
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Environmentally friendly and facile synthesis of Rh nanoparticles at room temperature by alkaline ethanol solution and their application for ethanol electrooxidation†
Fuhai Lia,
Hanqin Wenga,
Yun Shanga,
Zuoming Dinga,
Zheng Yang a,
Sheng Chengb and
Mingzhang Lin*ac
a,
Sheng Chengb and
Mingzhang Lin*ac
aSchool of Nuclear Science and Technology, University of Science and Technology of China, Hefei 230026, P. R. China. E-mail: gelin@ustc.edu.cn
bInstrumental Analysis Center, Hefei University of Technology, Hefei 230009, P. R. China
cInstitute of Nuclear Energy Safety Technology, Chinese Academy of Sciences, Hefei 230031, P. R. China
First published on 13th January 2017
Abstract
Rhodium (Rh) is one of the most important catalyst constituents in many industrial catalytic processes. The synthesis of rhodium nanoparticles (RhNps) has received considerable interest for decades. In this paper, we report an environmentally friendly and facile synthesis route of RhNps through direct reduction of RhCl3 by ethanol in an alkaline medium in the presence of PVP at room temperature, confirmed by UV-vis spectroscopy, TEM, XRD and XPS. The effects of the concentrations of OH− and ethanol, as well as the nature of the alcohols, which greatly affected the formation rate of RhNps, were systematically investigated to understand the mechanism. Our results support that ethoxy anions generated from the deprotonation of ethanol were highly active and responsible for the formation of RhNps. Electrochemical tests have shown that Rh/C displayed zero activity in an acid medium, while in an alkaline medium Rh/C was highly active and displayed enhanced catalytic activity towards ethanol oxidation compared to commercial Rh/C. The process presented in this work is expected to be useful for preparing Rh-based nanocatalysts at room temperature, which may be extended to other nanocatalysts.
Introduction
Noble-metal nanoparticles have received considerable attention over the past decades due to their remarkable properties and potential applications in photonics, electronics, sensing, biomedical research and catalysis, which are significantly different from their bulk form.1–6 It has been reported that rhodium nanoparticles (RhNps) can be used as catalysts in so many reactions such as the oxidation of carbon monoxide, hydrocarbonylation, hydroformylation, hydrogenation, methanation of carbon dioxide and Fischer–Tropsch reactions.7–11 Especially, noble-metal nanoparticles have been extensively employed as electrocatalysts for direct fuel cell applications. Compared to the acidic-electrolyte direct ethanol fuel cell (DEFC), direct alkaline ethanol fuel cells (DAEFCs) possess several advantages: faster reaction kinetics, higher energy densities, easy handling of the liquid fuels and no need for fluorinated polymers.12–15 Great efforts have been devoted to the study of DAEFCs especially the development of electrode catalyst materials. However, it remains particularly difficult to break the C–C bond in alcohols except methanol during electrochemical oxidation.16–18 Therefore the research and development of catalyst materials highly active towards the break of C–C bond plays a pivotal role. Recently several works reported the enhancement of complete electrooxidation of ethanol to CO2 by the addition of Rh to Pt catalysts in acid medium, without a significant enhancement of the overall kinetics.19–24 Kowal et al.25 and Li et al.23 obtained remarkable ethanol oxidizing efficiency employing PtRhSnO2/C as electrocatalyst. Rh was found to play a significant role in facilitating C–C bond cleavage. Rh alone was also found to be quite active towards ethanol electrooxidation in alkaline medium,26,27 while in acid medium it is practically inactive.23,25 Suo et al.26 found Rh/C showed a higher activity for ethanol oxidation at low potentials than PdRh/C and Pd/C in alkaline medium. Fontes et al.27 observed a remarkable good activity of Rh/C for ethanol oxidation in the first minutes. These results demonstrated that Rh-based catalysts are promising catalyst candidates for complete ethanol oxidation with excellent properties at low potentials in alkaline medium.A variety of methods have been employed in the production of RhNps since Toshima et al.28 first reported the synthesis of RhNps in 1978 in methanol–water solution in the presence of poly(vinyl alcohol) (PVA) at 79 °C. The polyol methods29–33 have been most widely employed in the preparation of RhNps, which require harsh reducing conditions, typically consist in refluxing a mixture of a precursor salt (e.g., RhCl3 or Na3RhCl6), a polyalcohol (e.g., ethylene glycol) and a stabilizing agent [e.g., poly(vinyl pyrrolidone) (PVP) or PVA] at high temperatures (60–190 °C) for 1–24 h, which is tedious and not-easy-operating, energy and time consuming. A photochemical approach34 has also been reported for the generation of shape selective RhNps. The photochemical approach brings the inconvenience of demanding continuous irradiation from a light source for 6 h, although it was conducted at room temperature. RhNps have been produced based on the chemical reduction methods using borohydride as a reducing agent.35,36 However, borohydride should be carefully treated since it is toxic to health and unstable.
In this work, we report for the first time a facile and environmentally friendly synthesis route of RhNps through direct reduction of RhCl3 by ethanol in alkaline medium in the presence of PVP under aerobic conditions at room temperature (20 °C). Compared with other room temperature reducing agents such as BH4−, ethanol is more eco-friendly due to its biomass origin and easy biodegradation. The reaction mechanism was discussed by studying the effects of various parameters such as the concentrations of OH− and ethanol, as well as the nature of alcohols, which greatly affect the formation rate of RhNps. Moreover, the electrocatalytic activities of carbon-supported RhNps in both acid and alkaline medium were also studied.
Experimental
Chemicals
Rhodium chloride hydrate (RhCl3·xH2O, 99.98%) was purchased from Sigma-Aldrich Co., Ltd. Poly(vinylpyrrolidone) (PVP, K-30), sodium hydroxide (NaOH, 96%), methanol (CH3OH, 99.7%), ethanol (C2H5OH, 99.7%), isopropanol ((CH3)2CHOH, 99.7%) and tert-butyl alcohol ((CH3)3COH, 99.7%) were obtained from Sinopharm Chemical Reagent Co., Ltd. Commercial Rh/C (5 wt%) was purchased from Aladdin Co., Ltd. All the above chemicals were of analytical grade and used without further purification. Vulcan XC-72 carbon was purchased from E-TEK Co., Ltd. Nafion solution (5%) was purchased from Dupont Co., Ltd. Ultrapure water (18.25 MΩ cm), generated by a Kertone Lab VIP®, was used in all experiments.Synthesis of colloidal and carbon-supported RhNps
In a typical synthesis of colloidal RhNps, PVP (0.229 g) was dissolved in water (22.5 mL). Then RhCl3 aqueous solution (2.5 mL, 20 mM) was pipetted into the above solution. In a separate flask, determined quantities of ethanol and NaOH were dissolved in water (22.5 mL). The ethanol–NaOH solution was then poured into the RhCl3–PVP solution under stirring to yield a mixture. The ethanol concentration was varied from 0.1 M to 2.0 M. The NaOH concentration was varied from 0.2 M to 1.0 M. The reddish RhCl3–PVP solution finally turned into a dark brown solution after consecutive color changes. Then the RhNps were collected by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, F = 9280 × g, 20 min) and washed with water three times to remove NaOH and excess PVP.
000 rpm, F = 9280 × g, 20 min) and washed with water three times to remove NaOH and excess PVP.
Carbon-supported RhNps were prepared through the same process, but in which XC-72 carbon was first dispersed in water (22.5 mL) under ultrasonification instead of PVP. In this way the RhNps were produced directly onto Vulcan carbon without stabilization by PVP. The final concentrations were 1 mM Rh(III), 1 M ethanol, 1 M NaOH. Finally the product was filtered, washed with water and dried at 65 °C for 12 h.
Characterizations
The UV-visible (UV-vis) spectra were recorded in a spectrophotometer (UV-2600, Shimadzu). The absorbance variation at a specific wavelength as a function of time was recorded by fixing the wavelength and measuring the absorbance variations every 10 s immediately after mixing the reactants directly in the UV cuvette. The morphology of the synthesized RhNps was examined with high resolution transmission electron microscopy (HR-TEM) (JEOL 2010) at 200 kV. The energy dispersive X-ray spectrum (EDX) was recorded with an INCA energy system (Oxford Instruments) connected with the TEM. The samples were prepared by pipetting RhNps colloidal suspensions onto carbon coated Cu grids and then dried at room temperature. The metallic loading of Rh/C was measured by ICP-MS (X Series 2, Thermo Fisher Scientific). The powder X-ray diffraction (XRD) patterns of the samples were collected on a powder diffraction system (TTR-III, Rigaku) using a Cu Kα radiation source (λ = 1.54178 Å) operating at 40 kV and 200 mA. The composition and valence state of the synthesized nanoparticles were identified by X-ray photoelectron spectroscopy (XPS) analysis (ESCALAB 250, Thermo-VG Scientific) with monochromatic Al Kα irradiation (hν = 1486.6 eV). Energy normalization was accomplished by placing the major C 1s peak at 284.6 eV.Electrocatalytic performance of Rh/C toward ethanol oxidation
Cyclic voltammetry (CV) and amperometric I–t measurements were conducted using a CHI 660E potentiostat (Shanghai Chenhua Device Company, P. R. China) in a three-electrode cell with a glassy-carbon (GC) electrode (working electrode), a platinum foil (counter electrode) and a saturated Ag/AgCl electrode (reference electrode). The glassy carbon substrate was polished using alumina slurry (0.05 μm) prior to use. To prepare the working electrode, Rh/C (2 mg) was dispersed ultrasonically in ethanol (1 mL) containing Nafion (5%, 10 μL). The suspension (10 μL) was deposited onto the GC electrode (d = 5 mm). The solvent was then evaporated at room temperature. The electrolyte was deaerated by bubbling N2 for 30 min prior to each experiment. A flow of N2 was maintained over the electrolyte during the measurements. At least 10 cycles were performed before collecting the CV data with a scan rate of 10 mV s−1 or 50 mV s−1. All the currents are normalized by the surface area of the glassy carbon substrate (0.196 cm2) and are given in unit of mA cm−2.Results and discussion
Synthesis and characterization of RhNps
Rhodium(III) chloride exists in the form of a variety of chloroaquorhodium(III) complexes ([RhCl6−n(H2O)n](n−3)+) when dissolved in water.37 Fig. 1a shows the UV-vis spectra of RhCl3 solution in the presence of PVP and in the presence of PVP and ethanol, which are nearly the same. No formation of RhNps was observed in the absence of NaOH. The reddish solutions (inset in Fig. 1a) showed a strong absorption band at around 220 nm due to the ligand-to-metal charge transfer (LMCT) of the Rh(III) complex38 and the contribution of PVP. Two absorption maxima were also observed at 373 and 484 nm corresponding to the d–d electron transition of Rh(III) ions in the chloroaquorhodium(III) complexes.38 Colloidal RhNps were simply synthesized by the direct reduction of Rh(III) ions by alkaline ethanol solution in the presence of PVP under aerobic conditions at room temperature (20 °C). Fig. 1b shows the time evolution of UV-vis spectra of the reaction mixture during the synthesis process. The spectra during the first 10 min are shown in Fig. 1c to clearly display the absorbance changes at around 645 nm. The absorption maxima for Rh(III) complex was blueshifted immediately after the addition of ethanol–NaOH solution which is probably due to the substitution of Cl− by OH−. Two new absorption bands at around 260 and 646 nm appeared as the color of the mixture changed from reddish to blue. The absorbances at 260 and 646 nm increased and reached their maximal value 4 min after the mixture. Then they decreased as the reaction time went on. The reactant turned from blue to pale yellow as the two new absorption bands disappeared 9 min after the mixture, which indicates the appearance and disappearance of transient species.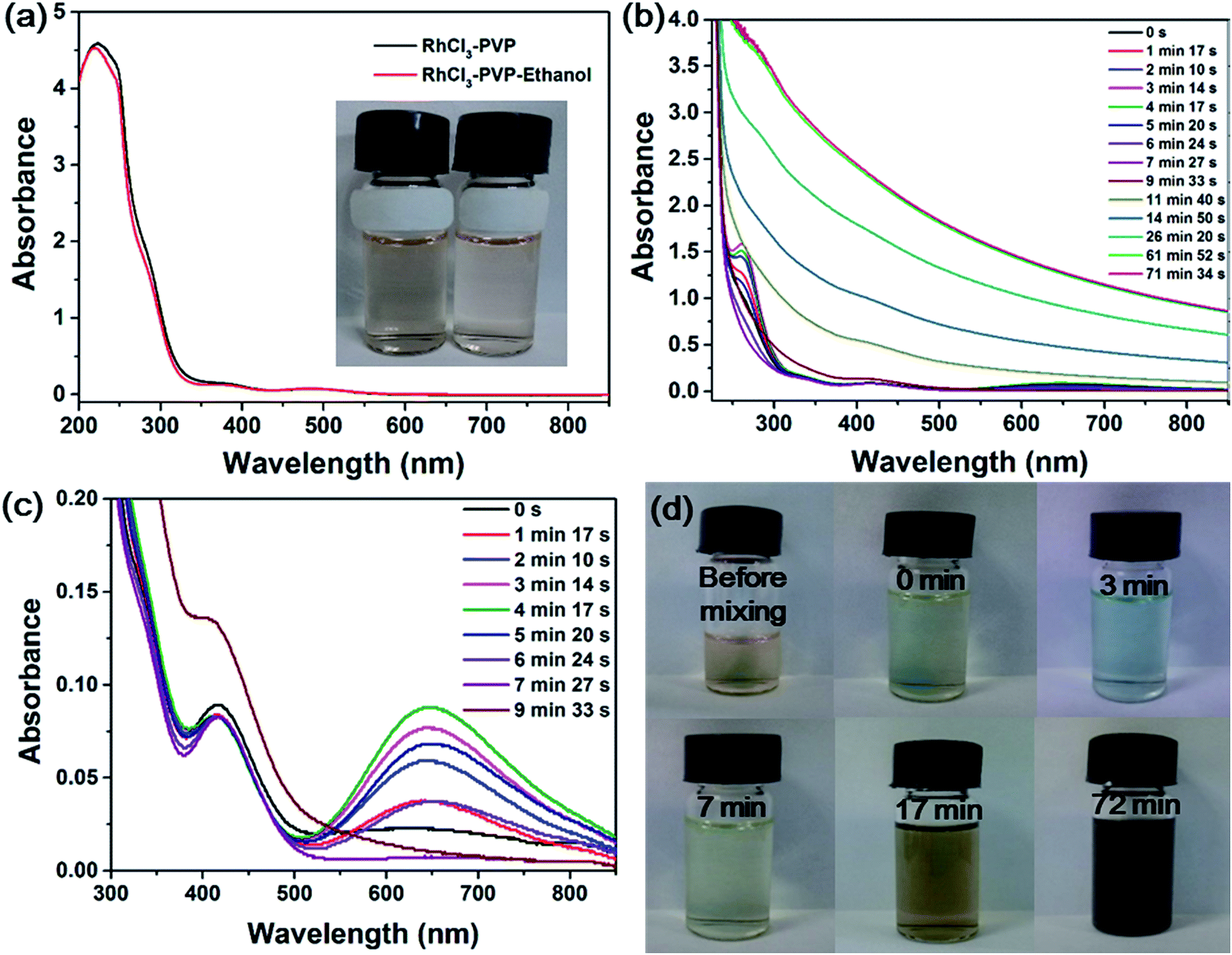 | ||
| Fig. 1 (a) UV-vis spectra of the solution containing 1 mM RhCl3 and 50 mM PVP in the absence and presence of 1 M ethanol. The inset shows the color of the solution. Time evolution of the UV-vis spectra of the mixture (b) during the whole process and (c) during the first 10 min. (d) Photographs of the mixture taken at different stages of the reaction: before mixing and 0, 3, 7, 17, 72 min after mixing. Reaction condition of (b–d): 1 mM RhCl3, 50 mM PVP, 1 M ethanol and 1 M NaOH at 20 °C in aerobic environment. | ||
As the reaction progressed, the whole spectra gradually rised up especially in the visible range, indicating the formation of Rh(0) particles. After the reduction of Rh(III) ions was completed (61 min after mixing), there is no remarkable change in the spectrum, as shown in Fig. 1b. The color of the solution turned from pale yellow to dark brown, as the featureless absorption in the visible range increased, indicating that the band structure of Rh particles was formed.38,39 Fig. 1d shows the photographs of the reactant mixture taken at different stages of the reaction. The color of the reactant mixture clearly changed from reddish to blue, pale yellow and dark brown as described above.
Fig. 2a and b show the representative TEM images of RhNps synthesized in 1 M ethanol solution at 20 °C. RhNps tend to aggregate due to the small size and extremely high surface free energy. The morphologies of RhNps are not uniform. The sizes of the nanoparticles were estimated to be 2–5 nm. The HRTEM images of typical single RhNps were shown in Fig. 2c. The lattice fringes of each nanoparticle are along the same direction, suggesting the nature of the as-synthesized RhNps are single crystal. The interplanar crystal spacing was examined to be 0.22 nm, which is very close to the (111) lattice spacing of the face centered cubic (fcc) rhodium, implying that the surface of RhNps were covered by the (111) lattice. The XRD pattern of as-synthesized products is shown in Fig. 2d. It exhibits four peaks in the 2θ range 20–100° corresponding to diffraction from the (111), (200), (220) and (311) lattice planes and matched with fcc rhodium (JCPDS 88-2334). Therefore the XRD pattern confirmed the formation of fcc RhNps. The average size of RhNps was calculated to be 3.5 nm using Scherrer equation, which is consistent with TEM images. Fig. 2e shows the Rh 3d XPS of RhNps after being deposited on Al foil. The Rh 3d region is characterized as a doublet, which arises from the spin–orbit coupling splitting corresponding to 3d3/2 and 3d5/2 electrons respectively. The Rh(0) 3d3/2 and Rh(0) 3d5/2 peaks appeared at binding energies of 312.01 and 307.37 eV, confirming the formation of RhNps as reported in the literature.40,41 After deconvolution the percentage of the reduced Rh(0) was estimated to be 68.6%. The XPS spectrum of O 1s confirmed the existence of unreduced Rh(III) (Fig. S1†). Two peaks were observed for the O 1s spectrum of RhNps formed in the absence of PVP at 533.72 eV and 531.31 eV, of which the latter corresponded to the oxygen atoms of oxidized Rh(III). EDX was also employed to investigate the valence state of RhNps (Fig. S2†). The atomic ratio of Rh to O was measured to be 81.7%:18.3% illustrating the existence of unreduced Rh(III).
 | ||
| Fig. 2 (a and b) TEM and (c) HRTEM images of the colloidal RhNps. (d) XRD pattern of the RhNps. (e) XPS of the RhNps. Condition of synthesis: 1 mM RhCl3, 50 mM PVP, 1 M ethanol and 1 M NaOH at 20 °C. | ||
To characterize the origin of the two transient absorption bands observed in Fig. 1, the absorbance variations at 260 and 646 nm as a function of time were measured, as shown in Fig. 3. The absorbance at 260 nm reached its maximal after 200 s reaction and decreased to its minimum after 370 s reaction, while the absorbance at 646 nm reached its maximal after 300 s reaction and decreased to its minimum after 530 s reaction. It is clear that the variation of the absorbance at 646 nm falls behind that at 260 nm. Thus we can speculate that the two absorption bands at 260 and 646 nm are originated from two different transient species.
 | ||
| Fig. 3 Absorbance variations at 260 and 646 nm of the solution containing 1 mM RhCl3, 50 mM PVP, 1 M ethanol and 1 M NaOH at 20 °C as a function of time. | ||
The spectra properties of Rh(II) and Rh(I) complexes, of which the life time varies greatly, have been studied by many literatures.42–47 Stable Rh(II) and Rh(I) complexes, such as (RhII(CH3COO)2)2 and (RhI(CO)2Cl)2, exist at room temperature. A new absorption peak at 265 nm was observed by Lilie et al.42 in the experiment of pulse radiolytic reduction of Rh(NH3)5H2O3+ in the presence of isopropanol and oxygen. It was identified as a dioxygen adduct of RhII(NH3)42+, O2RhII(NH3)4H2O2+, which is stable for several hours. Mulazzani et al.43 and Schwarz et al.44 both observed a new absorption band at around 260 nm in a pulse radiolysis study of Rh(bpy)33+, which was assigned to the generation of RhII(bpy)32+. Generally Rh(II) complex exhibits strong absorption bands at around 260 nm. As for Rh(I) complex, at least four forms of Rh(I) species, with their absorption bands located in the visible region at 485–565 nm, were clearly identified.43 The nature of the Rh(I) species, designated as “Rh(bpy)2+”, depends upon the pH of the solution, the concentration of Rh(I) as well as the nature and concentration of the counter-ion. A short-lived species absorbing at 520 nm was assigned to a Rh(I)Rh(II) complex by Derai et al.45 Some long-lived Rh(I)Rh(II) complexes are known to absorb in the 550 nm region.46,47 The transient absorption band at 260 nm is assigned to intermediate Rh(II) complex, which corresponds well with other literatures.42–44 Rh(I) complex generally exhibits absorption bands in the visible region, which depends on the ligand that stabilize the complex.43 In this work the variation of the absorption band at 646 nm lags behind that at 260 nm. Thus it is reasonable the broad absorption band at 646 nm was assigned to Rh(I) complex.
Oxygen was found to affect the synthesis process markedly. Colloidal RhNps were also obtained when dissolved oxygen was removed from both RhCl3–PVP solution and ethanol–NaOH solution. The time evolution of the UV-vis spectra was monitored during the reaction, as shown in Fig. 4. Compared with the situation in aerobic conditions (Fig. 1b), the whole spectra rised up immediately after the mixing. No transient absorption bands at around 260 and 646 nm were observed, which means no intermediate Rh(II) or Rh(I) complex was observed. Rh(0) particles were generated more quickly in this situation. It can be concluded oxygen here played a role in stabilizing the Rh(II) and Rh(I) complexes, which are easy to be further reduced without enough stabilization of ligands or oxidizing agents. The nature of Rh(II) and Rh(I) complexes may be a dioxygen adduct as reported by Lilie et al.42
 | ||
| Fig. 4 Time evolution of the UV-vis spectra of the mixture containing 1 mM RhCl3, 50 mM PVP, 1 M ethanol and 1 M NaOH at 20 °C in the absence of O2. | ||
Kinetic study and mechanism of RhNps formation
The effect of PVP was also investigated. Fig. S3† shows the UV-vis spectra of RhCl3–PVP solution in the presence of different concentrations of NaOH. There is only very slight difference. A weak band at around 415 nm due to substitution of Cl− by OH− was observed. It was reported that Rh(III) is dominated by hydroxylated complexes [Rh(OH)6]3− at high pH (pH ≥ 12).48 The yellow solutions (see inset in Fig. S3†), which were stable for at least 24 hours, also indicate the different species of Rh(III) complex from the reddish neutral RhCl3 solution (see inset in Fig. 1a). No formation of colloidal RhNps was observed in the presence of PVP and in the absence of ethanol. In our system PVP cannot reduce Rh(III) ions and only served as a stabilizing agent here.As discussed above, colloidal RhNps can only be obtained in alkaline medium. The only possible reducing species that are capable of reducing Rh(III) ions to Rh(0) at 20 °C is ethanol in alkaline medium. The effect of OH− concentration on the formation rate of RhNps was studied. Fig. 5a shows the UV-vis spectra of RhCl3–PVP–ethanol solution with low concentrations of NaOH. The absorbance increase in the visible range was insignificant unless the concentration of NaOH reached 0.08 M (pH = 12.75). Therefore to obtain colloidal RhNps the pH of the solution has to be higher than 12.75 (critical pH).
 | ||
| Fig. 5 (a) UV-vis spectra of the solution containing 1 mM RhCl3, 50 mM PVP, 1 M ethanol and 0.06–0.10 M NaOH. (b) Absorbance variations at 646 nm of the solution containing 1 mM RhCl3, 50 mM PVP, 1 M ethanol and 0.2–1.0 M NaOH as a function of time. | ||
Alcohol is a kind of weak acid and deprotonates at high pH, as shown in reaction (1):
| HR–OH ⇆ HR–O− + H+ | (1) |
| CH3CH2–OH ⇆ CH3CH2–O− + H+ | (2) |
Kwon et al.49 argued that ethoxy anion is the main reactive species at high pH (pH ≥ 11), with its concentration steeply increasing with higher pH. Ethoxy anion is believed to be significantly more reactive than ethanol,49–51 which is much easier for this species to give electrons and be oxidized to acetaldehyde, as shown in reaction (3):
| CH3CH2–O− → CH3CHO + H+ + 2e− | (3) |
We propose that ethoxy anion may be responsible for the formation of RhNps in alkaline medium at room temperature. A similar mechanism has been proposed by Gasparotto et al. to explain the formation of AuNps, AgNps and AuAgNps, with glycerol in alkaline medium at room temperature.52–55 The alkoxide formed from glycerol in high-pH medium was considered as the true reducing species.
The formation rates of RhNps at 0.2–1.0 M NaOH were examined by following the absorbance variation at 646 nm as a function of time (Fig. 5b). The rate of nanoparticle formation and the final absorbance was increased with a higher NaOH concentration (0.2–0.5 M). The reaction came to an end in 15 min with 0.5 M NaOH. This can be explained by accounting ethoxy anion as the true reducing species. More ethoxy anion is generated by deprotonation of ethanol with higher concentration of NaOH (at higher pH), thus accelerating the formation of nanoparticles. However, as the concentration of NaOH further increased (0.6–0.8 M), the formation rate did not increase at all. It even decreased when the concentration of NaOH reached 1.0 M. With the increase of NaOH concentration, more OH− can coordinate with Rh(III) ions and Rh(0) clusters,48 thus impede the reduction of unreduced Rh(III) ions by ethoxy anion. Standard electrode potential of Rh(III) is closer to zero at pH = 13.996 than at pH = 0,56 indicating that Rh(III) is more difficult to be reduced under the stabilization of OH− at high pH. The acceleration effect of OH− on the deprotonation of ethanol plays a dominant role, when the concentration of NaOH is relatively low, while the impediment effect will be dominant when the concentration of NaOH is too high. It is noteworthy that the existing time of Rh(I) complex increased with increasing NaOH concentration. OH− here also played a role in stabilizing intermediate Rh(I) complex.
The proposed mechanism was supported by electrochemical tests. The cyclic voltammograms (CV) of GC electrode in neutral and alkaline ethanol solutions are shown in Fig. 6. In neutral medium the oxidation current density was very low indicating that ethanol was difficult to be oxidized on this condition. Ethanol oxidation was slightly enhanced in 0.1 M NaOH solution. On both conditions the onset potentials were nearly the same at −0.3 V. In 1.0 M NaOH solution, the peak current density was much higher with the onset potential shifted 200 mV to more negative potentials, illustrating that ethanol is a stronger reducing agent at higher pH. This is due to the fast increase of ethoxy anions as the pH of electrolyte increases.
 | ||
| Fig. 6 Cyclic voltammograms (CVs) of GC electrode in N2 saturated solution containing 1.0 M ethanol and 0, 0.1, 1.0 M NaOH at 25 °C with a scan rate of 10 mV s−1. | ||
The effect of ethanol concentration on the formation rate of RhNps was also studied. Fig. 7 depicts the absorbance variation at 646 nm as a function of time with the concentration of ethanol varying from 0.1–2.0 M. The rate of RhNps formation was accelerated with increasing ethanol concentration as expected. A higher concentration of ethoxy anion is generated with higher ethanol concentration at the same pH, thus promoting the reduction of Rh(III) ions and the formation of RhNps. The existing time of Rh(I) complex decreased with increasing ethanol concentration, because it was further reduced more quickly.
 | ||
| Fig. 7 Absorbance variations at 646 nm of the solution containing 1 mM RhCl3, 50 mM PVP, 0.5 M NaOH and 0.1–2.0 M ethanol as a function of time. | ||
The possibility of preparing RhNps by other alcohols at room temperature in alkaline media was examined. RhNps were also obtained in 1.0 M NaOH solution in the presence of isopropanol with a much slower kinetics (Fig. 8). It is more difficult for isopropanol to deprotonate into its corresponding alkoxide through reaction (1) because of its higher pKa (17.1, 25 °C). The critical pH for isopropanol to produce RhNps at room temperature is 13.05 and higher than that of ethanol (12.75). These results strongly support the mechanism proposed above. Methanol, with a pKa of 15.5, would be expected to be more reactive than ethanol. However, no RhNps were produced in 1.0 M NaOH solution in the presence of methanol in 24 h. This anomaly may be caused by the different leaving ability of the α hydrogen (Hα),49 which means the different ability for alkoxides to give electrons. The bond energy of C–Hα in methanol is higher than the C–Hα in ethanol57 (no similar data for the corresponding alkoxides has ever been reported as far as we know), meaning that corresponding alkoxide for methanol is less reactive than that for ethanol. Tert-butyl alcohol, with a high pKa of 18.0, is expected to be less active than isopropanol. Moreover, there is no Hα for tert-butyl alcohol. No wonder that no RhNps were observed in alkaline tert-butyl alcohol solution at room temperature.
 | ||
| Fig. 8 Absorbance variations at 646 nm of the solution containing 1 mM RhCl3, 50 mM PVP, 1.0 M NaOH and 1 M ethanol/isopropanol as a function of time. | ||
Electrocatalytic performance of Rh/C towards ethanol oxidation
The electrocatalytic activity of commercial and as-synthesized Rh/C toward ethanol oxidation in alkaline medium was evaluated, as shown in Fig. 9. The inset in Fig. 9b shows the TEM image of as-synthesized RhNps deposited on Vulcan carbon. The metallic loading was 5.32 wt% determined by ICP-MS. Fig. 9a shows the CV curve of Rh/C in N2-saturated 0.1 M NaOH solution. Oxygen adsorption was observed at high potentials in the positive scan, while oxygen desorption occurred at low potentials in the negative scan.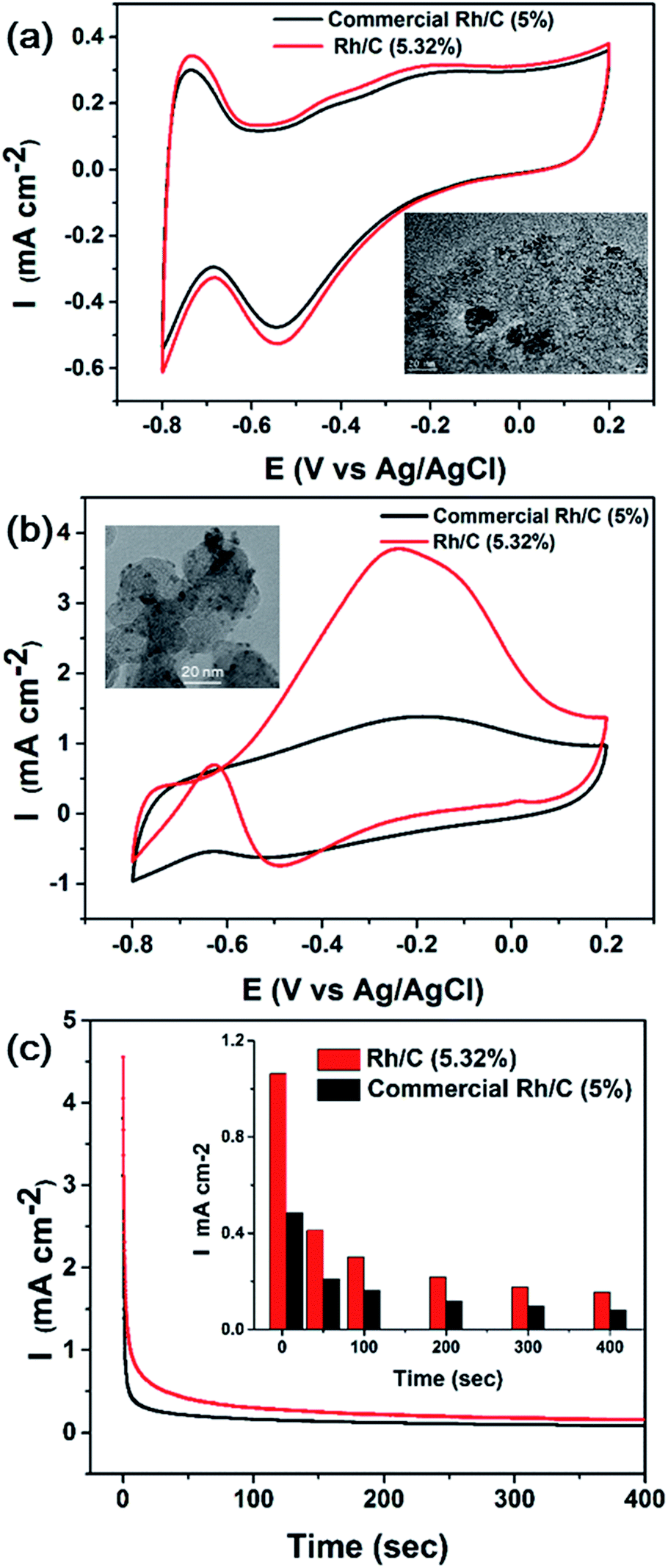 | ||
| Fig. 9 Cyclic voltammograms (CVs) of commercial and as-synthesized Rh/C at 25 °C with a scan rate of 50 mV s−1 (a) In 0.1 M NaOH solution. (b) In 1 M ethanol and 0.1 M NaOH solution. (c) Amperometric I–t curves of as-synthesized and commercial Rh/C in 1 M ethanol and 0.1 M NaOH solution. The inset in (a) shows the TEM image of commercial Rh/C. The inset in (b) shows as-synthesized Rh/C. | ||
The electrocatalytic performance of commercial and as-synthesized Rh/C was investigated in a 1 M ethanol + 0.1 M NaOH solution at 25 °C (Fig. 9b). In the positive scan, the oxidation of ethanol started from low potentials, and a broad oxidation peak occurred at −0.4 to 0.1 V (vs. Ag/AgCl) as potential went positively, which is consistent with literatures.26,58 After peak potential the current decreased due to the formation of oxidized species blocking the active sites.26
In the negative scan, the oxidation current was negligibly low at high potentials. At low potentials an oxidation peak was observed at −0.63 V as the oxidized species were removed exposing the active sites. Compared with commercial Rh/C, the as-synthesized Rh/C displayed enhanced catalytic activity towards ethanol oxidation. Note that the metallic loadings were nearly the same. And the current density was much higher than commercial Rh/C and the onset potential was more negative. The stability tests of the as-synthesized and commercial Rh/C were carried out as well. Amperometric I–t curves at −0.2 V were shown in Fig. 9c. Fig. S4† showed I–t curves at −0.5 and 0 V. Compared with commercial Rh/C, the as-synthesized Rh/C displayed comparable or even better stability. This is due to the difference in dispersity of RhNps on carbon. The nanoparticles of commercial Rh/C either were too small or agglomerated into big clusters, as we can see from inset in Fig. 9a.
The electrocatalytic activity of Rh/C toward ethanol oxidation in acid medium was also studied, as shown in Fig. S5.† It is obvious that Rh/C showed no activity for ethanol electrooxidation in acid medium. The alkaline environment is key to making Rh an active catalyst for ethanol oxidation. It is generally acknowledged that the difficulty of Hα dehydrogenation of ethanol in acid medium results in nearly zero activity of Rh due to the lack of hydroxide species,59 which is also applicable here. In our opinion, however, the role of ethoxy anions generated from the deprotonation of ethanol by OH− may be overlooked, since they are significantly more reactive than ethanol as has been demonstrated in this work with its concentration steeply increasing with higher pH. This idea helps us fully understand the role of OH− in ethanol oxidation in alkaline medium, which is important for the development of DAEFCs.
Conclusions
We have for the first time demonstrated a facile and environmentally friendly synthetic route of RhNps through direct reduction of RhCl3 by alkaline ethanol solution in the presence of PVP under aerobic conditions at room temperature. The reaction can be fairly fast. Two transient absorption bands at 260 and 646 nm were observed in the presence of oxygen and assigned to intermediate Rh(II) and Rh(I) complexes respectively, which were stabilized by oxygen and OH−. Ethoxy anions generated from the deprotonation of ethanol at high pH were responsible for the formation of RhNps. The formation rate of RhNps was increased with higher concentration of ethanol. A proper concentration of OH− would accelerate while too much OH− would impede the formation of RhNps. The low pKa and C–Hα energy of the alcohol was favorable for the formation of RhNps. Moreover, the carbon-supported nanoparticles were highly active and displayed enhanced catalytic activity towards ethanol electrooxidation than commercial Rh/C in alkaline medium. The significance of this work is to offer a new facile synthetic route of preparing Rh-based nanocatalysts at room temperature, which may be extended to other nanocatalysts.Acknowledgements
We acknowledge financial support for this research from the National Natural Science Foundation of China (No. 21377122 and 31501576), the Fundamental Research Fund for the Central Universities (WK2140000009), and China Postdoctoral Science Foundation (2016M592069).References
- C. Z. Zhu, D. Du, A. Eychmuller and Y. H. Lin, Chem. Rev., 2015, 115, 8896–8943 CrossRef CAS PubMed.
- L. Zhang, Z. X. Xie and J. L. Gong, Chem. Soc. Rev., 2016, 45, 3916–3934 RSC.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- M. R. Jones, K. D. Osberg, R. J. Macfarlane, M. R. Langille and C. A. Mirkin, Chem. Rev., 2011, 111, 3736–3827 CrossRef CAS PubMed.
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- Z. X. Fan and H. Zhang, Chem. Soc. Rev., 2016, 45, 63–82 RSC.
- L. O. Nindakova, N. M. Badyrova, V. V. Smirnov and S. S. Kolesnikov, J. Mol. Catal. A: Chem., 2016, 420, 149–158 CrossRef CAS.
- Y. Yuan, N. Yan and P. J. Dyson, ACS Catal., 2012, 2, 1057–1069 CrossRef CAS.
- J. R. Renzas, Y. W. Zhang, W. Y. Huang and G. A. Somorjai, Catal. Lett., 2009, 132, 317–322 CrossRef CAS.
- W. H. Chiou, Y. W. Wang, C. L. Kao, P. C. Chen and C. C. Wu, Organometallics, 2014, 33, 4240–4244 CrossRef CAS.
- M. H. Tan, G. H. Yang, T. J. Wang, T. Vitidsant, J. Li, Q. H. Wei, P. P. Ai, M. B. Wu, J. T. Zheng and N. Tsubaki, Catal. Sci. Technol., 2016, 6, 1162–1172 CAS.
- E. Antolini and E. R. Gonzalez, J. Power Sources, 2010, 195, 3431–3450 CrossRef CAS.
- Q. G. He, W. Chen, S. Mukerjee, S. W. Chen and F. Laufek, J. Power Sources, 2009, 187, 298–304 CrossRef CAS.
- S. Pai-Cheng, C. Hong-Shuo, C. Tsan-Yao, L. Chen-Wei, L. Chih-Hao, L. Jyh-Fu, C. Ting-Shan and W. Kuan-Wen, Int. J. Hydrogen Energy, 2013, 38, 4474–4482 CrossRef.
- E. H. Yu, U. Krewer and K. Scott, Energies, 2010, 3, 1499–1528 CrossRef CAS.
- S. Q. Song, W. J. Zhou, Z. H. Zhou, L. H. Jiang, G. Q. Sun, Q. Xin, V. Leontidis, S. Kontou and P. Tsiakaras, Int. J. Hydrogen Energy, 2005, 30, 995–1001 CrossRef CAS.
- C. Coutanceau, S. Brimaud, C. Lamy, J. M. Leger, L. Dubau, S. Rousseau and F. Vigier, Electrochim. Acta, 2008, 53, 6865–6880 CrossRef CAS.
- A. O. Neto, L. A. Farias, R. R. Dias, M. Brandalise, M. Linardi and E. V. Spinace, Electrochem. Commun., 2008, 10, 1315–1317 CrossRef.
- J. P. I. de Souza, S. L. Queiroz, K. Bergamaski, E. R. Gonzalez and F. C. Nart, J. Phys. Chem. B, 2002, 106, 9825–9830 CrossRef CAS.
- K. Bergamaski, J. F. Gomes, B. E. Goi and F. C. Nart, Ecletica Quim., 2003, 28, 87–92 CAS.
- K. Bergamaski, E. R. Gonzalez and F. C. Nart, Electrochim. Acta, 2008, 53, 4396–4406 CrossRef CAS.
- F. H. B. Lima, D. Profeti, W. H. Lizcano-Valbuena, E. A. Ticianelli and E. R. Gonzalez, J. Electroanal. Chem., 2008, 617, 121–129 CrossRef CAS.
- M. Li, W. P. Zhou, N. S. Marinkovic, K. Sasaki and R. R. Adzic, Electrochim. Acta, 2013, 104, 454–461 CrossRef CAS.
- F. H. B. Lima and E. R. Gonzalez, Electrochim. Acta, 2008, 53, 2963–2971 CrossRef CAS.
- A. Kowal, M. Li, M. Shao, K. Sasaki, M. B. Vukmirovic, J. Zhang, N. S. Marinkovic, P. Liu, A. I. Frenkel and R. R. Adzic, Nat. Mater., 2009, 8, 325–330 CrossRef CAS PubMed.
- Y. G. Suo and I. M. Hsing, J. Power Sources, 2011, 196, 7945–7950 CrossRef CAS.
- E. H. Fontes, R. M. Piasentin, J. M. S. Ayoub, J. C. M. Silva, M. H. M. T. Assumpcao, E. V. Spinace, A. O. Neto and R. F. B. Souza, Materials for Renewable and Sustainable Energy, 2015, 4, 1–10 CrossRef.
- H. Hirai, Y. Nakao and N. Toshima, Journal of Macromolecular Science: Part A–Chemistry, 1978, 12, 1117–1141 CrossRef.
- J. D. Hoefelmeyer, K. Niesz, G. A. Somorjai and T. D. Tilley, Nano Lett., 2005, 5, 435–438 CrossRef CAS PubMed.
- Y. W. Zhang, M. E. Grass, J. N. Kuhn, F. Tao, S. E. Habas, W. Y. Huang, P. D. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 5868–5869 CrossRef CAS PubMed.
- N. Zettsu, J. M. McLellan, B. Wiley, Y. D. Yin, Z. Y. Li and Y. N. Xia, Angew. Chem., Int. Ed., 2006, 45, 1288–1292 CrossRef CAS PubMed.
- H. Zhang, W. Y. Li, M. S. Jin, J. E. Zeng, T. K. Yu, D. R. Yang and Y. N. Xia, Nano Lett., 2011, 11, 898–903 CrossRef CAS PubMed.
- Z. X. Wu, W. L. Chen, H. Y. Liu, P. Zhai, C. X. Xiao, D. S. Su, H. C. Liu and D. Ma, Catal. Sci. Technol., 2015, 5, 4116–4122 CAS.
- S. Kundu, K. Wang and H. Liang, J. Phys. Chem. C, 2009, 113, 18570–18577 CAS.
- Y. Nakao and K. Kaeriyama, J. Colloid Interface Sci., 1989, 131, 186–191 CrossRef CAS.
- M. Marin-Almazo, J. A. Ascencio, M. Perez-Alvarez, C. Gutierrez-Wing and M. Jose-Yacaman, Microchem. J., 2005, 81, 133–138 CrossRef CAS.
- W. C. Wolsey, C. A. Reynolds and J. Kleinberg, Inorg. Chem., 1963, 2, 463–468 CrossRef CAS.
- M. Harada and Y. Inada, Langmuir, 2009, 25, 6049–6061 CrossRef CAS PubMed.
- M. Harada, N. Tamura and M. Takenaka, J. Phys. Chem. C, 2011, 115, 14081–14092 CAS.
- J. Hedman, R. Nilsson, S. A. Nemnonov, C. Nordling, V. A. Trapezni, M. F. Sorokina, O. I. Kljushni and M. Klasson, Phys. Scr., 1971, 4, 195–201 CrossRef CAS.
- F. Li, Q. Qian, F. Yan and G. Yuan, Mater. Chem. Phys., 2008, 107, 310–316 CrossRef CAS.
- J. Lilie, M. G. Simic and J. F. Endicott, Inorg. Chem., 1975, 14, 2129–2133 CrossRef CAS.
- Q. G. Mulazzani, S. Emmi, M. Z. Hoffman and M. Venturi, J. Am. Chem. Soc., 1981, 103, 3362–3370 CrossRef CAS.
- H. A. Schwarz and C. Creutz, Inorg. Chem., 1983, 22, 707–713 CrossRef CAS.
- R. Derai, H. Remita and M. O. Delcourt, Radiat. Phys. Chem., 1991, 38, 483–486 CrossRef CAS.
- K. R. Mann, N. S. Lewis, V. M. Miskowski, D. K. Erwin, G. S. Hammond and H. B. Gray, J. Am. Chem. Soc., 1977, 99, 5525–5526 CrossRef CAS.
- I. S. Sigal and H. B. Gray, J. Am. Chem. Soc., 1981, 103, 2220–2225 CrossRef CAS.
- A. Cobelo-Garcia, Environ. Sci. Pollut. Res., 2013, 20, 2330–2339 CrossRef CAS PubMed.
- Y. Kwon, S. C. S. Lai, P. Rodriguez and M. T. M. Koper, J. Am. Chem. Soc., 2011, 133, 6914–6917 CrossRef CAS PubMed.
- J. Szammer, M. Jaky and O. V. Gerasimov, Int. J. Chem. Kinet., 1992, 24, 145–154 CrossRef CAS.
- V. R. Gangwal, J. van der Schaaf, B. F. M. Kuster and J. C. Schouten, J. Catal., 2005, 229, 389–403 CrossRef CAS.
- L. H. S. Gasparotto, A. C. Garcia, J. F. Gomes and G. Tremiliosi, J. Power Sources, 2012, 218, 73–78 CrossRef CAS.
- A. C. Garcia, L. H. S. Gasparotto, J. F. Gomes and G. Tremiliosi, Electrocatalysis, 2012, 3, 147–152 CrossRef CAS.
- A. G. Garcia, P. P. Lopes, J. F. Gomes, C. Pires, E. B. Ferreira, R. G. M. Lucena, L. H. S. Gasparotto and G. Tremiliosi, New J. Chem., 2014, 38, 2865–2873 RSC.
- J. F. Gomes, A. C. Garcia, E. B. Ferreira, C. Pires, V. L. Oliveira, G. Tremiliosi and L. H. S. Gasparotto, Phys. Chem. Chem. Phys., 2015, 17, 21683–21693 RSC.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- R. Luo, in CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CD-ROM Version, CRC Press/Taylor and Francis, Boca Raton, 90th edn, 2010, pp. 9–71 Search PubMed.
- Q. He, S. Mukerjee, B. Shyam, D. Ramaker, S. Parres-Esclapez, M. J. Illan-Gomez and A. Bueno-Lopez, J. Power Sources, 2009, 193, 408–415 CrossRef CAS.
- G. Cui, S. Song, P. K. Shen, A. Kowal and C. Bianchini, J. Phys. Chem. C, 2009, 113, 15639–15642 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26591j |
| This journal is © The Royal Society of Chemistry 2017 |