
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmulsion centrifugal spinning for production of 3D drug releasing nanofibres with core/shell structure
Matej Buzgo *bc,
Michala Rampichovaac,
Karolina Vocetkovaabc,
Vera Sovkovaab,
Vera Lukasovaabc,
Miroslav Doupnikc,
Andrea Mickovaac,
Franco Rustichellia and
Evzen Amlerabc
*bc,
Michala Rampichovaac,
Karolina Vocetkovaabc,
Vera Sovkovaab,
Vera Lukasovaabc,
Miroslav Doupnikc,
Andrea Mickovaac,
Franco Rustichellia and
Evzen Amlerabc
aInstitute of Experimental Medicine, Czech Academy of Sciences, v.v.i., Vídeňská 1083, 142 20 Prague 4, Czech Republic
bDepartment of Biophysics, 2nd Faculty of Medicine, Charles University in Prague, V Úvalu 84, 150 06 Prague 5, Czech Republic. E-mail: buzgo@labdemo.cz
cUniversity Center of Energetically Efficient Buildings, Czech Technical University, Třinecká 1024, 273 43 Buštěhrad, Czech Republic
First published on 4th January 2017
Abstract
Herein we describe the core/shell centrifugal spinning process to deliver susceptible bioactive molecules. The fibres are produced from water-in-oil (W/O) emulsion, where poly-ε-caprolactone (PCL) dissolved in chloroform serves as the continuous phase and Pluronic F-68 (PF-68) dissolved in ethanol serves as the droplet phase. The successful core/shell fibre formation and discontinuous morphology of the core was identified by confocal microscopy. Encapsulation of a model enzyme resulted in protection of enzymatic activity and release during the first 7 days. The feasibility for tissue engineering applications was demonstrated by the incorporation of platelet lyophilisates as a source of growth factors. The cultivation of 3T3 fibroblasts and MG63 osteoblasts resulted in improved metabolic activity and fostered proliferation. Results of the study indicate that the proposed scaffold combines the 3D structure of scaffolds produced by centrifugal spinning with the drug delivery of growth factors.
Introduction
Tissue engineering has drawn much attention in recent years due to its potential to solve complex healthcare problems unsolvable using current methods.1,2 The regeneration capacity of the adult body is limited and further decreases with ageing. In order to achieve tissue regeneration, tissue engineering employs diverse scaffolds and protocols. Scaffolding platforms may be based on hydrogels, foams, 3D printed matrices and fibrous materials. The key properties for tissue engineering scaffolds are biocompatibility, mechanical and stereological properties respecting the target tissue and the capacity to integrate into the surrounding tissues.1,2Fibrous scaffolds have ideal properties for regeneration of a broad scale of target tissue. The nano- and microfibrous structure mimics the natural extracellular matrix and provides numerous contact points for cell adhesion. In addition, fibrous scaffolds have good mechanical properties, high porosity and pore interconnection.3,4 Due to this unique property, nanofibrous scaffolds offer numerous contact points for cells. The key strategy in producing fibrous scaffolds is electrospinning. Electrospinning enables the formation of a submicron fibrous mesh with a random or ordered structure.5 Electrospinning was successfully employed to encapsulate bioactive molecules for diverse tissue engineering applications. The release of encapsulated bioactive molecules stimulates and regulates cell recruitment, proliferation and differentiation in the site of injury. Sahoo et al.6 prepared coaxial nanofibres poly(lactic-co-glycolic acid) (PLGA) loaded with basic fibroblast growth factor (bFGF), and blend nanofibres with PLGA and bFGF. Jia et al.7 showed PLGA nanofibres loaded by dextran core with vascular endothelial growth factor (VEGF) for vascular tissue engineering applications. Tian et al.8 prepared VEGF-loaded poly(lactic acid-co-caprolactone) PLCL nanofibres by emulsion electrospinning. The scaffold enabled the sustained release of VEGF and stimulated mesenchymal stem cell (MSC) metabolic activity in a 20 day in vitro assay. In a recent study, improved cardiac differentiation of MSC on VEGF loaded scaffolds was shown.9
However, electrospun layers suffer from poor cellular penetration caused by small pore size and limited thickness. Several attempts were made to improve cellular infiltration to electrospun scaffolds. Mean pore size could be increased by a combination of fibres with leachable particles i.e. sacrificial co-fibres,10,11 salt12 or ice crystals.13 In addition, the organization of fibres highly depends on the collecting electrode.14 Rampichova et al.15 prepared a patterned 2D/3D collector, which produced a fibrous layer with loosely packed areas. The cells were able to penetrate through these areas and proliferate. Blakeney et al.16 produced fluffy 3D fibres using a special collector. Cocoon-like structures showed higher mean pore size and efficient cell penetration. Despite the possibility to increase the mean pore size, the thickness of an electrospun fibrous layer is also limited.33
Centrifugal spinning technology is an alternative approach to produce fibrous scaffolds from polymeric solutions and melts.17 In the centrifugal spinning process the polymeric solution or melt is placed in a rotating chamber with a thin orifice. When the applied centrifugal speed overcomes the surface tension of the solution, it is ejected from the orifice. Emerged jets are stretched and deposited on a collector. The produced fibres form fluffy meshes with a fibre diameter in the range from hundreds of nanometres to tens of micrometres. Centrifugal spinning was successfully applied for scaffold manufacturing from PLGA,18 polylactic acid,19 gelatin20 and polycaprolactone (PCL).20–22 However, the proposed scaffolds were rather a passive cell culture substrate. In order to stimulate cell proliferation and differentiation, bioactive molecules should be delivered using an optimal scaffold. So far, centrifugal spinning technology had been used for drug delivery applications only by Mary et al.23 In the study of tetracycline loaded PCL blended polyvinyl pyrrolidone (PVP) fibres were used as a drug delivery vehicle. The results showed rapid release of tetracycline within 24 h. To the best of our knowledge, no studies on delivering protein-based active molecules have yet been performed.
The aim of the study is to develop a new technique for delivering growth factors based on centrifugal spinning. In order to protect the proteins from denaturation in organic solvents we will develop core/shell centrifugal spinning technology. The proposed technology is based on emulsion co-spinning, where a continuous phase will give rise to a shell and droplet phase to the core of the fibres. The combination of the 3D structure of scaffolds produced by centrifugal spinning with drug delivery function will result in the formation of novel bioactive and biomimicking scaffolds.
Results and discussion
Development and characterization of Pluronic F-68 based (W)/PCL (O) emulsions
The proposed approach is based on the formation of a protein water-in-oil (W/O) emulsion, which is subsequently processed via a centrifugal spinning process. The technology is based on utilizing Pluronic F-68 (PF-68) as a protein surfactant stabilizing the W/O emulsion and protecting the proteins from denaturation. The polar (W) droplet phase is based on PF-68 dissolved in ethanol. An analysis of dynamic surface tension showed that the addition of PF-68 to pure ethanol increases the surface tension. Upon the addition of 0.1% PF-68 and 0.5% PF-68 the surface tension increased, indicating incomplete occupation of the surface by PF-68 molecules. PF-68 is an amphiphilic polymer containing polar polyethylene oxide (PEO) segments and non-polar polypropylene oxide (PPO) segments.24 Upon interaction with polar solvents, the polymer aggregates to thermodynamically stable particles with a minimal interaction of PPO with the polar solvent. The process therefore results in the self-assembly of particles with PEO coronas and PPO cores.25 Above 1% PF-68 concentration the surface tension was not further changed (2.5% PF-68, 5% PF-68, 7.5 PF-68, 10% PF-68 and 20% PF-68) (Fig. 1a). The result indicates that all formulations above 1% PF-68 were above the critical micellar concentration (CMC). An analysis by laser scattering showed the formation of PF-68 micelles with a size of about 5 nm. An increase in the PF-68 concentration resulted in the formation of particles with a size of about 500 nm (Fig. 1b). The highest number of large PF-68 micelles was found in the 20% PF-68 sample. The average size for 5% PF-68 was 3.7 nm and polydispersity index was 0.2. For 10% PF-68 the average diameter was 86 nm and polydispersity index was 0.33. For 20% PF-68 the polydispersity index was very high (0.69) indicating multiple peaks and non-stable emulsion properties. The observed micelle distribution is in accordance with literature.26,27 The small micelles correspond to pure PF-68 micelles. The large structures correspond to clusters containing aqueous components.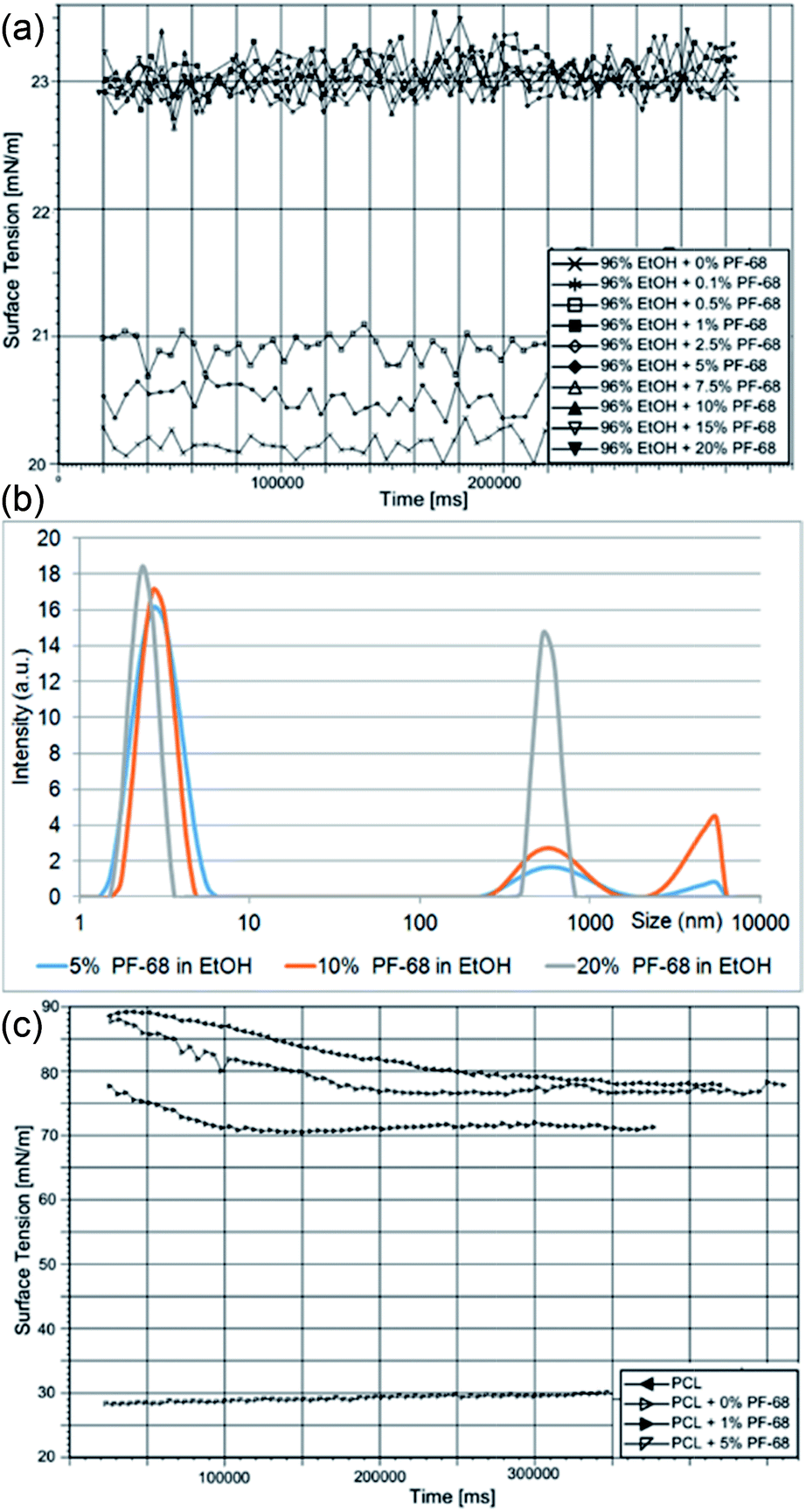 | ||
| Fig. 1 Characterization of PF-68 (W)/PCL (O) emulsions. (a) Effect of PF-68 on surface tension of EtOH. (b) Size of PF-68 micelles in 70% EtOH. (c) Effect of W/O emulsification on surface tension of PCL solution. | ||
The continuous phase of W/O emulsion was formed by 40% polycaprolactone (MW 45 kDa) dissolved in chloroform. The emulsification with the PF-68 ethanol/water phase resulted in the formation of a W/O emulsion. The emulsification changed the properties of the PCL solution. The addition of the polar phase containing 0% PF-68, 1% PF-68 and 5% PF-68 resulted in a decrease of surface tension (Fig. 1c). This decrease of surface tension is attributed to the accumulation of water/ethanol on the liquid surface. The decrease in case of 0% was low and decreased the tension from 80 mN m−1 to 77 mN m−1. The addition of surfactant in a concentration of 1% (wPF-68/wPCL) resulted in a further decrease to values about 70 mN m−1. The further increase in PF-68 concentration to 5% (wPF-68/wPCL) resulted in a significant decrease of surface tension (30 nN m−1). Such decrease may be caused by the higher concentration of PF-68 than CMC. The excess PF-68 occupied the surface of the polymeric liquid and resulted in a decrease of surface tension.
The mixing of PF-68 dissolved in 70% ethanol with 40% PCL in chloroform resulted in the formation of an emulsion formed by PF-68 particles. Upon mixing the ethanolic solution of PF-68 with non-polar solvent, the ethanol is readily mixed with chloroform. Such particles expose hydrophobic PPO segments to the non-polar solvent and polar PEO segments minimize contact with the non-polar solvent. As a result, the hydrophilic segments of PF-68 form a polar cavity and stimulate the rearrangement of PF-68 to particles to inverse micellar particles. Ben Henda et al.25 studied PF-68 micelles in polar and non-polar solvents. In case of binary mixtures, the hydrophobic solvents (phenol and p-xylene) resulted in the inhibition of micelle self-assembly or formation of various types of inverse micelles. However, in case of a ternary mixture with non-polar solvent and water the PF-68 behaviour became more complex. PF-68 was shown to form either inverse micelles or W/O emulsions with separated phases covered by surfactant molecules. Similar results were obtained for PF-127 in polar and non-polar solvents.28 In our experiment, the laser scattering measurement showed that the W/O emulsion contained PF-68 particles. The sample emulsified without PF-68 contained a water phase (PCL + 0% PF-68) with a size of 787 nm and polydispersity index was 0.72. The sample containing 5% PF-68 as droplet phase showed a particle size of about 355 nm and polydispersity index 0.3. With a further increase of PF-68 concentration the size was lower (170 nm for 10% PF-68 and polydispersity index of 0.5). Highly concentrated 20% PF-68 showed higher particle size (1476 nm and polydispersity index 0.39). This observation may be related to the aggregation of PF-68 particles and the formation of flocculated particle clusters. The size of PF-68 indicated that the particles were rather formed by a separated aqueous phase and non-polar phase covered by PF-68. Nevertheless, the experimental conditions are further complicated by the presence of 40% (w/v) PCL in the continuous phase. Polycaprolactone is insoluble in water and the formation of an emulsion results in altered rheological properties. Phase separation is the result of PLC interacting with a non-solvent (i.e. water, DMSO). The polymer-rich segments are dispersed in solvent and the polymer poor phase is occupied by a non-solvent.29 Similarly, phase separation occurs during the formation of W/O emulsions. The viscosity of polymeric liquids was measured in samples containing 40% PCL as a continuous phase and 0%, 1%, 5%, 10% and 20% PF-68 in 70% ethanol as an emulsion phase. The addition of an emulsion phase increased the viscosity of samples. The viscosity of 40% PCL without an emulsion phase was 790 ± 112 mPa s. The addition of an emulsion phase – 70% ethanol (PCL + 0% PF-68) resulted in an increase to 993 ± 92 mPa s. The addition of 5% PF-68 in 70% ethanol resulted in an increase to 1132 ± 112 mPa s. Nevertheless, the samples with 10% and 20% had even higher viscosity – 1370 ± 98 mPa s and 1610 ± 102 mPa s. The increase in viscosity may be caused by limited movement of PCL polymeric chains.
The centrifugal spinning process is connected with applying high centrifugal forces. The stabilization of the emulsion is therefore essential to obtain improved results of the encapsulation process. The stability of different formulations containing PF-68 were analysed by evaluating phase-separation during centrifugation. The emulsions were loaded to 2 ml tubes and centrifugation under 3000 × g was performed. The water phase was partially separated from the continuous nonpolar phase. The analysis by DLS showed that in case of a sample without PF-68 (PCL + 0% PF-68) the mean size was highly increased (from 787 nm to 1560 nm). The centrifugation of PCL + 5% PF solution resulted in an increase from 355 nm to 522 nm. Similarly, in case of PCL + 10% PF-68 the size increased from 170 nm to 332 nm. The increase of size was lower than a control sample without PF-68 and indicates the stabilization of the water phase by PF-68. Interestingly, for a PCL + 20% PF-68 solution, the size after centrifugation decreased from 1796 nm to 469 nm. This may be caused by the separation of aggregated particles after the partial separation of immiscible phases. The results clearly demonstrate that PF-68 was able to create water-phase particles upon mixing with PCL in chloroform. In addition, these particles were stable after centrifugation forces observed in the fibre preparation process.
Centrifugal spinning of coaxial fibres
The centrifugal spinning process was performed using Forcespinning™ technology (Fig. 2). Three experimental sets were performed by centrifugal core/shell technology: (1) core/shell fibres loaded with fluorescently labelled BSA, (2) core/shell fibres loaded with horseradish peroxidase and (3) core/shell fibres loaded with growth factors (platelet lyophylisates). All samples were prepared from W/O emulsions. First, the bioactive protein was mixed with PF-68 in 70% EtOH. When the concentration of PF-68 was above CMC (higher than wt 1%), PF-68 micelles were formed. The micelles had hydrophobic PPO cores and hydrophilic PEO coronas. Since the proteins are amphiphilic, part of the protein was probably embedded inside the PF-68 micelles. In the following step, PF-68 phase (W) was mixed with 40% PCL in chloroform as the oil phase (O). The samples were based on 40 ml of 40% PCL emulsified with 10 ml of PF-68 in 70% ethanol. The emulsification resulted in the formation of water/PF-68/protein droplets. The emulsion was further processed by centrifugal spinning to solid fibrous mesh. The fibres were spun using a rotating emitter with a G30 orifice. The rotation produces centrifugal force and draws the polymeric solution from the orifice in the form of a thin fibrous jet. The fibres are collected on the collector as a dry fibrous mesh. The yield of emulsion centrifugal spinning is 40 ± 7% as measured for basic PCL/10% PF-68 emulsions. However, the production yield is connected with emulsion stability and varies across formulations.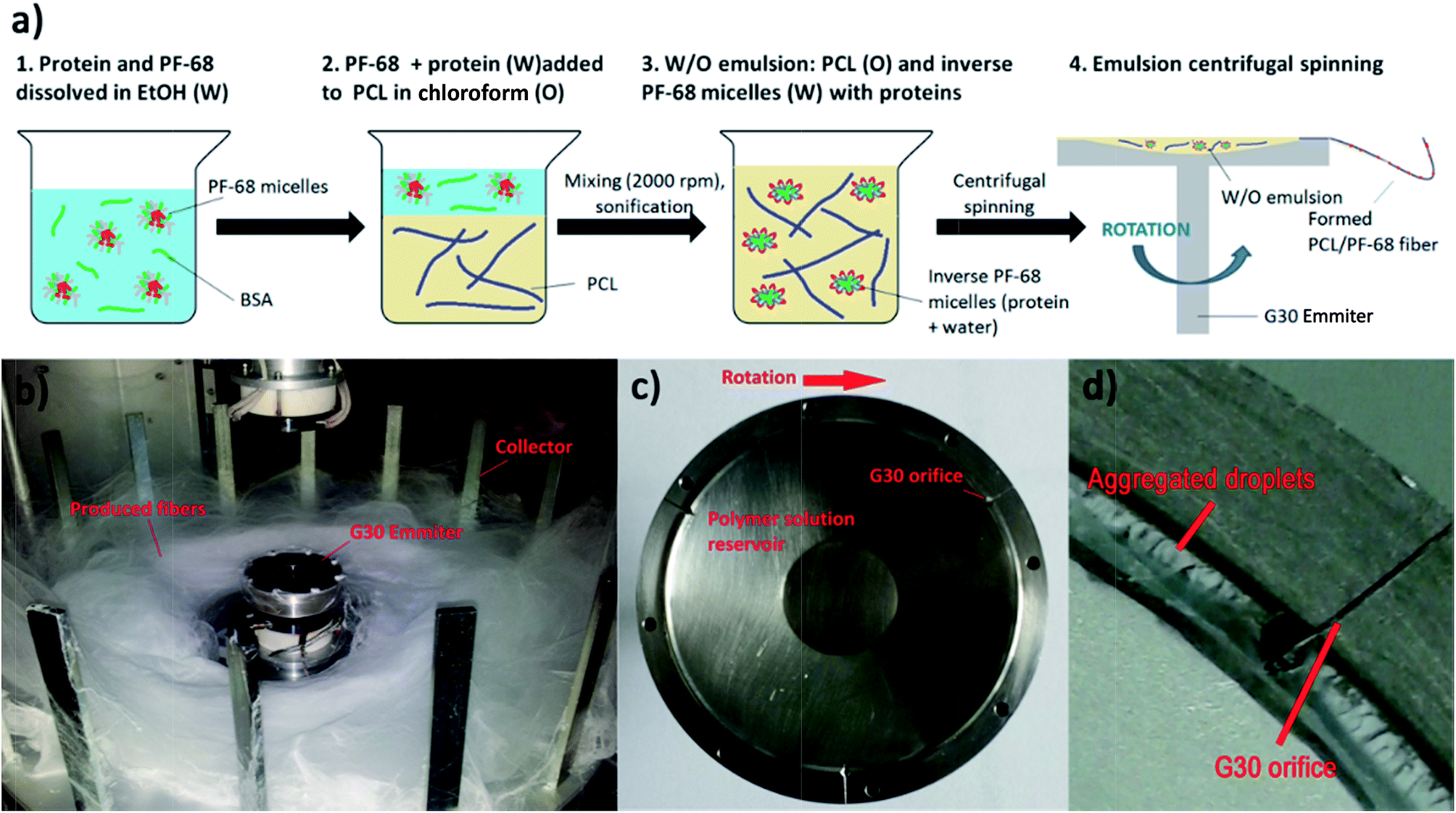 | ||
| Fig. 2 Scheme of emulsion centrifugal spinning process. (a) Description of emulsification process (b) centrifugal spinning apparatus. (c) G30 emitter structure. (d) The emulsion in sites of G30 orifice is not forming creamed emulsion aggregates. | ||
Scanning electron microscopic (SEM) analysis showed that the centrifugal spinning process resulted in the formation of a fibrous web (Fig. 3a–e). 40% PCL with blend encapsulated active molecules (LYO and FITC-BSA) showed fibrous morphology with microfibres (2.7 ± 1.5 μm) and nanofibres (338 ± 90 nm). Besides fibres, the mesh contained a large number of non-fibrous defects. The mean pore size is 47 ± 56 μm2 and the pores larger than 10 μm2 made up 36% of all pores. The samples based on emulsification by 70% ethanol (0% PF-68) showed more beaded morphology than blend PCL fibres. The fibrous mesh contained microfibres (1.6 ± 0.6 μm) and nanofibres (440 ± 110 nm) mixed with non-fibrous defects. The higher number of non-fibrous defects may be attributed to the separation of the hydrophobic PCL phase and the hydrophilic water based phase. The mean pore size was 38 ± 47 μm2 and the pores larger than 10 μm2 made up 32% of all pores. The sample based on emulsification with 5% PF-68 showed a lower number of non-fibrous droplets. The microfibres showed a higher mean size (2.4 ± 1.9 μm). The number of nanofibres was higher and showed a mean size of 470 ± 103 nm. The mean pore size was 30 ± 29 μm2 and pores >10 μm2 made up 24% of all pores. With a further increase of the PF-68 concentration the amount of defects was further lowered. The mean fibre size (2.1 ± 0.9 μm for microfibres and 453 ± 95 nm for nanofibres) and mean pore size (32 ± 21 μm2) was higher. Similarly, the samples with the highest concentration of PF-68 (20%) had similar morphology. The number of defects was low and the mean fibre size was 2 ± 1.2 μm for microfibres and 537 ± 168 nm for nanofibres. The mean pore size was 29 ± 39 μm2 and the number of pores larger than 10 μm2 was 21%. The observed data are consistent with the fibre sizes measured in our previous article.21
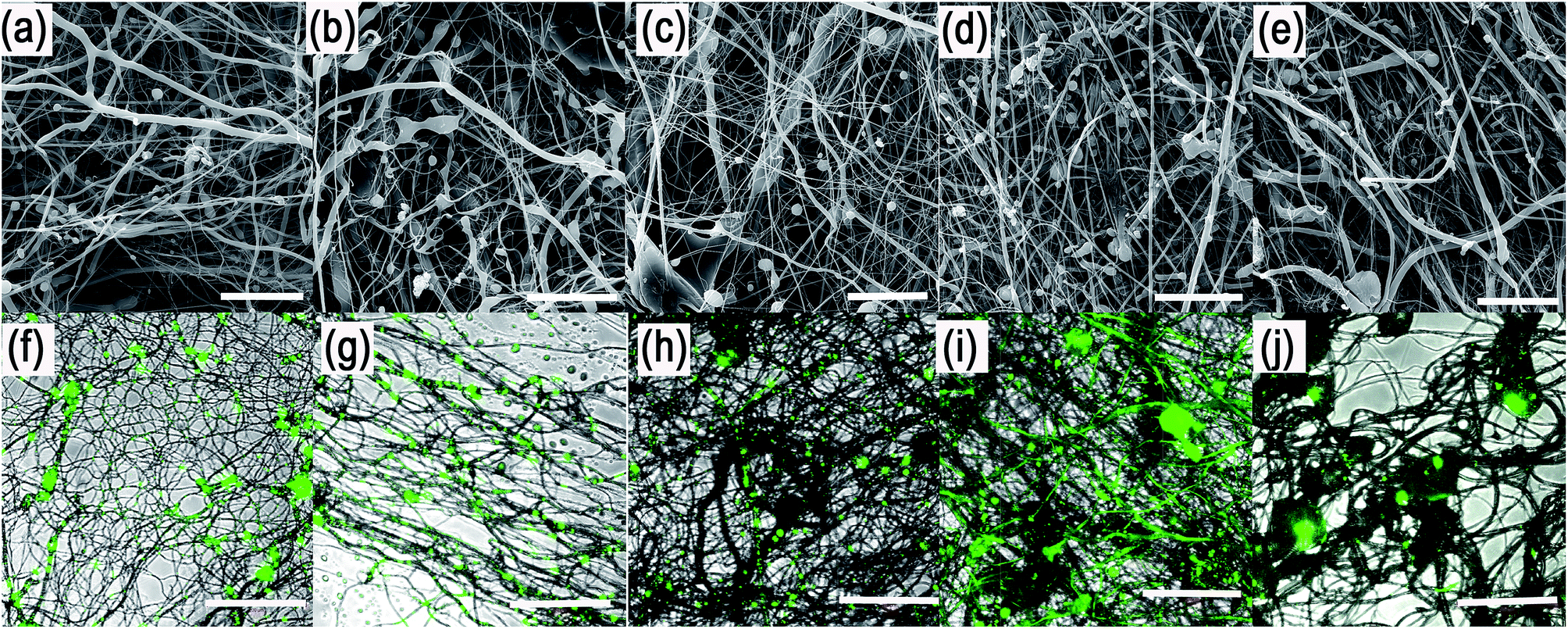 | ||
| Fig. 3 Morphology of core/shell fibres. (a) SEM image of 40% PCL + 20% PF-68 + LYO. (b) SEM image of 40% PCL + 10% PF-68 + LYO. (c) SEM image of 40% PCL + 5% PF-68 + LYO. (d) SEM image of 40% PCL + 0% PF-68 + LYO. (e) SEM image of 40% PCL blend with LYO. (f) Confocal image of 40% PCL + 20% PF-68 + FITC-BSA. (g) Confocal image of 40% PCL + 10% PF-68 + FITC-BSA. (h) Confocal image of 40% PCL + 5% PF-68 + FITC-BSA. (i) Confocal image of 40% PCL + 0% PF-68 + FITC-BSA. (j) Confocal image of 40% PCL blend with FITC-BSA Scale bar in all SEM samples 50 μm and in all confocal samples 200 μm. | ||
In order to evaluate the internal morphology, the fluorescently labelled FITC-BSA was encapsulated to the fibres and the localization was detected by confocal microscopy (Fig. 3f–j). The analyses showed that the FITC-BSA was localized outside the fibres in the PCL blend mesh. The localization of fluorescence was predominantly in defects and non-fibrous areas. Similarly, the sample based on emulsion without PF-68 (PCL + 0% PF-68) showed distribution in non-fibrous areas. However, the protein was also localized along the fibres. In case of samples with PF-68 the localization was dominantly along the fibres. The localization of FITC-BSA was in the case of 5%, 10% and 20% PF-68 samples in distinct areas. As the concentration of PF-68 rises, the number of droplets along the fibres also rises. However, in case of 20% PF-68 the droplets were merged and formed bigger droplets. The localization indicates that the fibres with PF-68 were formed by regions containing zones with fluorescent molecules and zones without fluorescent signal. Such structure produces core/shell fibres with an island-in-the-see structure. The structure of the centrifugal spun fibres resembles the structure of core/shell fibres produced by emulsion electrospinning.30–32 The chemical principle of both methods is similar. However, the fabrication process uses a different physical principle for fibre formation. Electrospinning is based on the steady-state drawing of fibres from the polymeric solution by electrostatic forces. On the other hand, the proposed emulsion centrifugal spinning technique is a dynamic process. The centrifugation draws fibres from the polymeric solution and simultaneously changes the properties of the polymer solution. In the present study we employed the emulsification method with PF-68 as the core phase. PF-68 may form inverse micelles and protect the aggregation of micelles upon centrifugation, as indicated by our results from centrifugation of W/O emulsion and the structure of core droplets observed by confocal microscopy. On the other hand, the centrifugation process results in partial separation of the aqueous phase resulting in decreased efficacy of the process. Therefore, future research should be focused on increasing the stability of the emulsion to decrease this negative effect. Nevertheless, the G30 emitter morphology with only 3 orifices results in the aggregation of droplets on the walls of the emitter. A construction change to a linear form of emitter minimizing aggregation on the walls may be beneficial for improved fibre quality.
Encapsulation of HRP PF-68-based core/shell fibres improves protection of enzymatic activity during encapsulation
In order to evaluate the internal morphology, the fluorescently labelled FITC-BSA was encapsulated to the fibres and the localization was detected by confocal microscopy (Fig. 3f–j). The analyses showed that the FITC-BSA was localized outside the fibres in the PCL blend mesh. The localization of fluorescence was predominantly in defects and non-fibrous areas. Similarly, the sample based on emulsion without PF-68 (PCL + 0% PF-68) showed distribution in non-fibrous areas. However, the protein was also localized along the fibres. In case of samples with PF-68 the localization was dominantly along the fibres. The localization of FITC-BSA was in the case of 5%, 10% and 20% PF-68 samples in distinct areas. As the concentration of PF-68 rises, the number of droplets along the fibres also rises. However, in case of 20% PF-68 the droplets were merged and formed bigger droplets. The localization indicates that the fibres with PF-68 were formed by regions containing zones with fluorescent molecules and zones without fluorescent signal. Such structure produces core/shell fibres with an island-in-the-see structure. The structure of the centrifugal spun fibres resembles the structure of core/shell fibres produced by emulsion electrospinning.30–32 The chemical principle of both methods is similar. However, the fabrication process uses a different physical principle for fibre formation. Electrospinning is based on the steady-state drawing of fibres from the polymeric solution by electrostatic forces. On the other hand, the proposed emulsion centrifugal spinning technique is a dynamic process. The centrifugation draws fibres from the polymeric solution and simultaneously changes the properties of the polymer solution. In the present study we employed the emulsification method with PF-68 as the core phase. PF-68 may form inverse micelles and protect the aggregation of micelles upon centrifugation, as indicated by our results from centrifugation of W/O emulsion and the structure of core droplets observed by confocal microscopy. On the other hand, the centrifugation process results in partial separation of the aqueous phase resulting in decreased efficacy of the process. Therefore, future research should be focused on increasing the stability of the emulsion to decrease this negative effect. Nevertheless, the G30 emitter morphology with only 3 orifices results in the aggregation of droplets on the walls of the emitter. A construction change to a linear form of emitter minimizing aggregation on the walls may be beneficial for improved fibre quality.Encapsulation of HRP PF-68-based core/shell fibres improves protection of enzymatic activity during encapsulation
The classical approach for preparing nanofibre based drug delivery systems is blend spinning.33 The polymer/drug composite is prepared by a simple mixing of miscible solutions and is often enhanced by mechanical or ultrasound dispergation. The main constraint of using blend spinning for delivering protein-based therapeutics is their denaturation in organic solvents. Biocompatible polymers (such as PCL, PLGA, PU, etc.) supporting adhesion of cells are typically hydrophobic and soluble in organic solvents. Therefore, mixing proteins with organic solvents decreases their activity. Such a disadvantage could be overcome by eliminating the contact with non-polar solvents. In our previous research we have shown that the encapsulation of proteins to liposomes embedded in fibres produced by coaxial electrospinning eliminated the degradation and improved the bioactivity of HRP.34 In addition, emulsion electrospinning is an alternative approach for producing core/shell fibres. Due to the high interfacial tension between the core and shell phases, the mobility of molecules is low and contact between susceptible bioactive molecules and nonpolar solvent is significantly reduced. In order to evaluate the hypothesis, the biological activity of a model enzyme – HRP was assessed. We prepared 5 samples with a distinct concentration of PF-68 in the emulsion phase. Blend encapsulation of HRP was used as a control to the emulsion approach. The morphology of fibres showed similar morphology as samples with embedded lyophilisates and BSA-FITC. The samples contained nano/microfibres with droplets. The detailed micrographs in Fig. 4 show the smooth surface of fibres in the case of 5%, 10% and 20% PF-68. The analysis of HRP activity was performed by analysing the activity of released HRP (Fig. 5a). The samples with 20%, 10% and 5% PF-68 (>60% activity) showed significantly higher activity compared with samples without PF-68 (<30% activity). The protective activity of PF-68 on the protein structure is well known.35 Due to the amphiphilic structure and high flexibility, PF-68 interacts with both the hydrophilic and hydrophobic segments of polymers and stabilizes the protein structure in a harsh environment. For instance, PF-68 is used as a protein stabilization agent for vaccines.36 The difference in protective activity between different concentrations of PF-68 may be related to phase transition during W/O emulsification. We hypothesize that in samples with a lower PF-68 concentration upon mixing with the chloroform phase the PF-68 molecules changed conformation. Upon transition from PPO-core micelles to PEO-core micelles the HRP may be in contact with chloroform. In samples with a higher PF-68 concentration, the phase transition may occur on the surface of the PF-68 particle structure and does not affect the structure inside the micelle. The protection of enzyme activity was observed in several works on emulsion electrospinning. Li et al.37 prepared PLA–PEG composite nanofibres loaded by protease K by emulsion electrospinning. The released proteinase K accelerated the biodegradation of the scaffold. Briggs et al.38 evaluated the effect of sonication and surfactant on lysozyme activity. The results showed that both addition of polar polymer to droplet and sonication lead to higher lysozyme activity after release from the nanofibres (Fig. 6). | ||
| Fig. 4 Morphology of core/shell fibres visualized by SEM. (a) 40% PCL + 20% PF-68 + HRP. (b) 40% PCL + 10% PF-68 + HRP. (c) 40% PCL + 5% PF-68 + HRP. (d) 40% PCL + 0% PF-68 + HRP. (e) 40% PCL blend with HRP. Scale bar 5 μm. | ||
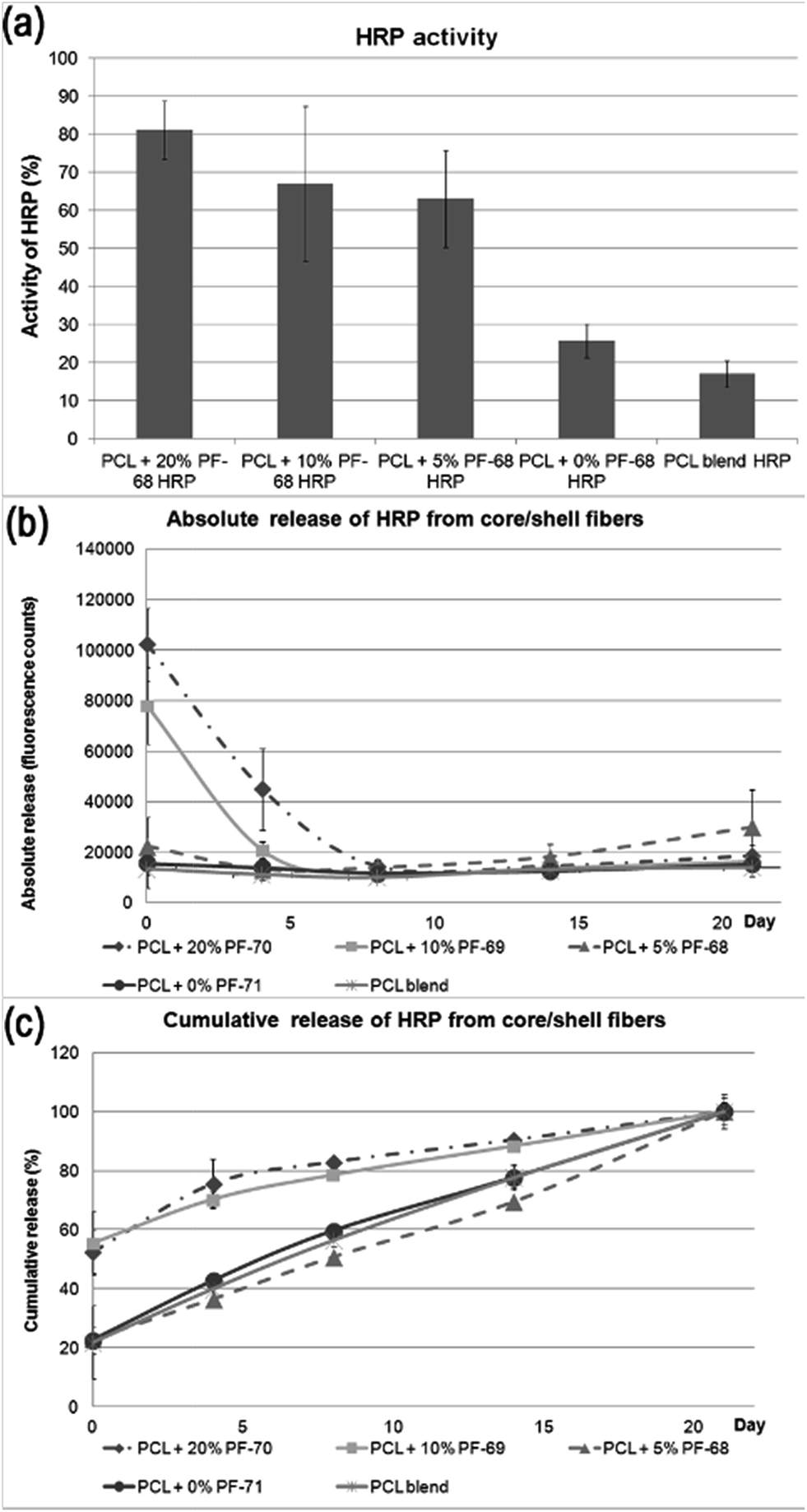 | ||
| Fig. 5 Release kinetics of HRP from core/shell fibers. (a) Activity of HRP released from core/shell fibers. (b) Absolute release of HRP from core/shell fibers. (c) Cumulative release of HRP from core/shell fibers. | ||
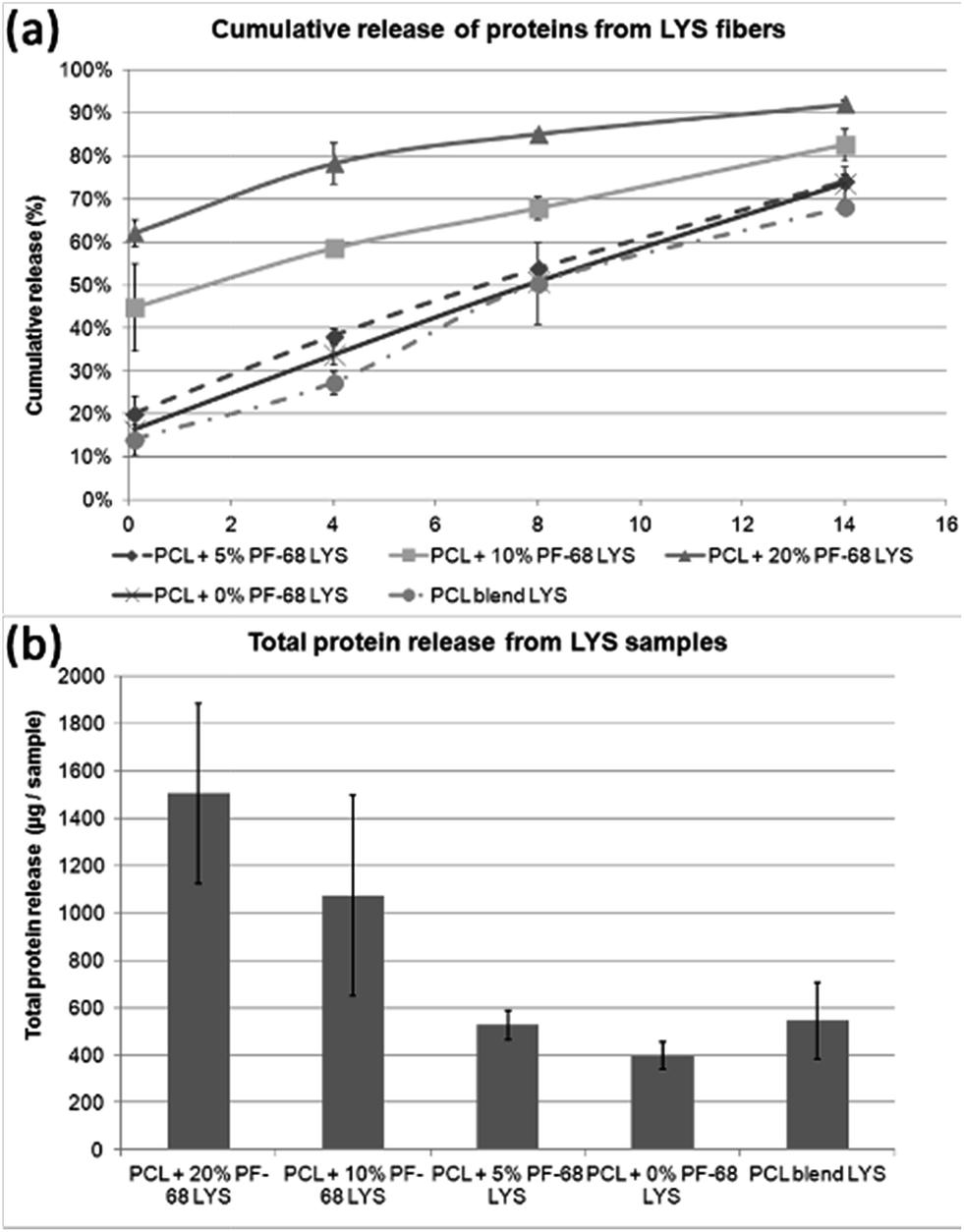 | ||
| Fig. 6 Release kinetics of lyophilisate (LYO) from core/shell fibers. (a) Absolute release of proteins from LYO core/shell fibers. (b) Cumulative release of proteins from LYO core/shell fibers. | ||
Beside analysis of the enzyme activity, we focused on HRP release kinetics from fibres (Fig. 5b and c). The analysis of HRP release showed that release from core/shell fibres was faster compared to non-coaxial samples. The absolute release showed that in case of PF-68 the encapsulation efficacy was much higher with an increase in PF-68 concentration. The reason for such observation may be in the higher number of PF-68 micelles. The samples not exhibiting morphology with the droplet phase (PCL blend and PCL + 0% PF-68) showed slow release and a much lower concentration of the embedded HRP. The higher number of PF-68 micelles in 20% and 10% PF-68 was also detected by confocal microscopy of BSA-FITC loaded samples. The results indicate that with the increase of micelle content the encapsulation efficacy is improved and is demonstrated by a higher total release of the embedded protein. In addition, with the increase of PF-68 concentration, a higher number of micelles are exposed to the surface of fibres. Therefore, upon incubation with aqueous solution, the micelles dissolve in aqueous solution and support the release of proteins. These simultaneous effects may have important consequences for the biological applications of the system.
Growth factor release stimulates fibroblast metabolic activity and proliferation
Similarly, the release and encapsulation efficacy of samples containing platelet lyophilisates was evaluated. Platelet lyophilisates were harvested from human platelets and contained a broad spectrum of active agents. Multiplexed immunoassay was performed for quantification of bioactive proteins.The content of cytokines, chemokines and growth factors was evaluated (Table 1). The analysis showed a higher presence of anti-inflammatory cytokines such as IL-1ra and IL-10. The IL-4 and IL-13 were detected in concentrations below 20 pg ml−1. The pro-inflammatory cytokines were present in higher concentrations (IL-17, IL-8, IL-9, INF-γ) and TNF-α were detected in higher concentrations. The concentration of IL-1b, IL-2, IL-5, IL-6, IL-7 was below 50 pg ml−1. RANTES was the dominant chemokine present in platelets. In addition, MIP-1b, eotaxin, IP-10 and MCP-1 were present in concentration above 50 pg ml−1. However, from the tissue engineering point of view, the growth factors have the highest importance for stimulating cell proliferation and differentiation. PDGF-bb was the most abundant growth factor identified by X-MAP assay. In addition VEGF and bFGF were present in higher concentration. G-CSF and GM-CSF were also present in concentration above 100 pg ml−1. In addition, ELISA was performed for quantification of additional growth factors. EGF, HGF, KGF and SDF-1α had the highest concentration.
| Mediator | Averaged value, pg mL−1 | SD, pg mL−1 |
|---|---|---|
| Anti-inflammatory cytokines | ||
| IL-1ra | 236 | 3.3 |
| IL-4 | 15 | 0.3 |
| IL-10 | 58 | 0.9 |
| IL-13 | 9 | 0.5 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Pro-inflammatory cytokines | ||
| IL-1b | 7 | 0.1 |
| IL-5 | 8 | 0.1 |
| IL-6 | 21 | 2.3 |
| IL-7 | 46 | 1.2 |
| IL-8 | 116 | 5.0 |
| IL-9 | 130 | 3.6 |
| IL-12 | 128 | 12.8 |
| IL-17 | 1043 | 48.4 |
| IFN-γ | 165 | 6.8 |
| TNF-α | 145 | 8.5 |
|
||
| Chemokines | ||
| RANTES | 11132 |
939.5 |
| MIP-1a | 13 | 0.6 |
| MIP-1b | 157 | 3.9 |
| Eotaxin | 78 | 1.6 |
| IP-10 | 315 | 1.8 |
| MCP-1(MCAF) | 58 | 16.9 |
|
||
| Growth factors | ||
| VEGF | 510 | 5.2 |
| PDGF-bb | 14656 |
247.1 |
| FGF basic | 274 | 6.9 |
| G-CSF | 139 | 18.1 |
| GM-CSF | 98 | 3.8 |
| EGF | 591 | 123.0 |
| HGF | 514 | 49.2 |
| KGF | 50 | 5.5 |
| SDF1-a | 339 | 55.9 |
The release was tested from samples containing embedded platelet lyophilisates (4.5 μgLYO mgPCL−1) by either blend centrifugal spinning (PCL blend LYO) or emulsion centrifugal spinning. The emulsion centrifugal spinning was performed with 0%, 5%, 10% and 20% PF-68. The release was analysed by quantification of the released proteins. The results showed that similar to the case of HRP, the release from PF-68 containing fibres was fostered as the PF-68 concentration was higher. PCL + 20% PF-68 showed fast release during the first 7 days. In the case of PCL + 10% PF-68 the release was more sustained and showed release over the overall 14 day period. The samples with low PF-68 content (PCL + 5% PF-68 and PCL + 0% PF-68) showed slow release similar to the blend fibres. The reason may be in limited desorption from the core/shell fibres. In the case of lower PF-68 content the core droplets may be hidden in the non-degradable PCL structure. The release is then governed by limited diffusion from non-eroding PCL bulk polymeric matrix. The slower release is also demonstrated by low total release during the incubation period. The samples with LYO embedded in 10% and 20% PF-68 showed total release in order of 15 μg. On the other hand, release from samples with slow release showed only release of 5 μg of protein during the incubation period.
The bioactivity of samples was tested on a model of 3T3 murine fibroblasts in a 14 day experiment. Cells adhered similarly to all samples and also cell proliferation was similar on all samples. Differences were seen only on day 7, when the lowest cell number and metabolic activity was detected on the PCL blend sample. The results indicate that the release of platelet lyophilisates stimulated cell metabolic activity (Fig. 7a). However, the proliferation of cells on the blended sample was only slowed down and no significant difference was visible on day 14 (Fig. 7b). The measured data was confirmed using confocal microscopy (Fig. 8).
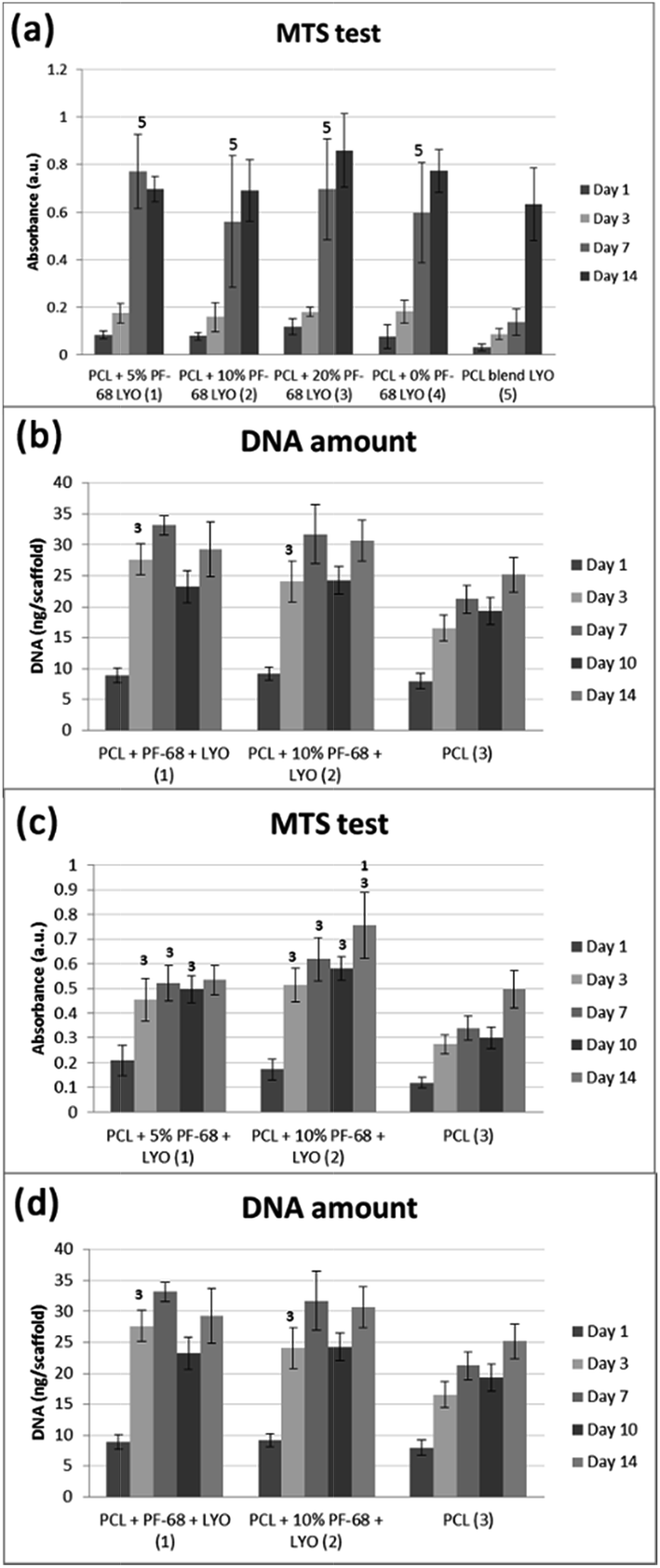 | ||
| Fig. 7 Results of cell culture studies. (a) Cell metabolic activity of 3T3 fibroblasts detected using MTS assay. (b) Cell proliferation of 3T3 fibroblasts evaluated using quantification of DNA. (c) Cell metabolic activity of MG63 osteoblasts detected using MTS assay. (d) Cell proliferation of MG63 osteoblasts evaluated using quantification of DNA. * indicates significant differences (p < 0.05). | ||
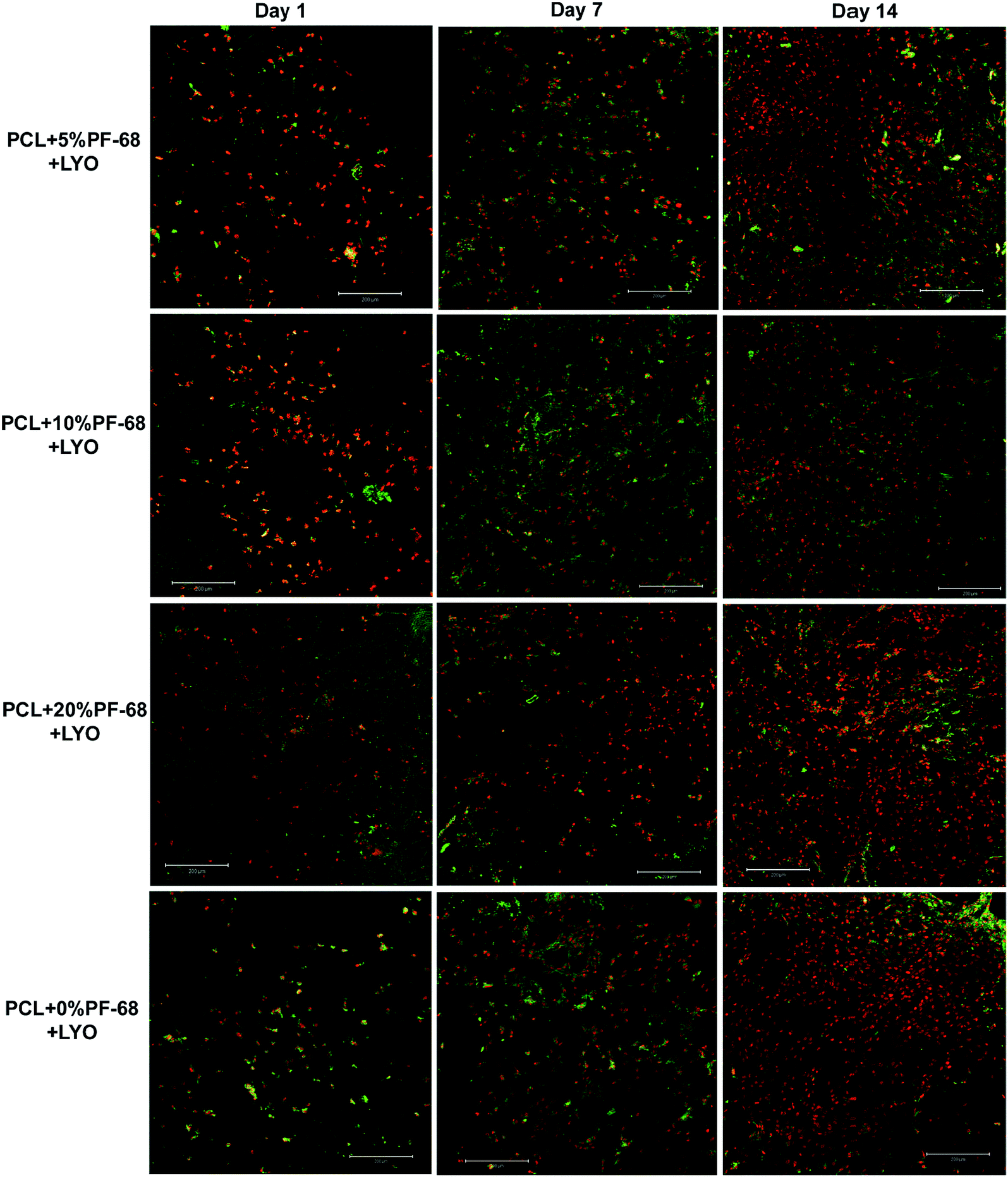 | ||
| Fig. 8 Confocal microscopy of coaxial nanofibers seeded with 3T3 fibroblasts. Cells were stained with DiOC (green colour) and propidium iodide (red colour) and visualized using confocal microscopy on day 1, 7 and 14. | ||
The cell culture study showed that release of platelet lyophilisates from core/shell fibres fosters cell proliferation and metabolic activity. However, the effect was rather transient and observed during the first 7 days. The reason may be connected with burst release from fibres, which was confirmed both in HRP and lyophilisates release experiments. The stability of proteins is rather limited and released proteins are rapidly degraded. For instance, the in vivo half-life of PDGF is only about 2 minutes (ref. 39) and EGF about 8 minutes.40 Therefore, the effect of release in the first days is diminished. The release may be prolonged by utilizing a polymer with slower degradation6 or by changing the ratio of degradable and non-degradable components.41 In addition, the amount of released active molecules may not be sufficient. The embedded amount of the most abundant growth factor – PDGF was about 350 pg per sample. Therefore, the increase in protein concentration may improve the efficacy of cell stimulation. Platelet-derived formulations were shown as a source of growth factors stimulating wound regeneration. Barsotti et al.42 showed platelet lyophilisates were able to promote proliferation of cells involved in wound healing in concentrations about 10–20% (v/v). However, we found in our previous study43 that upon encapsulation the lower concentrations of released platelet growth factors are sufficient to promote cell response compared to non-encapsulated platelets. This effect might be caused by the proximity of released growth factors to cells and formed bioactive molecule gradients. Similarly, Sell et al.44 prepared platelet lyophilisates-enriched nanofibres and showed improved cell colonization. Recombinant growth factors could be used to increase the loading of fibres and the amount of active molecules released. Yang et al.45 prepared core/shell nanofibres loaded by bFGF. The fibrous dressing showed improved wound regeneration in vivo compared to control samples without bFGF delivery. Similarly, Choi et al. prepared EGF enriched electrospun nanofibres and showed improved regeneration of chronic wounds.46 Up to date, only few growth factors are approved for clinical use, including bFGF, BMP-4 and BMP-7, which were successfully tested in clinical trials.47 However, significant change in formulation results in further clinical tests and the produced growth factors are not easily accessible on the market. Therefore, we believe that natural sources of growth factors from clinically well characterized sources, such as platelets, are the most promising source of growth factors for tissue engineering applications. Autologous PRP has already been used in clinical applications for their stimulating effect on cells and tissue healing, even without cell therapy.48
The proposed scaffold is designed for tissue engineering applications. Fibres produced by centrifugal spinning have fibrous morphology mimicking the ECM and high pore interconnection. The key advantage of electrospun scaffolds is less compacted scaffold morphology with larger pores. The higher pore size allows cells to penetrate deeply into the scaffold. The importance of penetration was demonstrated by Pham et al.49 Notably, the optimal pore size differs significantly for different types of cells. Pore sizes >300 μm have been recommended for vascularization in bone tissue engineering,50 whereas fibroblasts have been demonstrated to prefer a pore size of 6–20 μm.51
The centrifugal spinning process produces fibrous scaffolds enabling efficient penetration of cells to the scaffold. Recently, we compared the penetration to PCL scaffolds prepared by electrospinning and centrifugal spinning.21 The structure of the fibrous scaffold prepared by centrifugal spinning technology is optimal for application in skin tissue engineering. In the present work we have shown that the produced core/shell fibres fostered fibroblasts proliferation and enhanced metabolic activity. Nanofibrous scaffolds prepared by electrospinning have been shown as wound dressing.52,53 In addition, Plencner et al.54 recently showed the application of PCL fibres for stimulation of internal wound healing.
However, the application of the proposed system is not limited just to the field of skin tissue engineering. The platelet lyophilisates enriched core/shell fibres produced by centrifugal spinning have potential for bone tissue engineering. The potential for bone tissue engineering was evaluated by comparing core/shell samples (PCL with 5% and 10% PF-68 and embedded lyophilisates) with non-functionalized PCL fibres. The MTS assay showed significantly higher metabolic activity on samples functionalized by platelet lyophilisates than on non-functionalized samples during the first week (Fig. 7c). In the case of 5% PF-68 rapid growth of metabolic activity was observed between day 1–3. Similarly in the 10% PF-68 sample the rapid growth of metabolic activity was between days 1 and 3. However, the metabolic activity was further increased in subsequent days and steadily rose until day 14. Cell proliferation assay confirmed the trend in the MTS assay. However, the cell number was significantly higher on day 3 (Fig. 7d). The cell number on the following day was stable and control PCL achieved a comparable level on day 7. Confocal microscopy showed that cell number on day 7 was higher on samples with platelet lyophilisates and on day 14 similar on all scaffolds (Fig. 9).
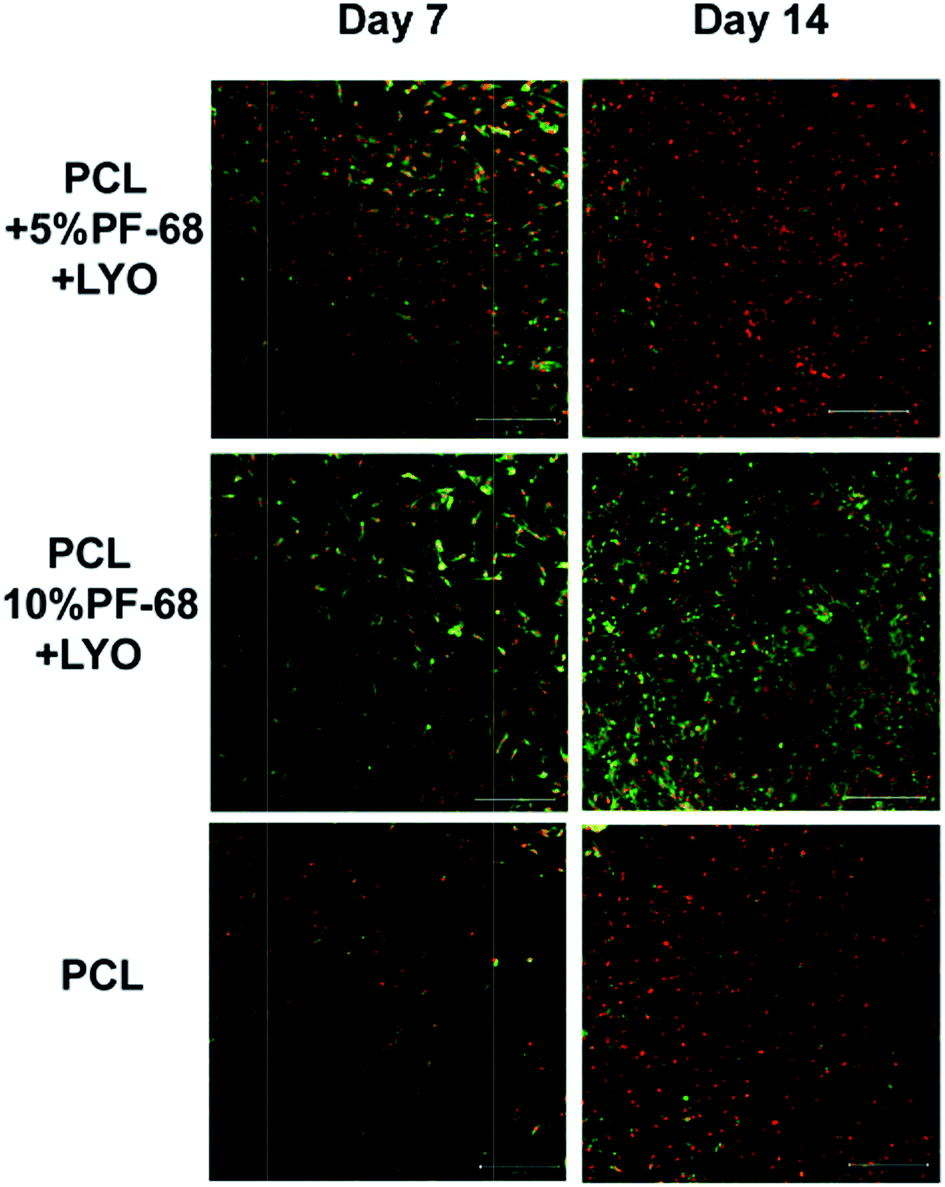 | ||
| Fig. 9 Confocal microscopy of coaxial nanofibers seeded with MG-63 cells. Cells were stained with DiOC (green colour) and propidium iodide (red colour). | ||
The positive effect of platelet-based formulations on bone tissue regeneration has been presented in numerous publications.55–57 The proposed technology of core/shell centrifugal spinning combines the advantages of 3D scaffolds with drug delivery structure. In addition, the soft-nature of the scaffold and degradation in time-frame of tissue regeneration ensures proper osteoinductive and osteoconductive properties.
The current study introduced novel core/shell centrifugal spinning technology. The results indicate that PF-68 based W/O emulsification is efficient in protecting bioactive proteins and enables the formation of stable emulsions processable to core/shell fibres by centrifugation spinning. In addition we have demonstrated the encapsulation of platelet lyophilisates to the core/shell fibres and biocompatibility with fibroblasts and osteoblasts. However, it is important to state that the presented results rather demonstrate the feasibility of core/shell centrifugal spinning technology. The new concept of core/shell centrifugal spinning is connected with numerous drawbacks. Work focusing on the stability of emulsion is essential for efficient encapsulation of diverse active molecules. The stabilization of the core phase and elimination of burst release will prolong the release times to achieve long-term protein release. Nevertheless, the biological properties of the system should be carefully validated on primary and stem cell models and relevant in vivo models.
Experimental
Emulsion centrifugal spinning
The emulsion centrifugal spinning technology is based on a double emulsification procedure followed by centrifugal spinning processing. First, 30% (w/v) Pluronic F-68 (PF-68, Sigma-Aldrich) dissolved in 90% ethanol is mixed with encapsulated protein dissolved in 50% ethanol and pure ethanol to achieve solution of 0.1, 1, 5, 10 or 20% PF-68 and encapsulated protein in 70% ethanol. The emulsification process is performed by drop-wise mixing of components followed by sonification. The emulsion of PF-68 and proteins is subsequently emulsified with 50% polycaprolactone (PCL, 45000 Da, Sigma-Aldrich) dissolved in chloroform to obtain 40% PCL dissolved in chloroform:ethanol. The ratio of the chloroform phase to the ethanol phase is 4:1. The ethanol phase is added drop-wise under stirring. The prepared emulsion is immediately processed by centrifugal spinning. The centrifugal spinning was performed by a Forcespinning process on a Cyclone L-1000 MD (Fiberio). All samples were processed using G30 needles at 11000 rpm and collector distance 10 cm. The fibres were collected on a vacuum assisted deposition system to prepare more condensed samples.
Evaluation of PF-68 emulsified in PCL using tensiometry and laser scattering
The effect of Pluronic F-68 in ethanol on surface tension was evaluated using a bubble-pressure tensiometre (BP50, Kruss). The samples with 0%, 1%, 5%, 10% and 20% PF-68 in ethanol or mixed with PCL were analysed using stable surface age mode. The results were interpreted as dependence of surface tension on time. The size distribution and zeta-potential of PF-68 droplets was analysed using a Zetasizer ZS (Malvern). The zeta-potential was evaluated using 40% PCL in chloroform as the continuous phase and PF-68 in 70% ethanol:water mixture as the droplet phase. Additional measurement focused on evaluating PF-68 in ethanol. The average droplet diameter was measured by dynamic light scattering (DLS) at 25 °C and an angle of 90° using a 5 mW He/Ne laser at 633 nm.
Centrifugal spinning of PCL/PF-68 coaxial samples for evaluation of core morphology
Coaxial fibers are based on mixture of two polymeric phases. The shell phase is supports cell adhesion and core is utilized for delivery of active molecules. In order to evaluate the possibility to prepare core/shell fibers by centrifugal spinning, morphology of the core phase, FITC-labelled bovine serum albumin (FITC-BSA) was used as a model drug. The samples were prepared as described previously and contained 300 μg ml−1 FITC-BSA in the core of the fibres. We prepared samples containing 0% PF-68 and FITC-BSA dispersed in 40% PCL (PCL/0% PF-68 (FITC-BSA)), 5% PF-68 and FITC-BSA dispersed in 40% PCL (PCL/0% PF-68 (FITC-BSA)), 10% PF-68 and FITC-BSA dispersed in 40% PCL (PCL/10% PF-68 (FITC-BSA)), 20% PF-68 and FITC-BSA dispersed in 40% PCL (PCL/20% PF-68 (FITC-BSA)). The control sample was prepared by directly dissolving a corresponding amount of FITC-BSA in 40% PCL solution (PCL blend FITC-BSA). The samples were centrifugally spun at 11000 rpm using a G30 emitter and static voltage of 48 V.
The morphology of fibres was evaluated using scanning electron microscopy (SEM). Air-dried samples of electrospun nanofibres were mounted on aluminium stubs and sputter-coated with a layer of gold using a Quorum Q150R. The samples were examined in Vega 3 SBU (Tescan) scanning electron microscope in the secondary electron mode at 15 kV. Mean fibre diameter was calculated by image analysis in the ImageJ program.
In order to examine the morphology of the core phase of emulsion nanofibres, confocal microscopy of the fluorescently labelled core was utilized. The distribution of FITC-BSA within the prepared fibres was observed using a Zeiss LSM 5 DUO confocal laser scanning microscope (FITC fluorescence, λex = 488 nm, λem = 520–560 nm).
Preparation of platelet lyophilisates
Fresh human leukocyte-depleted platelet concentrate derived from buffy coat in additive solution was obtained from a blood transfusion service (Sumperk, Czech Republic). The platelet concentration in the bag was 1020 × 109 platelets per l. The bag was prepared from the blood of 16 donors to minimize the inter-individual differences. The platelets were delivered on the day of production. Platelet lysate was prepared by the freeze–thaw method. The whole bag was frozen (−80 °C) and thawed (37 °C) three times in total to disrupt the cellular membranes. The solution was aliquoted and centrifuged (4100 g/15 min) to get rid of the cellular debris. Subsequently, the lysate was lyophilized under 480 mT (VirTis BenchTop Pro Freeze Dryer, SP Scientific, PA, USA) for 24 hours. The lyophilized platelet lysate was stored at −80 °C until use.Characterization of growth factor content
To quantify the concentrations of pro- and anti-inflammatory cytokines and growth factors the Bio-Plex 200 Multiplex System (Bio-Rad Laboratories, CA, USA) and enzyme-linked immunosorbent assay (ELISA, DuoSet®, R&D systems, USA) were used. To analyse the cytokine content of the platelet lysate, the commercially available cytokine panel (Bio-Plex Pro™ Human Cytokine 27-plex Assay, Bio-Rad Laboratories, CA, USA) was used in accordance with the manufacturer's instructions. The assay allows multiple cytokines to be quantified simultaneously in one well. Briefly, the platelet lysate was incubated with set of colour-coded magnetic beads, each conjugated with an antibody directed against a specific mediator. Biotinylated detection antibody was added and allowed to bind to streptavidin–phycoerythrin. To remove the unbound protein, thorough washing series were performed in between each step by an automatic wash station (Bio-Plex Pro™ II). Finally, the data were analysed using a BioPlex 200 instrument fitted with BioManager analysis software (5 parameter curve fitting). The concentration of growth factors (EGF, HGF, KGF and SDF1α) in the lyophilized platelet lysate was quantified by conducting the ELISA according to the manufacturer's instructions.Preparation of emulsion centrifugal 3D fibres with embedded platelet lyophilisates
The samples were prepared as described previously and contained 2 mg ml−1 platelet lyophilisates (LYO) in the core of the fibres. We prepared samples containing 0% PF-68 and platelet lyophilisate dispersed in 40% PCL (PCL/0% PF-68–LYO), 5% PF-68 and lyophilisate dispersed in 40% PCL (PCL/5% PF-68–LYO), 10% PF-68 and lyophilisate dispersed in 40% PCL (PCL/10% PF-68–LYO), 20% PF-68 and lyophilisate dispersed in 40% PCL (PCL/20% PF-68–LYO). The control sample was prepared by directly dissolving the corresponding lyophilisate in 40% PCL solution (PCL blend LYO). The samples were centrifugally spun at 11000 rpm using a G30 emitter and static voltage of 48 V.
Drug release studies on model of horseradish peroxidase (HRP) and platelet lyophilisates
The samples were prepared as described previously and contained 300 μg ml−1 HRP in the core of the fibres. We prepared samples containing 0% PF-68 and HRP dispersed in 40% PCL (PCL/0% PF-68 HRP), 5% PF-68 and HRP dispersed in 40% PCL (PCL/5% PF-68 HRP), 10% PF-68 and HRP dispersed in 40% PCL (PCL/10% PF-68 HRP), 20% PF-68 and HRP dispersed in 40% PCL (PCL/20% PF-68 HRP). The control sample was prepared by directly dissolving the corresponding HRP in 40% PCL solution (PCL blend HRP). The samples were centrifugally spun at 11000 rpm using a G30 emitter and static voltage of 48 V.
The samples containing HRP and LYO were subjected to a release experiment. The samples were cut into pieces (∼50 mg) and incubated with 1 ml of TBS buffer at room temperature. At specific intervals, the TBS buffer was withdrawn and replaced with fresh buffer. Release of HRP or platelet lyophilisates from the fibres was determined using a fluorescent Quant-IT Protein Assay Kit (Invitrogen). The assay was performed according to the manufacturer's guide. Briefly, 10 μl of sample or standard was mixed with 200 μl of fluorescent dye. The resulting fluorescence product was measured using a Synergy H1 multiplate fluorescence reader (Biotec, ex 470 nm, em. 570 nm). All values represent mean ± standard deviation of at least four independent samples.
The enzyme activity of the released HRP was measured on day 1 colorimetrically using tetramethylbenzidine (TMB, Sigma Aldrich) as the substrate. The enzyme substrate reaction was terminated with 50 μl of 2 N H2SO4 after 30 s. The enzyme activity was measured at 450 nm using an ELISA reader (Biotec, Synergy HT). The enzymatic activity was calculated as a percent ratio of enzyme concentration measured by the TMB assay (active enzyme, Sigma-Aldrich) and enzyme concentration measured by the Quant IT Protein Assay (overall protein, Life Technologies). All values represent mean ± standard deviation of at least ten independent experiments. Each experiment was performed from four independent samples.
Culture and seeding of human osteosarcoma cells (MG63) and murine 3T3 fibroblasts
Before cell seeding, scaffolds were cut into round patches of 6 mm diameter and sterilized using ethylene oxide. Samples were seeded with 10 × 103 MG-63 cells (Cell Lines Service GmbH, Germany) or alternatively with 3 × 103 3T3 fibroblasts (Sigma Aldrich, MO, USA) per well of the 96-well plate. Cells were left to adhere to the scaffolds for 2 hours in 30 μL of culture media (DMEM supplemented with 2% foetal bovine serum and penicillin/streptomycin). Subsequently, medium was added to the total volume of 300 μL per well.Metabolic activity determination
Metabolic activity was determined using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay (MTS assay, Promega). At 1, 3, 7, and 14 days, the scaffolds were transferred to a new 96-well plate containing 100 μL of fresh medium per well and 20 μL of CellTiter 96® Aqueous One Solution Reagent. The formazan absorbance in 100 μL of the solution was measured (λsample = 490 nm, λreference = 690 nm) after a 2 h incubation at 37 °C and 5% CO2 using an ELISA reader (Synergy HT; BioTek). The absorbance of the samples without cells was deducted from the cell-seeded samples.Quantification of DNA amount in samples
DNA content was determined using a Quant-iT™ dsDNA Assay Kit (Life Technologies) on days 1, 3, 7, and 14. To process the samples for the analysis of DNA content, 500 μL of cell lysis solution (0.2% v/v Triton X-100, 10 mM Tris (pH 7.0), 1 mM EDTA) was added to each sample. To prepare the cell lysate, the samples were processed through a total of 3 freeze/thaw cycles, i.e., the scaffold sample was first frozen at −70 °C and then thawed at RT. Between each freeze/thaw cycle, the scaffolds were roughly vortexed. The prepared samples were stored at −70 °C until analysis. Sample (10 μL) was mixed with 200 μL of reagent solution and the fluorescence intensity was measured on a multiplate fluorescence reader (Synergy HT, λex = 485 nm, λem = 525 nm). The data were processed using the calibration curve of the standards in the kit.Cell morphology determination by confocal microscopy
After 1, 7 and 14 days of culture, the scaffolds seeded with cells were fixed with frozen methanol (−20 °C), rinsed twice with PBS, incubated in 10 μg ml−1 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3); Invitrogen) for 45 min at RT, and then incubated in 5 μg ml−1 propidium iodide in PBS for 10 min. The scaffold was rinsed twice with PBS and scanned the same day. DiOC6 staining was used to visualize mitochondria and inner membranes, while propidium iodide staining was used to visualize cell nuclei. A Zeiss LSM 5 DUO confocal microscope at λex = 488 and 560 nm and λem = 505–550 and 575–650 nm was used for DiOC6 and propidium iodide detection, respectively.Statistical analysis
The quantitative data were presented as mean ± standard deviation (SD). The results were evaluated statistically using one-way analysis of variance (ANOVA) and the Student–Newman–Keuls test. The level of significance was set at 0.05.Conclusions
Core/shell centrifugation spinning technology enables the formation of fibres with embedded growth factors. The water-in-oil emulsification is based on the behaviour of Pluronic F-68 micelles enabling the encapsulation and protection of susceptible proteins. The results of the study are the formation of core/shell fibres containing the core phase in the form of distinct droplets. The release studies showed that an increased number of droplets is connected with improved encapsulation efficacy and prolonged release. However, due to the aqueous solubility of the core phase high burst release was observed. Testing of core/shell fibres with embedded platelet lyophilisates showed improved fibroblast and osteoblast metabolic activity and proliferation. The effect of released proteins was dominant during the first week of cultivation. Altogether, centrifugal electrospinning is a suitable method for fabricating protein loaded scaffolds for tissue engineering applications.Acknowledgements
This work has been supported by the Ministry of Education, Youth and Sports within the National Sustainability Programme I, projects No. LO1309, LO1508, and by project No. IPv6, and by the Czech Science Foundation, project No. 15-15697S, Operational Program – Prague Competitiveness CZ.2.16/3.1.00/21528, and the Grant Agency of Charles University (projects No. 1246314, 1228214, 1262414).References
- R. Langer and J. Vacanti, Science, 1993, 260, 920–926 CAS.
- D. W. Hutmacher, J. T. Schantz, C. X. Lam, K. C. Tan and T. C. Lim, J. Tissue Eng. Regener. Med., 2007, 1, 245–260 CrossRef CAS PubMed.
- L. Susan, et al., Biomed. Mater., 2006, 1, R45 CrossRef PubMed.
- W. J. Li, C. T. Laurencin, E. J. Caterson, R. S. Tuan and F. K. Ko, J. Biomed. Mater. Res., 2002, 60, 613–621 CrossRef CAS PubMed.
- D. Lukáš, A. Sarkar, L. Martinová, K. Vodseďálková, D. Lubasová, J. Chaloupek, P. Pokorný, J. Chvojka and M. Komárek, Text. Prog., 2009, 41, 59–140 CrossRef.
- S. Sahoo, L. T. Ang, J. C.-H. Goh and S.-L. Toh, J. Biomed. Mater. Res., Part A, 2010, 93, 1539–1550 Search PubMed.
- X. Jia, C. Zhao, P. Li, H. Zhang, Y. Huang, H. Li, J. Fan, W. Feng, X. Yuan and Y. Fan, J. Biomater. Sci., Polym. Ed., 2011, 22, 1811–1827 CrossRef CAS PubMed.
- L. Tian, M. Prabhakaran, X. Ding, D. Kai and S. Ramakrishna, J. Mater. Sci., 2012, 47, 3272–3281 CrossRef CAS.
- L. Tian, M. Prabhakaran, X. Ding, D. Kai and S. Ramakrishna, J. Mater. Sci.: Mater. Med., 2013, 24, 2577–2587 CrossRef CAS PubMed.
- B. M. Baker, A. O. Gee, R. B. Metter, A. S. Nathan, R. A. Marklein, J. A. Burdick and R. L. Mauck, Biomaterials, 2008, 29, 2348–2358 CrossRef CAS PubMed.
- A. Guimaraes, A. Martins, E. D. Pinho, S. Faria, R. L. Reis and N. M. Neves, Nanomedicine, 2010, 5, 539–554 CrossRef CAS PubMed.
- J. Nam, Y. Huang, S. Agarwal and J. Lannutti, Tissue Eng., 2007, 13, 2249–2257 CrossRef CAS PubMed.
- M. Simonet, O. D. Schneider, P. Neuenschwander and W. J. Stark, Polym. Eng. Sci., 2007, 47, 2020–2026 CAS.
- C. Vaquette and J. J. Cooper-White, Acta Biomater., 2011, 7, 2544–2557 CrossRef CAS PubMed.
- M. Rampichova, J. Chvojka, M. Buzgo, E. Prosecka, P. Mikes, L. Vyslouzilova, D. Tvrdik, P. Kochova, T. Gregor, D. Lukas and E. Amler, Cell Proliferation, 2013, 46, 23–37 CrossRef CAS PubMed.
- B. A. Blakeney, A. Tambralli, J. M. Anderson, A. Andukuri, D. J. Lim, D. R. Dean and H. W. Jun, Biomaterials, 2011, 32, 1583–1590 CrossRef CAS PubMed.
- N. E. Zander, J. Appl. Polym. Sci., 2015, 132, 41269 CrossRef.
- L. Wang, J. Shi, L. Liu, E. Secret and Y. Chen, Microelectron. Eng., 2011, 88, 1718–1721 CrossRef CAS.
- L. Ren, V. Pandit, J. Elkin, T. Denman, J. A. Cooper and S. P. Kotha, Nanoscale, 2013, 5, 2337–2345 RSC.
- A. M. Loordhuswamy, V. R. Krishnaswamy, P. S. Korrapati, S. Thinakaran and G. D. V. Rengaswami, Mater. Sci. Eng., C, 2014, 42, 799–807 CrossRef CAS PubMed.
- M. Rampichova, M. Buzgo, J. Chvojka, E. Prosecka, O. Kofronova and E. Amler, Cell Adhes. Migr., 2014, 8, 36–41 CrossRef PubMed.
- N. E. Zander, J. Appl. Polym. Sci., 2015, 132, 41269 CrossRef.
- L. A. Mary, T. Senthilram, S. Suganya, L. Nagarajan, J. Venugopal, S. Ramakrishna and V. R. G. Dev, EXPRESS Polym. Lett., 2013, 7, 238–248 CrossRef.
- E. V. Batrakova and A. V. Kabanov, J. Controlled Release, 2008, 130, 98–106 CrossRef CAS PubMed.
- M. Ben Henda, N. Ghaouar and A. Gharbi, J. Polym., 2013, 2013, 7 Search PubMed.
- W. Brown, K. Schillen and S. Hvidt, J. Phys. Chem., 1992, 96, 6038–6044 CrossRef CAS.
- W. Brown, K. Schillen, M. Almgren, S. Hvidt and P. Bahadur, J. Phys. Chem., 1991, 95, 1850–1858 CrossRef CAS.
- R. Ivanova, B. Lindman and P. Alexandridis, Langmuir, 2000, 16, 9058–9069 CrossRef CAS.
- K. A. G. Katsogiannis, G. T. Vladisavljević and S. Georgiadou, Eur. Polym. J., 2015, 69, 284–295 CrossRef CAS.
- S. K. Murase, L. P. Lv, A. Kaltbeitzel, K. Landfester, L. J. del Valle, R. Katsarava, J. Puiggali and D. Crespy, RSC Adv., 2015, 5, 55006–55014 RSC.
- Y. Yang, X. Li, L. Cheng, S. He, J. Zou, F. Chen and Z. Zhang, Acta Biomater., 2011, 7, 2533–2543 CrossRef CAS PubMed.
- J. Pal, S. Singh, S. Sharma, R. Kulshreshtha, B. Nandan and R. K. Srivastava, Mater. Lett., 2016, 167, 288–296 CrossRef CAS.
- T. J. Sill and H. A. von Recum, Biomaterials, 2008, 29, 1989–2006 CrossRef CAS PubMed.
- A. Mickova, M. Buzgo, O. Benada, M. Rampichova, Z. Fisar, E. Filova, M. Tesarova, D. Lukas and E. Amler, Biomacromolecules, 2012, 13, 952–962 CrossRef CAS PubMed.
- H. J. Lee, A. McAuley, K. F. Schilke and J. McGuire, Adv. Drug Delivery Rev., 2011, 63, 1160–1171 CrossRef CAS PubMed.
- S. Ohtake, R. A. Martin, A. Saxena, D. Lechuga-Ballesteros, A. E. Santiago, E. M. Barry and V. Truong-Le, J. Pharm. Sci., 2011, 100, 3076–3087 CrossRef CAS PubMed.
- X. Li, H. Zhang, H. Li, G. Tang, Y. Zhao and X. Yuan, Polym. Degrad. Stab., 2008, 93, 618–626 CrossRef CAS.
- T. Briggs and T. L. Arinzeh, J. Biomed. Mater. Res., Part A, 2014, 102, 674–684 CrossRef PubMed.
- D. F. Bowen-Pope, T. W. Malpass, D. M. Foster and R. Ross, Blood, 1984, 64, 458–469 CAS.
- D. P. Calnan, A. Fagbemi, J. Berlanga-Acosta, T. Marchbank, T. Sizer, K. Lakhoo, A. D. Edwards and R. J. Playford, Gut, 2000, 47, 622–627 CrossRef CAS PubMed.
- E. Prosecka, M. Buzgo, M. Rampichova, T. Kocourek, P. Kochova, L. Vyslouzilova, D. Tvrdik, M. Jelinek, D. Lukas and E. Amler, J. Biomed. Biotechnol., 2012, 2012, 428503 Search PubMed.
- M. Chiara Barsotti, P. Losi, E. Briganti, E. Sanguinetti, A. Magera, T. Al Kayal, R. Feriani, R. Di Stefano and G. Soldani, PLoS One, 2013, 8, e84753 Search PubMed.
- M. Buzgo, R. Jakubova, A. Mickova, M. Rampichova, E. Prosecka, P. Kochova, D. Lukas and E. Amler, Nanomedicine, 2013, 8, 1137–1154 CrossRef CAS PubMed.
- S. A. Sell, P. S. Wolfe, J. J. Ericksen, D. G. Simpson and G. L. Bowlin, Tissue Eng., Part A, 2011, 17, 2723–2737 CrossRef CAS PubMed.
- Y. Yang, T. Xia, W. Zhi, L. Wei, J. Weng, C. Zhang and X. Li, Biomaterials, 2011, 32, 4243–4254 CrossRef CAS PubMed.
- J. S. Choi, K. W. Leong and H. S. Yoo, Biomaterials, 2008, 29, 587–596 CrossRef CAS PubMed.
- K. Lee, E. A. Silva and D. J. Mooney, J. R. Soc., Interface, 2011, 8, 153–170 CrossRef CAS PubMed.
- E. Kon, G. Filardo, B. Di Matteo, F. Perdisa and M. Marcacci, Operat. Tech. Sports Med., 2013, 21, 108–115 CrossRef.
- Q. P. Pham, U. Sharma and A. G. Mikos, Biomacromolecules, 2006, 7, 2796–2805 CrossRef CAS PubMed.
- V. Karageorgiou and D. Kaplan, Biomaterials, 2005, 26, 5474–5491 CrossRef CAS PubMed.
- J. L. Lowery, N. Datta and G. C. Rutledge, Biomaterials, 2010, 31, 491–504 CrossRef CAS PubMed.
- K. T. Shalumon, K. H. Anulekha, K. P. Chennazhi, H. Tamura, S. V. Nair and R. Jayakumar, Int. J. Biol. Macromol., 2011, 48, 571–576 CrossRef CAS PubMed.
- S. M. Jung, G. H. Yoon, H. C. Lee and H. S. Shin, J. Biomater. Sci., Polym. Ed., 2015, 26, 252–263 CrossRef CAS PubMed.
- M. Plencner, B. East, Z. Tonar, M. Otahal, E. Prosecka, M. Rampichova, T. Krejci, A. Litvinec, M. Buzgo, A. Mickova, A. Necas, J. Hoch and E. Amler, Int. J. Nanomed., 2014, 9, 3263–3277 CrossRef CAS PubMed.
- R. Simman, A. Hoffmann, R. J. Bohinc, W. C. Peterson and A. J. Russ, Ann. Plast. Surg., 2008, 61, 337–344 CrossRef CAS PubMed.
- A. Malhotra, M. H. Pelletier, Y. Yu and W. R. Walsh, Arch. Orthop. Unfall-Chir., 2013, 133, 153–165 Search PubMed.
- A. Oryan, S. Alidadi and A. Moshiri, Expert Opin. Biol. Ther., 2016, 16, 213–232 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |