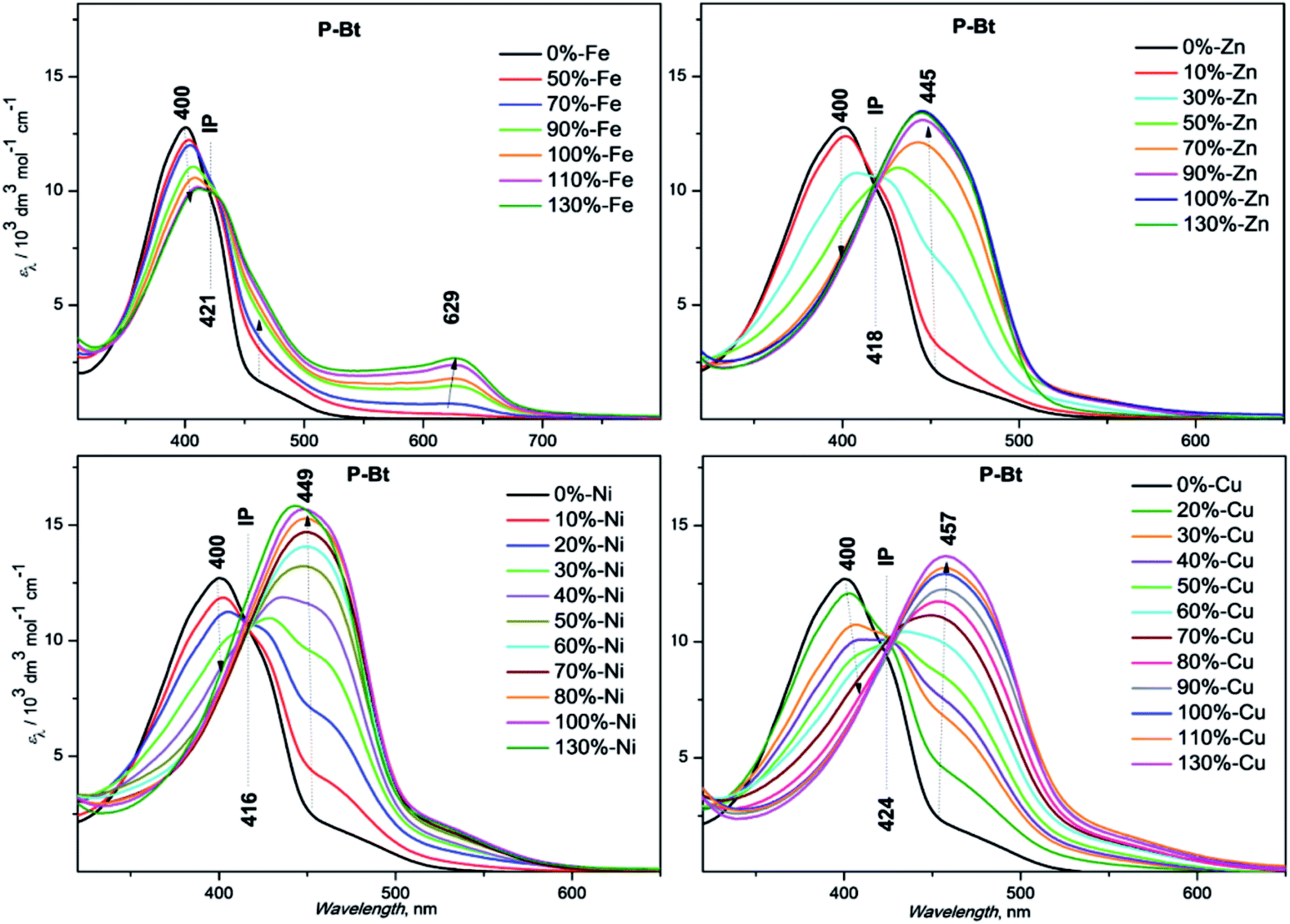 | ||
| Fig. 1 UV/vis titration spectra of P-Bt (2 × 10−4 M, based on the monomer unit), with ion couplers (2 × 10−2 M). | ||
The self-assembly process of unimers with ICs is accompanied by the appearance of new absorption bands. A UV/vis study of this process allows us to track the formation of MSPs. The absorption spectra obtained for studied systems can be analyzed by the three stages of assembly using different values of r. The first stage covers ratios of r from 0 to 0.5 and shows: (i) the negligible changes of band shape, (ii) the appearance of new absorption bands and (iii) the formation of isosbestic points (IP) indicating the transformation of free U species into other defined forms in solution. The stoichiometry hints that the new species could be the dimer (U–(IC)–U), analogous to the above-mentioned “butterfly dimer”.46 The second stage of the assembly process, from r 0.6 to 1, shows the shift of newly formed absorption bands to lower energies accompanied with an increase in the molar absorption coefficient (ε) (except Fe2+ IC, which shows a decrease of ε). The last stage (where r > 1) does not show significant changes in the absorption spectra and can be considered as a depolymerization process of the MSPs, where the pybox end moiety is end-capped by IC coordinated only from one side. According to the UV/vis analysis of all stages of the self-assembly process, MSPs gain the maximum length at a ratio (r) of about 1. As already mentioned above, the studied system is dynamic and the described model serves as a rough description of the presumed behavior. It is obvious that the various chemical species present in the mixture depends upon the stability constants of each compound. Consequently, at r = 0.5 (as an example) “butterfly dimers” are present in the mixture as well as longer polymeric fragments, so free IC and U could be expected in the reaction mixture. It can be concluded that, at selected overall concentrations of the components in the system, the model fits well the observed spectral behavior during its titration course. In summary, the polymerization degree is not dependent only on the values of r, but is also a function of the overall concentration and solvents used.
Absorption maximal positions differ in their dependence on the metal ion coupler used for the MSP preparation. The highest shifts to longer wavelengths (about 50 nm) compared to the corresponding free U is observed in the case of Cu2+ (see ESI, Fig. S5†). Maximal absorption shifts to longer wavelengths were detected during the titration experiment (only 20 nm) for Fe2+ at r = 1. But this MSP displays new absorption bands at around 630 nm that are attributed to the metal-to-ligand charge transfer transition (MLCT) (see Fig. 1, S3 and S4 in the ESI†).
Additionally, the influence of r on the absorption maximal position of newly formed bands corresponding to MSP formation has been characterized by means of graphical dependence (see ESI,† Fig. S6†). In all the cases, we observed a similar trend, with significant shifts to lower energies after the addition of the first portion of IC (r = 0.3). The stepwise increase of IC concentration leads to further shifts of absorption band positions to lower energies. The maximal absorption edges of MSPs with various ICs at r = 1 are summarized in Table 1. The Ni2+ ion coupler showed the lowest energy maximum position at r = 0.5. Further addition of the Ni2+ IC has practically no influence on the absorption maximum position. This observation indicates that the thermodynamically preferred motif in the case of the Ni2+ IC is the “butterfly dimer” U–IC–U. Further addition of the Ni2+ IC probably leads to the prolongation of the MSP chains formed. The rest of the ICs, Fe2+, Zn2+, Cu2+, exhibit a gradual shift of absorption bands to lower energies r (from 0.1 up to 1). In these cases, the IC–U moiety was formed in the first step (the ion coupler is coordinated only by one tridentate pybox moiety). The next step is represented by the dimer formation of a U–IC–U type, followed by MSP chain formation with increasing r. The longest MSP chains are expected at r equals unity in all the studied systems (see Table 1 above, a summary of all the basic optical properties). The further addition of IC ions probably leads to the shortening of MSP chains and to the formation of MSPs end-capped by an IC moiety on the chain-ends. All the systems are of a dynamic nature. It can be presumed that chemical relaxation (rate of reaching the equilibrium) is different in dependence on the U and IC character in each experiment. 1 day (24 h) was found to be sufficient for the equilibration of all the studied systems. It is presumed that 24 h is a sufficient time for chemical relaxation of all the studied systems and all the components are in an inner equilibrium. But it is worth mentioning that even after 24 h, supramolecular systems are still of a dynamic nature and the introduction of additional portions of IC or U leads to re-equilibration. This process is more precisely described below as an end-capping experiment.
The observed behavior indicates that a change in the overall concentrations of all the components in the studied system leads to immediate re-equilibration of the studied system in the solution. Unfortunately, this fact disallows a GPC/SEC study of the prepared MSPs. The injection of the studied mixture into GPC columns leads to several orders of dilution in the studied materials. Chemical relaxation (rate of reaching equilibrium) appears quickly enough to decompose the formed MSPs. Only free unimer is observed at the end of the GPC path in all cases. The ion coupler remains bound on the styragel-based GPC columns. Strong chelating reagents, like bipyridine or EDTA, must be used to release the metal ions from the column.
The end-capping experiment of the end-groups of MSP was performed based on P-Bt unimer and Zn2+ ion coupler (r = 1) by the addition of a Pybox-Th ligand. The addition of the Pybox-Th ligand to the solution of MSP leads to the appearance of a new absorption band with a maximum at 353 nm and a shoulder at 550 nm (yellow line, Fig. 2). The complexation study of the Pybox-Th ligand with the Zn2+ IC showed shifts to a lower energy with an absorption maximum at 353 nm. The fact that the Zn2+ ICs at the chain-ends of the MSP are end-capped by Pybox-Th ligands confirms the dynamic character of the studied MSPs in solution.
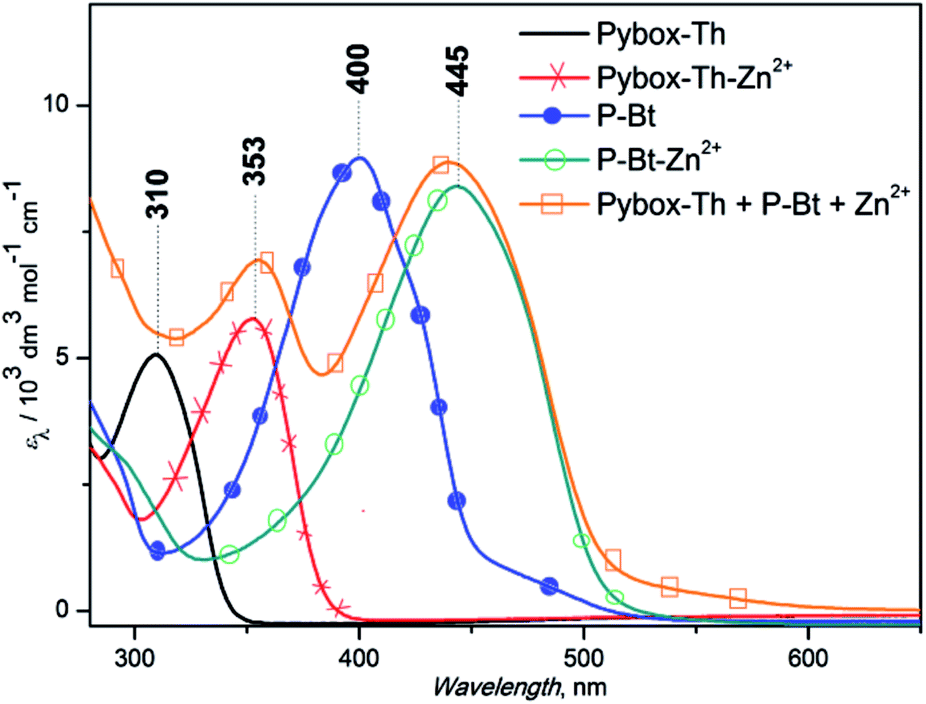 | ||
| Fig. 2 Absorption spectra of P-Bt end-capping experiment. | ||
| State | Unimer | MSP | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P-Th | P-Bt | P-Te | P-Th + Zn | P-Bt + Zn | P-Te + Zn | |||||||
| ϕ F, % | λ F nm | ϕ F, % | λ F nm | ϕ F, % | λ F nm | ϕ F, % | λ F nm | ϕ F, % | λ F nm | ϕ F, % | λ F nm | |
| Solution | 12.8 | 404 | 7.3 | 455 | 14.3 | 420 | 22.2 | 434 | 34.2 | 542 | 17.4 | 483 |
| Film | 1.57 | 580 | 1.17 | 603 | 1.45 | 554 | 2.02 | 614 | 1.99 | 635 | 1.51 | 618 |
The solid-state structure of unimers and their MSPs with the Cu2+ IC was investigated by powder X-ray diffraction (XRD), allowing insight concerning the crystallinity of the above-mentioned materials. Cu2+ was chosen as the best IC leading to the highest shifts of absorption maximum to lower energies ascribable to the formed MSPs (see Table 1). The narrow width of the peaks and the presence of higher order reflections at low angles of P-Bt and P-Te samples in the XRD patterns indicated the high degree of crystallinity of the unimers studied. XRD patterns of P-Bt showed an intense and sharp first-order reflection at a d spacing of 5.7 Å in the 2θ range of 16° (see Fig. 3, and S8 in the ESI†) corresponding to X-ray diffraction patterns of P-Th, P-Te and their Cu2+ based MSPs.
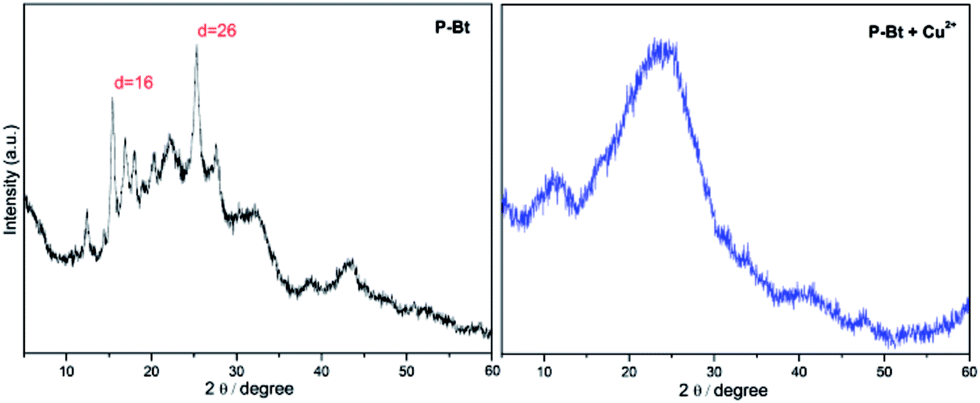 | ||
Fig. 3 X-ray diffraction pattern of P-Bt and its Cu2+ based MSP. The molar ratio between the unimer and Cu2+ is (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). :1). | ||
The second most intense peak occurred at 2θ = 26° and corresponded to a d spacing of 3.5 Å. Additionally, the relatively intense presence of reflections with a low intensity but high sharpness in the 2θ range of 15–25° was observed for the P-Bt unimer. This detection points to a high degree of crystallinity together with a high rigidity of the conjugated bithiophene linker, which was also confirmed by a UV/vis study of solid thin films. The XRD pattern of P-Te showed the two sharpest peaks at 2θ = 9° and 28°, corresponding to a d spacing of 10 Å and 3.3 Å, respectively. As a result, the packaging of P-Te is the same as for P-Bt unimer. Adversely, the P-Th unimer showed a negligible degree of crystallinity. Such observations, together with UV/vis solid-state results for the P-Th sample, are caused by less ordered configurations. The self-assembled systems, or MSPs, were also included in the XRD analysis. Their diffraction patterns demonstrated a lack of high order reflections in all the unimer based supramolecular systems. Only the broadening of peaks was observed, indicating the amorphous character of the synthesized (MSP)s.
X-ray photoelectron spectroscopy analysis (XPS) was used for the analysis of both free unimers and their MSPs with Cu2+ IC. This method was used as an additional tool for the analysis of MSP formations and also for establishing the correct coordination center of the oxazoline moiety (N or O atom). The N1s and O1s XPS spectra of the P-Bt unimer and corresponding Cu2+ based MSP are shown in Fig. 4. For all the unimers studied (P-Th, P-Bt and P-Te), a change of position of the peaks in the N1s spectra to a higher binding energy (1.1–2 eV) was observed. This is caused by the electron withdrawing capacity of Cu2+ ions, which remove more valence electrons of the nitrogen atoms in the pyridine and oxazoline groups, resulting in the enhancement of the effective nuclear charge experienced by the core-electron, thereby increasing the electron binding energy. Because the binding energy of O1s electrons changed only slightly (and to a lower binding energy), a predominant coordination of metal ions through the nitrogen atoms can be expected.
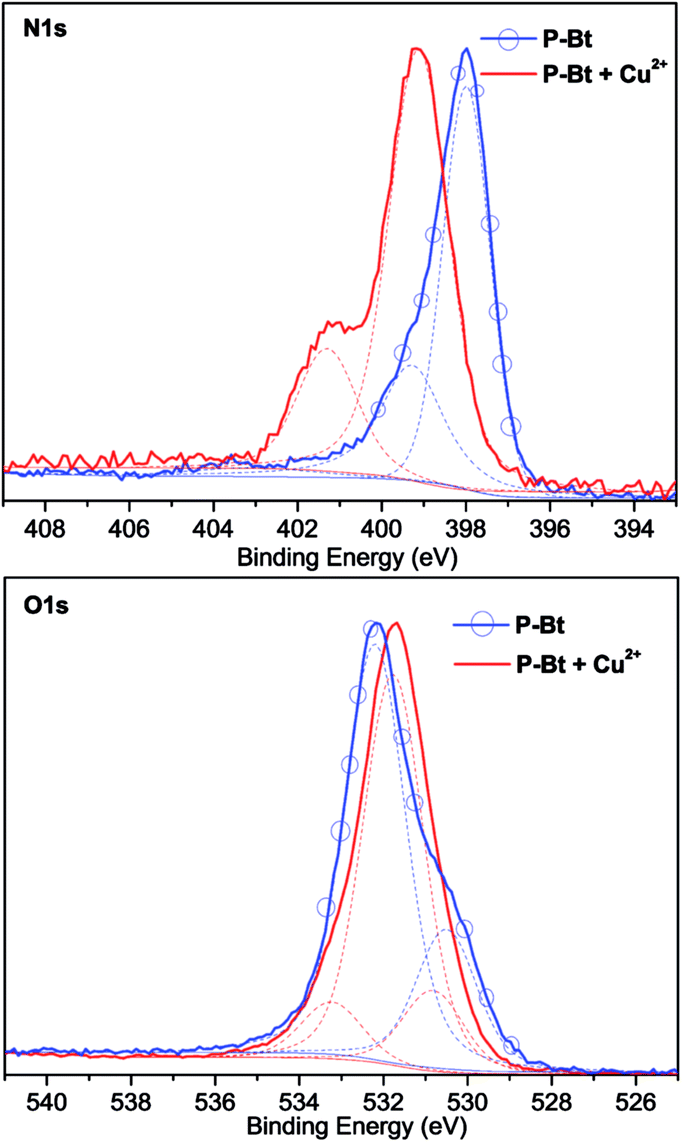 | ||
| Fig. 4 XPS analysis of P-Bt and corresponding Cu2+ based MSP. The molar ratio between the unimer and Cu2+ is (1:1). | ||
Luminescence study
:1) for the preparation of the samples in solution was used. Solid films were prepared by a spin coating method, using the same solvent system. The basic unimer emission characteristics (λF (nm) emission maxima and ϕF (%) fluorescence quantum yields) are shown in Table 2. The synthesized unimers showed a red shift of emission in solution in the following order: P-Th > P-Te > P-Bt. A deviation from the dependence measured in solution was observed in the thin film, with the following order: P-Te > P-Th > P-Bt. A shift of maximal fluorescence to a longer wavelength (of 50 nm for P-Te and 180 nm for P-Th) was observed. Such a large bathochromic shift in the solid state for P-Th, compared to P-Te and P-Bt, is the result of a lowest substituent hindrance (thiophene-linker) compared to the rest of the unimers, strongly influencing molecular packing in the solid state.
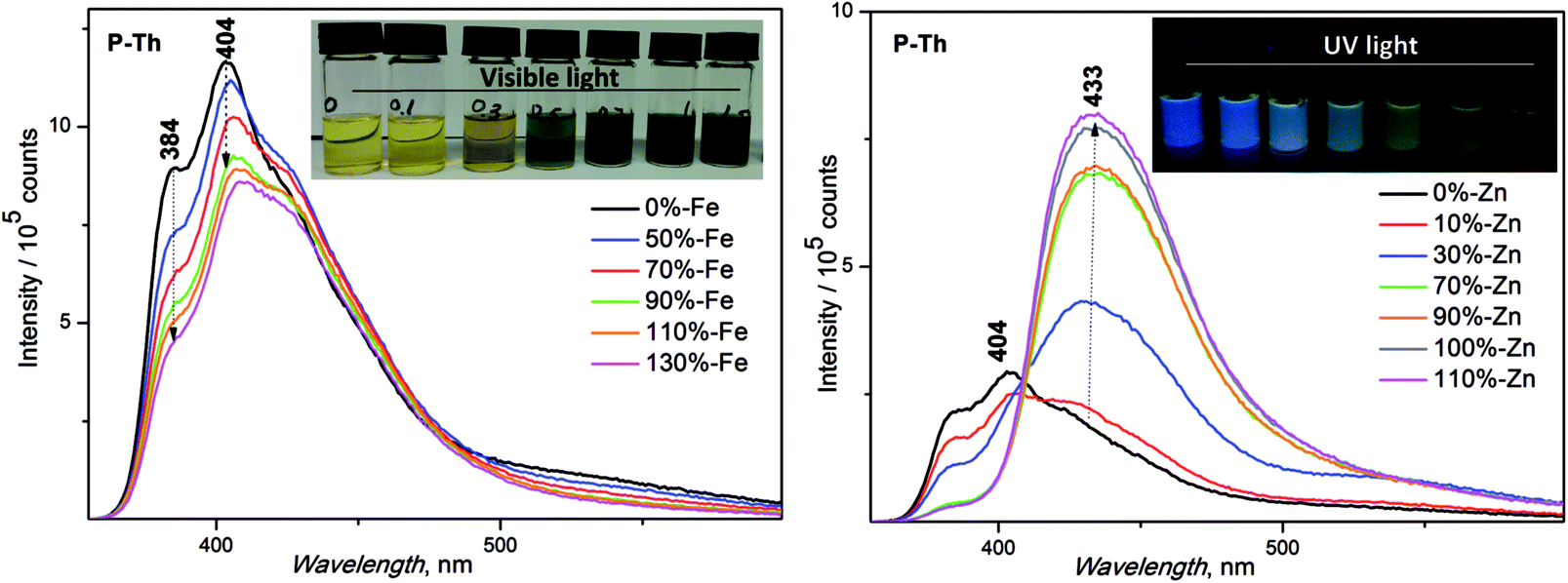 | ||
| Fig. 5 Emission spectra of P-Th with increasing amounts of Zn2+ and Fe2+ metal ions. Inset: captured pictures of samples, Zn2+-UV light, Fe2+-visible light. | ||
:1) in various r ratios clearly shows that the ion coupler used coordinates two ligands. The main changes in the NMR titration spectra were detected at r = 0.5, where the proton signals ascribed to thiophene, as well as those ascribed to oxazoline (CH2O) moieties, are shifted downfield (see Fig. S11 in the ESI†). The further addition of Zn2+ has no influence on the character of the NMR spectra. The corresponding NMR titration spectra of the Pybox-Th ligand correlates very well with the UV/vis spectra. NMR titration experiments with the unimers were performed using P-Th and the Zn2+ IC. The above-mentioned MSP system was chosen due to its better solubility of the P-Th unimer among other unimers and due to the diamagnetic character of the Zn2+ based MSP, allowing NMR analysis. The measurements were performed in DMSO-d6 using concentrations for P-Th of 0.02 M and for Zn2+ of 0.2 M. DMSO-d6 was chosen due to: (i) the good solubility of all the components in the system unimer, selected ion coupler and formed MSPs (as well as the supportive function of DMSO-d6 mentioned above); (ii) no overlap with the signals ascribable to the unimer and formed MSPs. The NMR titration spectra are summarized in Fig. S12 in the ESI.† The free uncoordinated unimer P-Th has proton signals in both the high and low field regions of the proton scale. The changes in both regions were observed after the addition of diamagnetic IC (Zn2+). The down-field signals ascribable to the aromatic rings (thiophene and pyridine) were shifted down from 8.08 to 8.15 ppm for thiophene and from 8.3 to 8.35 ppm for pyridine moiety.
Additionally, new signals close to the main signals of the aromatic moieties appeared and negligible broadening was also observed, indicating the presence of constitutional dynamics in solution.48 The second series of signals in the lower magnetic field corresponding to the oxazoline groups also shifted down-field from 4.5 to 4.6 ppm for CH2O and from 4.06 to 4.12 ppm for CH2N groups. However, the main changes upon complexation are seen when r is close to unity and a further addition (or increase) of Zn2+ IC concentration does not lead to shift changes in the NMR spectra. These results, together with the UV/vis study, revealed that the most stable and probably possessing the longest MSP chains are systems with r = 1. Besides that, the formation of MSPs using the P-Bt unimer and the Cu2+ IC with r = 1 was confirmed by a MALDI-TOF study. The mass m/z (z = 2) corresponding to the U–IC–U–IC sequence (found for C60Cu2H48N12O8S4, (658.8)), together with the peaks of different fragments with higher m/z values, indicate the certainty of MSP formation and the presence of high molecular oligomeric sequences that were observed (see Fig. S13, ESI†). Further deciphering of the MALDI-TOF spectrum is nearly impossible (also due to the presence of ClO4− counterions).
Experimental section
Materials
Tetrabutylammonium bromide, thionyl chloride, 2-chloroethylamine hydrochloride, triethylamine, 1,3-bis(diphenylphosphino)propane nickel(II) chloride [Ni(dppp)Cl2] (dpp stands for diphenylphosphino-propane), 2,5-bis(trimethylstannyl)thiophene, 5,5′-bis(trimethylstannyl)-2,2′-bithiophene, 2,5-bis(trimethylstannyl)-thieno[3,2-b]thiophene, zinc(II) perchlorate hexahydrate, iron(II) perchlorate hydrate, nickel(II) perchlorate hexahydrate, copper(II) perchlorate hexahydrate, (all from Sigma-Aldrich) and 4-hydroxypyridine-2,6-dicarboxylic (chelidamic) acid (1), were obtained from commercial sources (Sigma-Aldrich) and used without further purification.Solvents used for the palladium catalyzed synthesis of unimers and ligands such as toluene and tetrahydrofuran (THF) were dried and distilled by standard methods and stored under argon. Toluene (Lachner) was distilled under argon from sodium/benzophenone prior to use, THF (Aldrich) was distilled from LiAlH4 before use, methanol (Aldrich), dichloromethane and chloroform (both from Lachner), were used as obtained.
Measurements
1H and 13C NMR spectra were recorded on Varian UNITYINOVA 400 or Varian SYSTEM 300 instruments in CDCl3, d6-DMSO, CD3OD and referenced to the solvent signal (δ): 7.25 ppm (CDCl3), 2.50 ppm (d6-DMSO) or 3.33, 4.78 ppm (CD3OD) for 1H and 77.0 ppm (CDCl3), 49.15 ppm (CD3OD) ppm for 13C spectra. Coupling constants, J (in Hz), were obtained by a first-order analysis. UV/vis spectra were recorded on a Shimadzu UV-2401PC using a solvent mixture (CHCl3/CH3OH (1/1)) of the prepared compounds. Solid state samples were prepared using coating via a slow solvent evaporation technique on the surface of the quartz plate.Photoluminescence spectra were measured on a Fluorolog 3-22 Jobin Yvon Spex instrument (Jobin Yvon Instruments S. A., Inc., USA) in CHCl3 + CH3OH solutions using a four-window quartz cuvette (1 cm). The emission spectra were excited using the wavelength, λex, equal to the position of the absorption maxima of a particular compound. Quantum yields, ϕF, of photoluminescence were measured using an integration sphere Quanta-φ F-3029 Horriba Jobin Yvon. X-ray diffraction studies were performed using Bragg–Brentano geometry on a PanAlytical X'Pert PRO MPD X-ray diffraction system equipped with a PIXcel detector using a Cu-anode (Cu-Kα; λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS) measurements were carried out with a K-Alpha+ spectrometer (ThermoFisher Scientific, East Grinstead, UK). The samples were analyzed using a micro-focused, monochromated Al Kα X-ray source (400 μm spot size) at an angle of incidence of 30° (measured from the surface) and an emission angle normal to the surface. The kinetic energy of the electrons was measured using a 180° hemispherical energy analyzer operated in the constant analyzer energy mode (CAE) at a 50 eV pass energy for the high resolution spectra. Data acquisition and processing were performed using Thermo Advantage software. XPS spectra were fitted with Voigt profiles obtained by convolving Lorentzian and Gaussian functions. The analyzer transmission function, Scofield sensitivity factors, and effective attenuation lengths (EALs) for photoelectrons were applied for quantification. EALs were calculated using the standard TPP-2M formalism. All the spectra were referenced to the C1s sp2 peak at 284.2 eV. The BE scale was controlled by the well-known positions of the photoelectron C–C and C–H, C–O and C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O peaks of polyethylene terephthalate and Cu2p, Ag3d, and Au4f peaks of metallic Cu, Ag and Au, respectively. The BE uncertainty of the reported measurements and analysis is in the range of ±0.1 eV.
O)–O peaks of polyethylene terephthalate and Cu2p, Ag3d, and Au4f peaks of metallic Cu, Ag and Au, respectively. The BE uncertainty of the reported measurements and analysis is in the range of ±0.1 eV.
SEC records were obtained using a Spectra Physics Analytical HPLC pump P1000 with two SEC columns: Polymer Labs (Bristol, USA) Mixed-D and Mixed-E. The system was equipped with a Thermo UV6000 DAD detector. 0.05 M tetrabutylammoniumhexafluoro-phosphate in CHCl3/methanol (1/1, v/v, CHROMASOLV, RiedeldeHaen) was used as an eluent (0.7 mL min−1).
Molecular weights of the MSPs were measured by the following method. The raw samples were withdrawn directly from the polymerization mixture and dissolved in a 1/1 chloroform/methanol mixture to a concentration of 1 mg mL−1. Samples were mixed with a matrix solution (saturated solution of 10 mg mL−1 dithranol in 1/1 chloroform/methanol mixture) in a ratio of 1/100 (v/v). 1 μL of this mixture was deposited on a polished steel MALDI target (Bruker).
Mass spectra of the polymers were measured on an UltrafleXtreme TOF instrument (Bruker), equipped with a 355 nm smartbeam-2 laser, capable of a pulsing frequency of 1 kHz. The mass spectrometer was operated by FlexControl 3.3 software (Bruker). The acquired spectra were processed by FlexAnalysis 3.3 software (Bruker). The ionization laser power was adjusted to just above the threshold in order to produce charged species.
Synthesis of starting materials
Starting compounds dimethyl 4-hydroxypyridine-2,6-dicarboxylate (2), dimethyl 4-bromopyridine-2,6-dicarboxylate (3), 4-bromopyridine-2,6-dicarboxylic acid (4), 4-bromopyridine-2,6-dicarbonyl dichloride (5), 4-bromo-N2,N6-bis(2-chloroethyl)pyridine-2,6-dicarboxamide (6) and 4-bromo-2,6-bis(4,5-dihydro-2-oxazolyl)-pyridine (7) were prepared according to a modified previously reported synthetic procedure.49 For more detailed information (synthesis and spectra) see the ESI.†Synthesis of unimers
935 g mol−1), C, 60.19; H, 4.38; N, 14.04; S, 10.71, found: C, 59.87; H, 4.41; N, 14.52; S, 10.45.
Preparation of metallo-supramolecular polymers
A set of solutions of the unimers P-Th, P-Bt and P-Te at both a constant concentration (2 × 10−4 M) and volume (2 mL) was prepared in a chloroform/methanol mixture (1/1 by vol). Additionally, the solutions of Fe2+, Zn2+, Ni2+, and Cu2+ perchlorate hydrates (2 × 10−2 M) in the same solvent mixture were added into the solution of the particular unimer in ratios r from 0.1 eq. to 2.5 eq. The optical properties (UV/vis, photoluminescence) of all the solutions were measured after 24 hours in order to get an inner equilibrium of the solution for signal measurement.NMR titration study
NMR titration experiments of the MSPs were performed in DMSO-d6 solvent. The mixture of deuterated solvents, chloroform/methanol (1/1 by volume), applicable for all the other spectroscopic studies in solution in the current work was not used due to the insolubility of the unimer to the concentration (2 × 10−2 M) required for NMR study and a sufficient resolution of the NMR spectrum for interpretation. Therefore, DMSO-d6 solvent was used only for NMR titration of the P-Th unimer with the Zn2+ IC as a suitable solvent allowing the preparation of solutions at the desired concentrations for NMR analysis. The stock solution of P-Th (2 × 10−2 M) was prepared in the mentioned solvent and divided into 8 portions equal to 0.5 mL each. Simultaneously, Zn(ClO4)2 × 6H2O at a concentration of 0.2 M in deuterated DMSO was prepared and added to the P-Th solutions stepwise in the following r: 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2 eq. The prepared samples were left for 24 h to achieve inner equilibrium before NMR measurements.NMR experiments for the Pybox-Th ligand were performed in the same way as for P-Th, but using a deuterated mixture of chloroform and methanol (1:1) as a solvent.
End-capping experiment of MSPs based on P-Bt and Zn2+
Stock solutions of P-Bt (SS1) and Pybox-Th (SS2) (0.02 M for both) in the mixture of solvents (methanol and chloroform, 1:1) were prepared. The structure of Pybox-Th is outlined in Scheme 1. An aliquot of 20 μL of each solution (SS1 and SS2) was taken and dissolved to 2 mL. For convenience, dissolved P-Bt is called SA solution and Pybox-Th is called SB solution. The UV/vis spectra of the prepared samples were measured. The solution of Zn(ClO4)2 × 6H2O (0.02 M) was prepared and used as the IC during the synthesis of MSPs by the following method: to the SA solution was added Zn2+ metal ions (20 μL) and the resulting mixture, referred as C was stirred for 1 h and after that was left for 24 h in order to have the inner equilibrium in solution mentioned above. The UV/vis spectrum of the solution was measured in order to check the formation of the metallo-supramolecular polymer. Subsequently, 20 μL of Pybox-Th (SS2) was added to the SC solution. After that, the resulting solution was stirred for 1 h and subjected to UV/vis measurement. Additionally, 10 μL of Zn2+ ICs were added to the SB solution and the absorption spectrum was recorded.
Conclusions
In summary, the novel ditopic pybox based unimers, using cheap and available chelidamic acid as a precursor, have been successfully prepared and characterized. Upon treatment of the synthesized unimers with ion couplers, Fe2+, Zn2+, Ni2+, Cu2+, the metallo-supramolecular polymers were formed. Formation of the MSPs was investigated by UV/vis, fluorescence and NMR spectroscopies. The appearance of new absorption bands and isosbestic points confirmed the formation of the above-mentioned species. The incorporation of different metal ions into the polymer chain resulted in a difference in the optical properties of the MSPs formed. The highest shift of maximal absorption to lower energies, compared to free unimers, was observed for complexation/assembling with the Cu2+ IC. Adversely, the lowest shift of the maximal absorption position to lower energies was detected for the Fe2+ ion coupler for all the unimers in the studied group, but in this case new bands around 600 nm, ascribable to the metal to ligand charge transfer transition (MLCT), were observed. Complexation of unimers with Zn2+ leads to a significant increase in fluorescence intensity, contrary to all the other metals, which showed fluorescence quenching. However, the presence of uncoordinated metal centers in the metallo-supramolecular polymers makes them capable of interacting with certain tridentate monotopic ligands. This confirms the dynamic character of the prepared MSPs (constitutional dynamics between formed MSP chains and free unimer in solution).Acknowledgements
The authors are grateful to the Czech Science Foundation (project 15-22305S) and the COST Action CM1302 (SIPS) European Network on Smart Inorganic Polymers and the Grant Agency of Charles University (project number 876213 and 796713) for financial support.References
- J.-M. Lehn, Supramol. Chem.: Concepts and Perspectives, VCH, Weinheim, New York, 1995, vol. 8, pp. 89–138 Search PubMed.
- J. L. Atwood, J. E. D. Davies, D. D. MacNicol, F. Vögtle and J.-M. Lehn, Compr. Supramol. Chem., Pergamon, Oxford, 1996, vol. 6, pp. 643–690 Search PubMed.
- J.-M. Lehn, Makromol. Chem., Macromol. Symp., 1993, 69, 1–17 CrossRef CAS.
- J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS.
- J. Zhang, S. M. Chen and X. H. Bu, Dalton Trans., 2010, 2487–2489 RSC.
- X. Zhao, D. Sun, S. Yuan, S. Feng, R. Cao, D. Yuan, S. Wang, J. Dou and D. Sun, Inorg. Chem., 2012, 51, 10350–10355 CrossRef CAS.
- Z. Z. Lu, R. Zhang, Y. Z. Li, Z. J. Guo and H. G. Zheng, J. Am. Chem. Soc., 2011, 133, 4172–4174 CrossRef CAS.
- M. Cibian, S. Langis-Barsetti, F. G. D. Mendonça, S. Touaibia, S. Derossi, D. Spasyuk and G. S. Hanan, Eur. J. Inorg. Chem., 2015, 1, 73–82 CrossRef.
- V. A. Friese and D. G. Kurth, Coord. Chem. Rev., 2008, 252, 199–211 CrossRef CAS.
- M. Chiper, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2009, 30, 565–578 CrossRef CAS.
- M. Burnworth, L. M. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–338 CrossRef CAS.
- J. Yuan, X. Fang, L. Zhang, G. Hong, Y. Lin, Q. Zheng, Y. Xu, Y. Ruan, W. Weng, H. Xia and G. Chen, J. Mater. Chem., 2012, 22, 11515–11522 RSC.
- J. R. Kumpfer and S. J. Rowan, J. Am. Chem. Soc., 2011, 133, 12866–12874 CrossRef CAS.
- M. H. Demirel, S. Köytepe, A. Gültek and T. Seçkin, J. Polym. Res., 2014, 21, 345–355 CrossRef.
- J. Brassinne, J.-P. Bourgeois, C.-A. Fustin and J.-F. Gohy, Soft Matter, 2014, 10, 3086–3092 RSC.
- J. R. Kumpfer, J. Jina and S. J. Rowan, J. Mater. Chem., 2010, 20, 145–151 RSC.
- P. R. Andres and U. S. Schubert, Adv. Mater., 2004, 16, 1043–1068 CrossRef CAS.
- F. S. Han, M. Higuchi, Y. Akasaka, Y. Otsuka and D. G. Kurth, Thin Solid Films, 2008, 516, 2469–2473 CrossRef CAS.
- A. Maier, K. Cheng, J. Savych and B. Tieke, ACS Appl. Mater. Interfaces, 2011, 3, 2710–2718 CrossRef CAS PubMed.
- Y. Li, T. Ren and W.-J. Dong, J. Photochem. Photobiol., A, 2013, 251, 1–9 CrossRef CAS.
- R. Dong, Y. Zhou, X. Huang, X. Zhu, Y. Lu and J. Shen, Adv. Mater., 2014, 27, 498–526 CrossRef.
- P. Štenclová, K. Šichová, I. Šloufová, J. Zedník, J. Vohlídal and J. Svoboda, Dalton Trans., 2016, 45, 1208–1224 RSC.
- J. Svoboda, P. Štenclová, F. Uhlík, J. Zedník and J. Vohlídal, Tetrahedron, 2011, 67, 75–79 CrossRef CAS.
- T. Vitvarova, J. Zednik, M. Blaha, J. Vohlidal and J. Svoboda, Eur. J. Inorg. Chem., 2012, 2012, 3866–3874 CrossRef CAS.
- I. Šloufová, B. Vlčková, M. Procházka, J. Svoboda and J. Vohlídal, J. Raman Spectrosc., 2014, 45, 338–348 CrossRef.
- P. Bláhová, J. Zedník, I. Šloufová, J. Vohlídal and J. Svoboda, Soft Mater., 2014, 12, 214–229 CrossRef.
- D. Rais, M. Menšík, P. Štenclová-Bláhová, J. Svoboda, J. Vohlídal and J. Pfleger, J. Phys. Chem. A, 2015, 24, 6203–6214 CrossRef.
- N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dincă, J. Am. Chem. Soc., 2013, 135, 13326–13329 CrossRef CAS.
- W. H. Zhang, S. W. Chien and T. S. A. Hora, Coord. Chem. Rev., 2011, 255, 1991–2024 CrossRef CAS.
- R. Custelcean, J. Bosano, P. V. Bonnesen, V. Kertesz and B. P. Hay, Angew. Chem., Int. Ed., 2009, 48, 4025–4029 CrossRef CAS.
- S. Singh, J. Chaturvedi and S. Bhattacharya, Dalton Trans., 2012, 41, 424–431 RSC.
- E. Menéndez-Pedregal, M. Vaquero, E. Lastra, P. Gamasa and A. Pizzano, Chem.–Eur. J., 2015, 21, 549–553 CrossRef.
- C. Friebe, A. Wild, J. Perelaer and U. S. Schubert, Macromol. Rapid Commun., 2012, 33, 503–509 CrossRef CAS.
- A. Duerrbeck, S. Gorelik, J. Hobley, J. Wu, A. Hor and N. Long, Chem. Commun., 2015, 51, 8656–8659 RSC.
- B. M. Paterson and P. S. Donnelly, Chem. Soc. Rev., 2011, 40, 3005–3018 RSC.
- L. Alibabaei, M. Wang, R. Giovannetti, J. Teuscher, D. D. Censo, J.-E. Moser, P. Comte, F. Pucciarelli, S. M. Zakeeruddin and M. Gratzel, Energy Environ. Sci., 2010, 3, 956–961 RSC.
- S. N. MacMillan, H. W. Hill and J. C. Peters, Chem. Sci., 2014, 5, 590–597 RSC.
- T. M. Porter, G. B. Hall, T. L. Groy and R. J. Trovitch, Dalton Trans., 2013, 42, 14689–14692 RSC.
- F. Czerny, P. Döhlert, M. Weidauer, E. Irran and S. Enthaler, Inorg. Chim. Acta, 2015, 425, 118–123 CrossRef CAS.
- J. D. Nobbs, A. K. Tomov, R. Cariou, V. C. Gibson, A. J. P. White and G. J. P. Britovsek, Dalton Trans., 2012, 41, 5949–5964 RSC.
- J. Jankowska, J. Paradowska, B. Rakiel and J. Mlynarski, J. Org. Chem., 2007, 72, 2228–2231 CrossRef CAS.
- P. J. Brown, D. S. Thomas, A. Köhler, J. S. Wilson, J.-S. Kim, C. M. Ramsdale, H. Sirringhaus and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 064203 CrossRef.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef.
- T. Suzuki, T. Sato, J. Zhang, M. Kanao, M. Higuchi and H. Maki, J. Mater. Chem. C, 2016, 4, 1594–1598 RSC.
- J. Zhang, C.-Y. Hsu and M. Higuchi, J. Photopolym. Sci. Technol., 2014, 27, 297–300 CrossRef.
- P. Štenclová-Bláhová, J. Svoboda, I. Šloufová and J. Vohlídal, Phys. Chem. Chem. Phys., 2015, 17, 13743–13756 RSC.
- A. Bremer, U. Müllich, A. Geist and P. J. Panak, New J. Chem., 2015, 39, 1330–1338 RSC.
- T. Vermonden, W. M. de Vos, A. T. M. Marcelis and E. J. R. Sudholter, Eur. J. Inorg. Chem., 2004, 14, 2847–2852 CrossRef.
- A. De Bettencourt-Dias, S. Viswanathan and A. Rollett, J. Am. Chem. Soc., 2007, 129, 15436–15437 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26665g |
| This journal is © The Royal Society of Chemistry 2017 |