
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalf-sandwich chiral rare-earth metal complexes with linked tridentate amido-indenyl ligand: synthesis, characterization, and catalytic properties for intramolecular hydroamination†
Zhuo Chai,
Jiang Chu,
Yunyi Qi,
Mujun Tang,
Jinsong Hou and
Gaosheng Yang *
*
Key Laboratory of Functionalized Molecular Solids, Ministry of Education, Anhui Laboratory of Molecule-Based Materials, Institute of Organic Chemistry, College of Chemistry and Materials Science, Anhui Normal University, Wuhu, Anhui 241000, China. E-mail: gshyang@mail.ahnu.edu.cn
First published on 16th December 2016
Abstract
The enantioselective intramolecular hydroamination of alkenes with free amines has been a touchstone reaction in the development of many new chiral rare-earth (RE) metal complexes. In this article, we describe the synthesis and characterization of four novel rare-earth metal (Y, Sm, Lu, Sc) complexes, featuring a tridentate carbon-linked amido-indenyl ligand with a shortened linker between the indene ring and the chiral diamine moiety, as compared to our previously developed structurally similar silicon-linked ligand. Despite the generally lower enantioselectivities obtained in the current work, the structure–activity analyses of these complexes based on X-ray crystallographic data could be useful for further development of highly efficient chiral rare-earth metal complexes for asymmetric synthesis.
Introduction
Rare-earth metal complexes with various organic ligands belong to a unique class of compounds with both intriguing structures for coordination chemistry research and special reactivity for organic synthesis.1 In this field, the design and synthesis of new chiral ligands and their complexes with various rare-earth metals has been a research focus, as have their applications to various organic reactions as chiral (pre)catalysts. In particular, the enantioselective intramolecular hydroamination of unactivated alkenes with free amines, which represents the earliest and arguably most studied type of transformation with chiral rare-earth metal complexes, has become a touchstone reaction in the development of many new chiral rare-earth metal complexes.2 The extensive studies in this realm have not only led to the invention of several highly efficient rare-earth metal catalyst systems for this particular reaction,3–6 but also provided some useful mechanistic insight into the structure–reactivity relationship with important implications for catalyst design in general asymmetric catalysis.As epimerization have been identified as a crucial limiting factor to the chiral inducing power of ansa-lanthanocene complexes containing cyclopentadienyl group (Cp)-based C1-symmetric chiral sandwich ligands,3,4 chemists have developed numerous alternative non-Cp based chiral ligands that can circumvent this issue, including chiral BINOL7/BINAM8 based ligands, chiral vicinal diamine based ligands9 and others,10 with impressive advances being achieved. On the other hand, the development of half-sandwich rare-earth metal complexes with Cp ligands bearing a short side arm containing additional chelating functionalities, known as constrained geometry complexes (CGCs), has received much interest with some unique structures and reactivities being disclosed.11 However, the development of chiral CGCs of rare-earth metals, which could serve as precatalysts for asymmetric synthesis, has been rarely reported.
In this context, our group have recently demonstrated that chiral yttrium complex 1 bearing a tridentate silicon-linked amido-indenyl ligand could catalyse the intramolecular hydroamination of non-activated olefins with up to 97% ee (Fig. 1).6 However, the poor crystallinity of complexes of this ligand with other rare-earth metals hampers a thorough structure–reactivity study with these complexes. Moreover, similar to many other chiral rare-earth metal complexes examined in this reaction, the degrees of enantiocontrol achieved with this complex varied significantly from one substrate to another one, even in cases with only minor structural changes. It is presumed that replacing the silicon linker by a smaller carbon one might create a more compact chiral environment around the metal center, which may have significant influence on the reactivity and/or chiral induction power of the corresponding RE metal complexes.12 Herein, we report the results of our research efforts in this regard.
 | ||
| Fig. 1 Chiral half-sandwich RE metal complexes for intramolecular annulative hydroamination of unactivated olefins. | ||
Results and discussion
Syntheses and characterization
Our study began with the syntheses of the complexes 4 (Scheme 1). First, condensation of 2-(1H-inden-3-yl)acetic acid 1 with the commercially available chiral diamine provided chiral amide 2 in 83% yield, which was then reduced with lithium aluminium hydride to furnish the desired methylene-linked indene-chiral diamine ligand 3 in a yield of 69%. Subsequent treatment of this ligand with [(Me3Si)2N]3Ln(μ-Cl)Li(THF)3 (Ln = Y, Sm, Lu, Sc) in toluene furnished the corresponding chiral rare-earth complexes 4a–4d in excellent yields. The high overall yields for the preparation of this type of complexes provide an obvious advantage over our previously developed relevant silicon-linked complexes.6 All of the four complexes were characterized by NMR and elemental microanalysis. Moreover, the good crystallinities of these complexes allows for a more systematic single crystal X-ray crystallographic analyses, which could provide more useful insights to the structure–activity relationship (Fig. 2). | ||
| Scheme 1 Syntheses of the chiral rare-earth metal complexes 4. | ||
 | ||
| Fig. 2 ORTEP representation of RE complexes 4 (drawn with 30% probability ellipsoids). RE = Y, Sm, Lu or Sc. | ||
Description of crystal structures
The X-ray structures of the four complexes are very similar, differing in only the core rare-earth metal. They all crystallized as mononuclear structures in the monoclinic space group P21 with four molecules in the unit cell, and the rare-earth metal atoms are about in a tetrahedral coordination.As shown in Table 1, the M–C bond lengths of 4 (2.467(3)–2.770(3) Å) between the rare-earth metal atoms and the indenyl carbon atom correlate well with those of related complexes previously reported by us and others. The covalent M–N bond lengths (M–N2 and M–N3) are appreciably shorter than non-covalent ones (M–N1). Moreover, an apparent lanthanide contraction effect is observed on both the bond distances and bond angles: as the radii of the rare-earth metal atoms decreases, both the M–C and M–N bond lengths decreases while the bond angles of N–M–N bonds involving the two nitrogen atoms of the chiral ligand (N1 and N2) increases. Compared to our previous work with the chiral yttrium (Y) complex of the silicon-linked amido-indenyl ligand, the bond lengths of the two yttrium complexes, C9–C10 (1.512(6)) vs. Si–C (1.881(4)) and C9–N2 (1.471(6)) vs. Si–N (1.710(3)), would render the indene ring and the chiral diamine moiety closer as expected.
| 4a (Y) | 4b (Sm) | 4c (Lu) | 4d (Sc) | |
|---|---|---|---|---|
| C11–RE | 2.611(4) | 2.667(3) | 2.568(4) | 2.476(2) |
| C12–RE | 2.630(4) | 2.715(3) | 2.576(4) | 2.467(3) |
| C13–RE | 2.682(4) | 2.760(3) | 2.629(5) | 2.523(3) |
| C14–RE | 2.716(4) | 2.770(3) | 2.678(4) | 2.609(2) |
| C19–RE | 2.679(4) | 2.729(3) | 2.660(4) | 2.588(2) |
| N1–RE | 2.487(3) | 2.558(2) | 2.423(3) | 2.343(2) |
| N2–RE | 2.171(3) | 2.226(2) | 2.132(3) | 2.029(2) |
| N3–RE | 2.238(3) | 2.301(2) | 2.193(3) | 2.087(2) |
| C9–C10 | 1.512(6) | 1.520(5) | 1.526(6) | 1.512(5) |
| C9–N2 | 1.471(6) | 1.456(4) | 1.452(5) | 1.474(3) |
| N1–RE–N2 | 73.52(12) | 71.84(8) | 75.39(12) | 77.93(8) |
| N1–RE–N3 | 101.44(12) | 102.28(9) | 100.73(12) | 99.80(8) |
| N2–RE–N3 | 113.63(11) | 113.42(7) | 113.01(11) | 114.88(8) |
Another structural feature of complexes 4 isolated in this work resides in the planar chirality of the indene ring. In previous work with the rare-earth metal complexes of the chiral tridentate amido-indenyl ligand, we observed two different orientations of the indene ring in the complexes depending on the rare-earth metal ion (Sp for Y and Rp for Er). In the current work, only one is observed in all of the four complexes with the absolute configuration of the indene ring being Rp irrespective of the metal core. This observation suggests the carbon-linked chiral ligand might predominate in determining the coordination orientation of the indene ring in these complexes.
Catalysis of reaction
With the chiral rare-earth metal complexes in hand, we then evaluated their catalytic activities in the intramolecular annulative hydroamination of aminoalkene 5a (Table 2). Complex 4d with the smallest ion radii of the core metal (Sc) demonstrated the poorest activity and enantioselectivity, while complex 4b with the largest samarium ion is the most reactive one yet still with a very poor enantioselectivity. The absolute configuration of the product was assigned based on comparison of peak orders in HPLC assay on a chiral column with literature data.6
|
||||
|---|---|---|---|---|
| Entry | 4 (Ln) | T (°C) | t (h) | eeb (%) |
| a Unless otherwise noted, the reactions were conducted with 0.27 mmol of aminoalkene 5a in 0.5 mL of C6D6. All conversions are >95% based on 1H NMR analysis using ferrocene as the internal standard.b Determined by chiral HPLC after acylation of the product 6a to the corresponding amide 7a with 4-MeOC6H4COCl. | ||||
| 1 | 4a (Y) | 20 | 5.0 | 67 |
| 2 | 4b (Sm) | 20 | 2.0 | 34 |
| 3 | 4c (Lu) | 20 | 29.0 | 87 |
| 4 | 4d (Sc) | 80 | 50.0 | 30 |

The complex 4c of lutetium (Lu) displayed the highest enantioselectivity for the reaction, albeit with a moderate catalytic efficiency (entry 3). These results suggest that a good match of the sizes of the metal ion and the chiral ligand is crucial for both reactivity and enantioselectivity.
For a direct comparison of catalytic power between the carbon-linked complex and the previously developed silicon-linked one, we first probed the substrate scope of 4a, in spite of the moderate enantioselectivity obtained (Table 3). In terms of enantioselectivity, the carbon-linked yttrium complex 4a is invariably much inferior to the silicon-linked one, but they behaved quite similarly in responses to changes in the substrate structures. Moreover, their differences in catalytic efficiency varied from substrate to substrate, and in some cases 4a proved to be much more favoured in this regard (entries 1, 4 and 8).
| Entry | Substrate | Product | t (h) | eec (%) |
|---|---|---|---|---|
| a See footnote a of Table 2 for general reaction conditions. Reactions were run at 20 °C unless specified otherwise.b Data in parentheses refer to those obtained with the silicon-linked yttrium complex, see ref. 6.c Determined by chiral HPLC after acylation of the products 6 to the corresponding amides 7 with 4-MeOC6H4COCl.d Run at 60 °C. | ||||
| 1 | 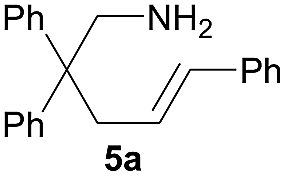 |
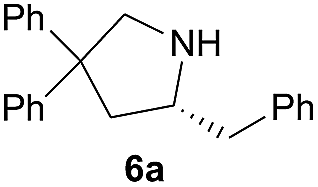 |
5.0 (9.6)b | 67 (97)b |
| 2 |  |
 |
3.0 (0.8) | 60 (85) |
| 3d | 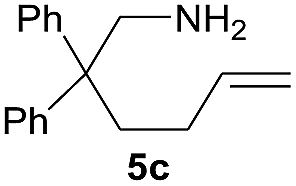 |
 |
16.0 (15.6) | 45 (69) |
| 4 |  |
 |
4.0 (13.0) | 56 (82) |
| 5d |  |
 |
50.0 (9.0) | 52 (80) |
| 6d |  |
 |
68.0 (9.6) | 41 (58) |
| 7 |  |
 |
0.6 (0.6) | 65 (75) |
| 8d | 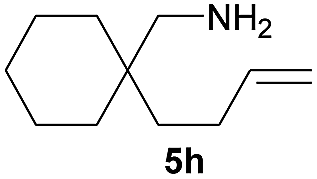 |
 |
10.0 (62.1) | 43 (72) |
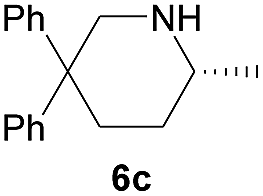
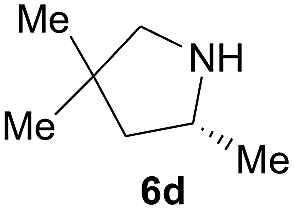
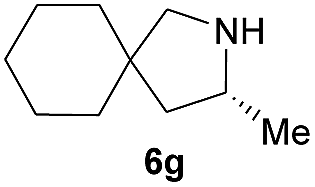
Next, we examined the substrate scope with complex 4c, the optimum precatalyst of the carbon-linked ligand series (Table 4). This complex provided higher enantioselectivity in the formation of five-membered pyrrolidines 6a and 6b (entries 1 and 2), as compared to the results obtained with the yttrium complex 4a, while a slightly lower ee value was obtained in the case of six-membered piperidine 6c (entry 3). Given the high enantioselectivity obtained with 5a, we then studied the substituent effect of the benzene ring on the reaction by examining several substituted styrene-type aminoalkenes. It is worth mentioning that systematic examination of this type of internal alkenes in rare-earth metal-catalysed asymmetric hydroamination reactions is unprecedented. Both the electronic nature and positions of the substituents on the benzene ring showed significant influence on the reaction rate and enantioselectivity. When the substituent is at the para position, an electron-withdrawing one, such as a halogen, is clearly favoured over an electron-donating one in terms of enantioselectivity (entries 5, 7 vs. entries 8, 9). The meta or ortho positions are not favoured when an electron-withdrawing substituent is present. Surprisingly, the meta position is mostly favoured in the case of 5p bearing a strongly electron-donating group (entry 11). The catalytic efficiency of the complex in these reactions, however, showed no consistent trend.
| Entry | Substrate | Product | t (h) | eeb (%) |
|---|---|---|---|---|
| a See footnote a of Table 2 for general reaction conditions. Reactions were run at 20 °C unless specified otherwise.b See footnote b of Table 2 for general reaction conditions. Reactions were run at 20 °C unless specified otherwise.c Run at 60 °C.d Run at 50 °C. | ||||
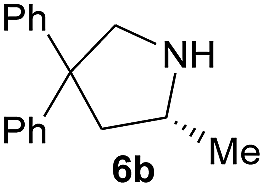
| 1 |  |
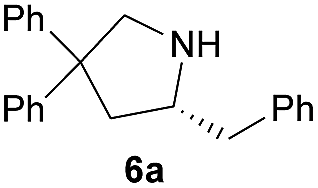 |
29.0 | 87 |
| 2 | 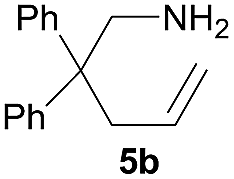 |
 |
1.5 | 68 |
| 3c |  |
 |
7.0 | 42 |
| 4 |  |
 |
1.5 | 65 |
| 5d |  |
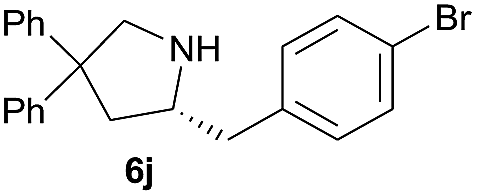 |
8.0 | 80 |
| 6c |  |
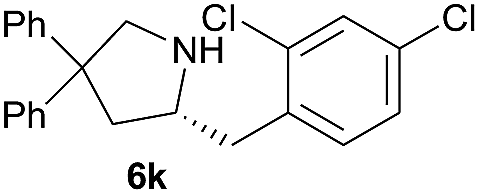 |
0.7 | 51 |
| 7 |  |
 |
3.0 | 80 |
| 8c |  |
 |
17.0 | 67 |
| 9 |  |
 |
6.3 | 28 |
| 10 |  |
 |
8.0 | 61 |
| 11 |  |
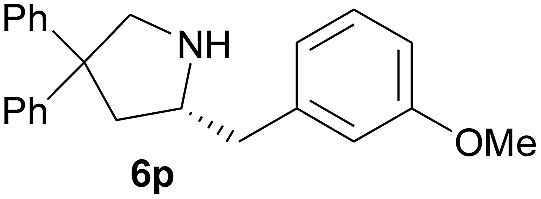 |
6.3 | 83 |
| 12 |  |
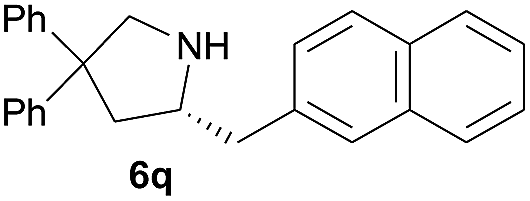 |
4.9 | 78 |
Mechanistic consideration
In view of the experimental results and previous relevant studies,2 a tentative reaction mechanism was proposed using the reaction of aminoalkene 5b as example (Scheme 2). First, upon the mixing of the chiral rare-earth metal complexes 4 and aminoalkene 5b, a fast ligand exchange occurred to provide the intermediate I, which would undergo intramolecular hydroamination to provide intermediate II. This intermediate would undergo protonation with the substrate 5b to release the cyclized product 6b while regenerating the intermediate I. The stereochemical results of the reaction might be explained by the transition state model TS, which is based on the X-ray structures of 4. In this model, the main chain of 5b adopts a chair-like conformation with the bulky diphenyl substituents far away from the chiral diamine backbone, and the nitrogen atom would attack the Si face of the alkene as mediated by the rare-earth metal centre to provide the R-configuration product. We suppose that the inferior enantiocontrolling power of these complexes, as compared to the silicon-linked one, might be partly attributed to the chiral planarity (Sp) of the indene ring. In the current case, the phenyl group of the indene ring is placed away from the chiral diamine backbone, which might be detrimental to the synergy of these two chiral elements of the complex in the enantiodifferentiating process. However, a more exact rationale for the highly fluctuating enantioselectivity of this reaction system requires further in-depth investigation. | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
In summary, we have synthesised and characterized four novel chiral half-sandwich rare-earth metal complexes featuring a carbon-linked tridentate indene-diamine ligand. When used as precatalysts for the intramolecular hydroamination of unactivated alkenes, these complexes demonstrated significantly varied catalytic activity to provide the chiral pyrrolidine and piperidine products in moderate to good enantioselectivities. Although such results are not up to our expectation for the new carbon-linked ligand featuring a shortened linker between the indene ring and the chiral diamine moiety as compared to our previously developed structurally similar silicon-linked tridentate indene-diamine ligand, the structure–activity analyses of these complexes based on X-ray crystallographic data provide some useful information for further development of highly efficient chiral rare-earth metal complexes for asymmetric catalysis. Efforts towards this are currently underway in our laboratories.Experimental section
General methods
All reactions with air- or moisture sensitive materials were performed in oven (120 °C) or flame-dried glassware under an inert atmosphere of argon, employing standard Schlenk and glovebox techniques. All solvents were distilled over either finely divided LiAlH4 or sodium benzophenone ketyl under argon prior to use unless otherwise noted. CDCl3 was dried over activated 4 Å molecular sieves. C6D6 was distilled from sodium/benzophenone ketyl. [(Me3Si)2N]3Ln(μ-Cl)Li(THF)3 was prepared by a literature method.13 (1R,2R)-N,N-Dimethylcyclohexane-1,2-diamine is commercially available or could be prepared by using literature method.14 All the aminoalkene substrates were synthesized according to literature protocols.15 Elemental analysis data were obtained with a Perkin-Elmer 2400 Series II elemental analyzer. 1H NMR and 13C NMR spectra for analyses of compounds were recorded with a Bruker AV 300 NMR or AVANCE-500 spectrometers in C6D6 or CDCl3. High performance liquid chromatography (HPLC) analyses were performed on a Agilent 1200 chromatograph using chiral Daicel Chiralcel® columns as noted.Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) afforded 2 (1.75 g, 83% yield) as a yellow solid. 1H NMR (500 MHz, CDCl3): δ 7.47 (d, J = 7.3 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.5 Hz, 1H), 7.22 (t, J = 7.4 Hz, 1H), 6.44 (s, 1H), 6.39 (brs, 1H), 3.56 (d, J = 16.1 Hz, 1H), 3.50 (d, J = 16.1 Hz, 1H), 3.46–3.32 (m, 3H), 2.56–2.48 (m, 1H), 2.16–2.06 (m, 1H), 1.97 (s, 6H), 1.82–1.68 (m, 2H), 1.67–1.58 (m, 1H), 1.34–1.23 (m, 1H), 1.19–1.05 (m, 2H), 1.04–0.92 (m, 1H). 13C NMR (125 MHz, CDCl3): δ 170.5, 144.4, 144.2, 138.6, 131.9, 126.2, 125.0, 123.7, 119.4, 66.4, 51.4, 39.6, 38.0, 36.9, 32.5, 25.3, 24.6, 21.3. HRMS (ESI) calcd for C19H27N2O ([M + H]+) calcd for 299.2123, found 299.2119.:1) to give 3 (0.39 g, 69% yield) as a yellow oil. [α]20D = −66.8 (c = 0.10, CH2Cl2). 1H NMR (500 MHz, C6D6): δ 7.35 (d, J = 7.5 Hz, 1H), 7.31 (d, J = 7.3 Hz, 1H), 7.24 (t, J = 7.4 Hz, 1H), 7.15 (t, J = 7.5 Hz, 1H), 6.10 (s, 1H), 3.18–3.12 (m, 1H), 3.09 (s, 2H), 2.88–2.69 (m, 4H), 2.32–2.21 (m, 2H), 2.18–2.11 (m, 1H), 2.01 (s, 6H), 1.66–1.52 (m, 3H), 1.23–1.14 (m, 1H), 1.13–1.00 (m, 2H), 0.99–0.87 (m, 1H). 13C NMR (75.5 MHz, CDCl3): δ 144.3, 143.4, 141.4, 127.6, 125.0, 123.5, 122.7, 117.9, 65.8, 57.6, 44.8, 38.9, 36.8, 30.9, 27.5, 24.4, 23.7, 19.6. HRMS (ESI) calcd for C19H29N2 ([M + H]+) calcd for 285.2331, found 285.2328.:1 at −10 °C. 1H NMR (500 MHz, C6D6): δ 7.76 (dd, J = 8.4, 0.9 Hz, 1H), 7.38 (dt, J = 8.3, 0.9 Hz), 6.95–6.89 (m, 1H), 6.87 (d, J = 3.2 Hz, 1H), 6.85–6.81 (m, 1H), 6.30 (d, J = 3.2 Hz, 1H), 3.93 (dd, J = 10.5, 7.7 Hz, 1H), 3.83 (td, J = 11.2, 6.0 Hz, 1H), 3.56 (dd, J = 14.2, 5.8 Hz, 1H), 3.43 (ddd, J = 14.2, 11.5, 7.9 Hz, 1H), 2.78–2.69 (m, 1H), 2.30–2.21 (m, 1H), 2.12 (td, J = 10.1, 3.4 Hz, 1H), 1.59–1.45 (m, 5H), 1.26–1.18 (m, 1H), 1.12–0.94 (m, 3H), 0.92 (s, 3H), 0.60 (qd, J = 12.1, 3.6 Hz, 1H), 0.39 (s, 9H), 0.17 (s, 9H). 13C NMR (125 MHz, C6D6): δ 127.4, 127.2, 123.5, 122.1, 121.7, 121.2, 120.7, 119.7, 93.3, 68.1, 67.0, 60.8, 43.5, 35.0, 32.2, 30.0, 26.1, 25.3, 20.7, 6.1, 4.4. Anal. calcd for C25H44N3Si2Y: C, 56.47; H, 8.34; N, 7.90; found: C, 56.53; H, 8.30; N, 7.82.:1 at −10 °C. 1H NMR (500 MHz, C6D6): δ 17.90 (s, 1H), 13.31 (d, J = 8.5 Hz, 1H), 10.75 (s, 1H), 6.77 (d, J = 6.8 Hz, 2H), 6.19 (s, 1H), 5.81 (d, J = 7.9 Hz, 1H), 4.09 (t, J = 7.0 Hz, 1H), 3.69 (d, J = 12.1 Hz, 1H), 3.56 (d, J = 13.1 Hz, 1H), 3.25 (s, 1H), 2.92–2.75 (m, 2H), 2.62 (d, J = 14.2 Hz, 1H), 2.49–2.38 (m, 1H), 1.95 (d, J = 12.7 Hz, 1H), 1.23–1.11 (m, 1H), 0.68 (s, 9H), 0.30 (s, 1H), 0.10 (s, 1H), −0.30 (s, 3H), −1.65 (s, 1H), −2.69 (s, 3H), −3.24 (s, 9H). 13C NMR (125 MHz, C6D6): δ 131.8, 130.5, 120.7, 117.8, 117.2, 116.9, 112.5, 92.6, 86.2, 85.3, 82.5, 43.8, 38.4, 32.1, 27.3, 25.6, 23.6, 23.4, 3.5, 2.2, 1.0. Anal. calcd for C25H44N3Si2Sm: C, 50.62; H, 7.48; N, 7.08; found: C, 50.68; H, 7.45; N, 7.02.:1 at −10 °C. 1H NMR (500 MHz, C6D6): δ 7.76–7.69 (m, 1H), 7.37 (d, J = 8.3 Hz, 1H), 6.94–6.88 (m, 1H), 6.87–6.80 (m, 2H), 6.29 (d, J = 3.2 Hz, 1H), 3.81–3.68 (m, 2H), 3.50–3.42 (m, 1H), 3.42–3.33 (m, 1H), 3.04–2.96 (m, 1H), 2.07–1.98 (m, 2H), 1.62 (s, 3H), 1.54–1.46 (m, 2H), 1.23–1.04 (m, 3H), 0.93–0.86 (m, 1H), 0.75 (s, 3H), 0.58–0.48 (m, 1H), 0.41 (s, 9H), 0.28 (s, 9H). 13C NMR (126 MHz, C6D6): δ 127.0, 126.4, 123.9, 122.2, 122.1, 121.8, 120.9, 120.2, 94.5, 67.0, 60.7, 44.4, 34.1, 31.0, 28.8, 25.5, 24.8, 20.2, 6.2, 6.0. Anal. calcd for C25H44N3Si2Sc: C, 61.56; H, 9.09; N, 8.61; found: C, 61.62; H, 9.03; N, 8.58.
:1) afforded 2 (1.75 g, 83% yield) as a yellow solid. 1H NMR (500 MHz, CDCl3): δ 7.47 (d, J = 7.3 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.5 Hz, 1H), 7.22 (t, J = 7.4 Hz, 1H), 6.44 (s, 1H), 6.39 (brs, 1H), 3.56 (d, J = 16.1 Hz, 1H), 3.50 (d, J = 16.1 Hz, 1H), 3.46–3.32 (m, 3H), 2.56–2.48 (m, 1H), 2.16–2.06 (m, 1H), 1.97 (s, 6H), 1.82–1.68 (m, 2H), 1.67–1.58 (m, 1H), 1.34–1.23 (m, 1H), 1.19–1.05 (m, 2H), 1.04–0.92 (m, 1H). 13C NMR (125 MHz, CDCl3): δ 170.5, 144.4, 144.2, 138.6, 131.9, 126.2, 125.0, 123.7, 119.4, 66.4, 51.4, 39.6, 38.0, 36.9, 32.5, 25.3, 24.6, 21.3. HRMS (ESI) calcd for C19H27N2O ([M + H]+) calcd for 299.2123, found 299.2119.:1) to give 3 (0.39 g, 69% yield) as a yellow oil. [α]20D = −66.8 (c = 0.10, CH2Cl2). 1H NMR (500 MHz, C6D6): δ 7.35 (d, J = 7.5 Hz, 1H), 7.31 (d, J = 7.3 Hz, 1H), 7.24 (t, J = 7.4 Hz, 1H), 7.15 (t, J = 7.5 Hz, 1H), 6.10 (s, 1H), 3.18–3.12 (m, 1H), 3.09 (s, 2H), 2.88–2.69 (m, 4H), 2.32–2.21 (m, 2H), 2.18–2.11 (m, 1H), 2.01 (s, 6H), 1.66–1.52 (m, 3H), 1.23–1.14 (m, 1H), 1.13–1.00 (m, 2H), 0.99–0.87 (m, 1H). 13C NMR (75.5 MHz, CDCl3): δ 144.3, 143.4, 141.4, 127.6, 125.0, 123.5, 122.7, 117.9, 65.8, 57.6, 44.8, 38.9, 36.8, 30.9, 27.5, 24.4, 23.7, 19.6. HRMS (ESI) calcd for C19H29N2 ([M + H]+) calcd for 285.2331, found 285.2328.:1 at −10 °C. 1H NMR (500 MHz, C6D6): δ 7.76 (dd, J = 8.4, 0.9 Hz, 1H), 7.38 (dt, J = 8.3, 0.9 Hz), 6.95–6.89 (m, 1H), 6.87 (d, J = 3.2 Hz, 1H), 6.85–6.81 (m, 1H), 6.30 (d, J = 3.2 Hz, 1H), 3.93 (dd, J = 10.5, 7.7 Hz, 1H), 3.83 (td, J = 11.2, 6.0 Hz, 1H), 3.56 (dd, J = 14.2, 5.8 Hz, 1H), 3.43 (ddd, J = 14.2, 11.5, 7.9 Hz, 1H), 2.78–2.69 (m, 1H), 2.30–2.21 (m, 1H), 2.12 (td, J = 10.1, 3.4 Hz, 1H), 1.59–1.45 (m, 5H), 1.26–1.18 (m, 1H), 1.12–0.94 (m, 3H), 0.92 (s, 3H), 0.60 (qd, J = 12.1, 3.6 Hz, 1H), 0.39 (s, 9H), 0.17 (s, 9H). 13C NMR (125 MHz, C6D6): δ 127.4, 127.2, 123.5, 122.1, 121.7, 121.2, 120.7, 119.7, 93.3, 68.1, 67.0, 60.8, 43.5, 35.0, 32.2, 30.0, 26.1, 25.3, 20.7, 6.1, 4.4. Anal. calcd for C25H44N3Si2Y: C, 56.47; H, 8.34; N, 7.90; found: C, 56.53; H, 8.30; N, 7.82.:1 at −10 °C. 1H NMR (500 MHz, C6D6): δ 17.90 (s, 1H), 13.31 (d, J = 8.5 Hz, 1H), 10.75 (s, 1H), 6.77 (d, J = 6.8 Hz, 2H), 6.19 (s, 1H), 5.81 (d, J = 7.9 Hz, 1H), 4.09 (t, J = 7.0 Hz, 1H), 3.69 (d, J = 12.1 Hz, 1H), 3.56 (d, J = 13.1 Hz, 1H), 3.25 (s, 1H), 2.92–2.75 (m, 2H), 2.62 (d, J = 14.2 Hz, 1H), 2.49–2.38 (m, 1H), 1.95 (d, J = 12.7 Hz, 1H), 1.23–1.11 (m, 1H), 0.68 (s, 9H), 0.30 (s, 1H), 0.10 (s, 1H), −0.30 (s, 3H), −1.65 (s, 1H), −2.69 (s, 3H), −3.24 (s, 9H). 13C NMR (125 MHz, C6D6): δ 131.8, 130.5, 120.7, 117.8, 117.2, 116.9, 112.5, 92.6, 86.2, 85.3, 82.5, 43.8, 38.4, 32.1, 27.3, 25.6, 23.6, 23.4, 3.5, 2.2, 1.0. Anal. calcd for C25H44N3Si2Sm: C, 50.62; H, 7.48; N, 7.08; found: C, 50.68; H, 7.45; N, 7.02.:1 at −10 °C. 1H NMR (500 MHz, C6D6): δ 7.76–7.69 (m, 1H), 7.37 (d, J = 8.3 Hz, 1H), 6.94–6.88 (m, 1H), 6.87–6.80 (m, 2H), 6.29 (d, J = 3.2 Hz, 1H), 3.81–3.68 (m, 2H), 3.50–3.42 (m, 1H), 3.42–3.33 (m, 1H), 3.04–2.96 (m, 1H), 2.07–1.98 (m, 2H), 1.62 (s, 3H), 1.54–1.46 (m, 2H), 1.23–1.04 (m, 3H), 0.93–0.86 (m, 1H), 0.75 (s, 3H), 0.58–0.48 (m, 1H), 0.41 (s, 9H), 0.28 (s, 9H). 13C NMR (126 MHz, C6D6): δ 127.0, 126.4, 123.9, 122.2, 122.1, 121.8, 120.9, 120.2, 94.5, 67.0, 60.7, 44.4, 34.1, 31.0, 28.8, 25.5, 24.8, 20.2, 6.2, 6.0. Anal. calcd for C25H44N3Si2Sc: C, 61.56; H, 9.09; N, 8.61; found: C, 61.62; H, 9.03; N, 8.58.Crystal structure determination
The single crystal X-ray diffraction data for complexes 4 were collected on a diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at room temperature. Saint program and SADABS program carried out the data integration. The structures were solved by a direct method and refined on F2 using SHELXTL suite of program. All non-hydrogen atoms were anisotropically refined by full-matrix least squares methods. All hydrogen atoms were geometrically generated and isotropically refined using a riding model. Crystallographic data in CIF files are provided in the ESI.† CCDC-1509830 (4a), 1509831 (4b), 1509832 (4c), and 1509833 (4d) contain the supplementary crystallographic data for this paper.Enantioselective annulative intramolecular hydroamination of olefins catalysed by complexes 4
In an argon-filled glovebox, 5 mol% of 4, Cp2Fe (6.0 mg, internal standard) and aminoalkene 5 (0.32 mmol) were weighed into a 5 mm NMR tube equipped with a Teflon valve (J-Young), and C6D6 (0.6 mL) was added. The hydroamination reaction was then monitored by 1H NMR analysis for estimate of both conversion and yield using ferrocene as the internal standard.Acknowledgements
Financial support of this research from the National Natural Science Foundation of China (NSFC No. 21672005 and 21472001) and the Special and Excellent Research Fund of Anhui Normal University are gratefully acknowledged.Notes and references
- For recent reviews: (a) P. W. Roesky, Molecular Catalysis of Rare-Earth Elements (Structure and Bonding), Springer, Berlin, 2010 Search PubMed; (b) D. Patel and S. T. Liddle, Rev. Inorg. Chem., 2012, 32, 1–22 CrossRef CAS; (c) F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657 RSC; (d) J. Lucas, P. Lucas, T. L. Mercier, A. Rollat and W. Davenport, Rare Earths, Elsevier, 2015 Search PubMed; (e) B. S. Soller, S. Salzinger and B. Rieger, Chem. Rev., 2016, 116, 1993 CrossRef CAS PubMed.
- For selected recent reviews: (a) P. S. Hanley and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 8510 CrossRef CAS PubMed; (b) J. Hannedouche and E. Schulz, Chem.–Eur. J., 2013, 19, 4972 CrossRef CAS PubMed; (c) J. Hannedouche, J. Collin, A. Trifonov and E. Schulz, J. Organomet. Chem., 2011, 696, 255 CrossRef CAS; (d) I. Aillaud, J. Collin, J. Hannedouche and E. Schulz, Dalton Trans., 2007, 5105 RSC; (e) G. Zi, Dalton Trans., 2009, 9101 RSC; (f) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed; (g) H. Pellissier, Coord. Chem. Rev., 2016, 324, 17 CrossRef CAS.
- (a) S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS PubMed; (b) J.-S. Ryu, T. J. Marks and F. E. McDonald, J. Org. Chem., 2004, 69, 1038 CrossRef CAS PubMed; (c) S. Hong, A. M. Kawaoka and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 15878 CrossRef CAS PubMed; (d) S. Hong and T. J. Marks, J. Am. Chem. Soc., 2002, 124, 7886 CrossRef CAS PubMed; (e) M. R. Douglass, M. Ogasawara, S. Hong, M. V. Metz and T. J. Marks, Organometallics, 2002, 21, 283 CrossRef CAS; (f) M. A. Giardello, V. P. Conticello, L. Brard, M. R. Gagné and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10241 CrossRef CAS; (g) M. A. Giardello, V. P. Conticello, L. Brard, M. Sabat, A. L. Rheingold, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10212 CrossRef CAS; (h) M. R. Gagné, L. Brard, V. P. Conticello, M. A. Giardello, C. L. Stern and T. J. Marks, Organometallics, 1992, 11, 2003 CrossRef.
- D. V. Vitanova, F. Hampel and K. C. Hultzsch, J. Organomet. Chem., 2007, 692, 4690 CrossRef CAS.
- K. Manna, M. L. Kruse and A. D. Sadow, ACS Catal., 2011, 1, 1637 CrossRef CAS.
- Z. Chai, D. Hua, K. Li, J. Chu and G. Yang, Chem. Commun., 2014, 50, 177 RSC.
- For examples: (a) D. V. Gribkov, K. C. Hultzsch and F. Hampel, Chem.–Eur. J., 2003, 9, 4796 CrossRef CAS PubMed; (b) D. V. Gribkov and K. C. Hultzsch, Chem. Commun., 2004, 730 RSC; (c) D. V. Gribkov, K. C. Hultzsch and F. Hampel, J. Am. Chem. Soc., 2006, 128, 3748 CrossRef CAS PubMed; (d) A. L. Reznichenko, F. Hampel and K. C. Hultzsch, Chem.–Eur. J., 2009, 15, 12819 CrossRef CAS PubMed; (e) A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984 CrossRef CAS PubMed; (f) D. V. Vitanova, F. Hampel and K. C. Hultzsch, J. Organomet. Chem., 2011, 696, 321 CrossRef CAS.
- For recent examples of chiral BINAM-based complexes: (a) A. L. Reznichenko and K. C. Hultzsch, Organometallics, 2013, 32, 1394 CrossRef CAS; (b) A. Aillerie, V. Rodriguez-Ruiz, R. Carlino, F. Bourdreux, R. Guillot, S. Bezzenine-Lafollée, R. Gil, D. Prim and J. Hannedouche, ChemCatChem, 2016, 8, 2455 CrossRef CAS; (c) S. Germain, E. Schulz and J. Hannedouche, ChemCatChem, 2014, 6, 2065 CrossRef CAS; (d) I. Aillaud, C. Olier, Y. Chapurina, J. Collin, E. Schulz, R. Guillot, J. Hannedouche and A. Trifonov, Organometallics, 2011, 30, 3378 CrossRef CAS; (e) Y. Chapurina, H. Ibrahim, R. Guillot, E. Kolodziej, J. Collin, A. Trifonov, E. Schulz and J. Hannedouche, J. Org. Chem., 2011, 76, 10163 CrossRef CAS PubMed; (f) K. Huynh, B. K. Anderson and T. Livinghouse, Tetrahedron Lett., 2015, 56, 3658 CrossRef CAS; (g) H. M. Lovick, D. K. An and T. Livinghouse, Dalton Trans., 2011, 7697 RSC.
- For examples of chiral vicinal diamine-based complexes: (a) N. Meyer, A. Zulys and P. W. Roesky, Organometallics, 2006, 25, 4179 CrossRef CAS; (b) P. Benndorf, J. Jenter, L. Zielke and P. W. Roesky, Chem. Commun., 2011, 2574 RSC; (c) Y. Zhang, W. Yao, H. Li and Y. Mu, Organometallics, 2012, 31, 4670 CrossRef CAS.
- (a) P. N. O'Shaughnessy, P. D. Knight, C. Morton, K. M. Gillespie and P. Scott, Chem. Commun., 2003, 1770 RSC; (b) P. N. O'Shaughnessy and P. Scott, Tetrahedron: Asymmetry, 2003, 14, 1979 CrossRef; (c) P. N. O'Shaughnessy, K. M. Gillespie, P. D. Knight, I. J. Munslow and P. Scott, Dalton Trans., 2004, 2251 RSC; (d) S. Hong, S. Tian, M. V. Metz and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 14768 CrossRef CAS PubMed; (e) Q. Wang, L. Xiang, H. Song and G. Zi, Inorg. Chem., 2008, 47, 4319 CrossRef CAS PubMed; (f) Q. Wang, L. Xiang, H. Song and G. Zi, J. Organomet. Chem., 2009, 694, 691 CrossRef CAS; (g) G. Zi, Q. Wang, L. Xiang and H. Song, Dalton Trans., 2008, 5930 RSC; (h) Q. Wang, F. Zhang, H. Song and G. Zi, J. Organomet. Chem., 2011, 696, 2186 CrossRef CAS.
- For reviews: (a) H. Braunschweig and F. M. Breitling, Coord. Chem. Rev., 2006, 250, 2691 CrossRef CAS; (b) J. Cano and K. Kunz, J. Organomet. Chem., 2007, 692, 4411 CrossRef CAS; (c) S. Arndt and J. Okuda, Chem. Rev., 2002, 102, 1953 CrossRef CAS PubMed; for recent examples: (d) N. K. Hangaly, A. R. Petrov, M. Elfferding, K. Harms and J. Sundermeyer, Dalton Trans., 2014, 43, 5930 RSC; (e) Z. Jian, N. K. Hangaly, W. Rong, Z. Mou, D. Liu, S. Li, A. Trifonov, J. Sundermeyer and D. Cui, Organometallics, 2012, 31, 4579 CrossRef CAS; (f) Z. Jian, A. R. Petrov, N. K. Hangaly, S. Li, W. Rong, Z. Mou, K. A. Rufanov, K. Harms, J. Sundermeyer and D. Cui, Organometallics, 2012, 31, 4276 Search PubMed; (g) Z. Chai, D. Hua, K. Li, S. Zhou, J. Chu and G. Yang, J. Organomet. Chem., 2014, 768, 136 CrossRef CAS; (h) L. N. Jende, C. O. Hollfelder, C. Maichle-Mössmer and R. Anwander, Organometallics, 2015, 34, 32 CrossRef CAS.
- For early works on linked Cp/indene-amido ligands: (a) J. Okuda, S. Verch, R. Stürmer and T. P. Spaniol, J. Organomet. Chem., 2000, 605, 55 CrossRef CAS; (b) J. Okuda, S. Verch, R. Stürmer and T. P. Spaniol, Chirality, 2000, 472 CrossRef CAS; (c) A. L. McKnight, M. A. Masood and R. M. Waymouth, Organometallics, 1997, 16, 2879 CrossRef CAS.
- (a) E. Sheng, S. Wang, G. Yang, S. Zhou, L. Cheng, K. Zhang and Z. Huang, Organometallics, 2003, 22, 684 CrossRef CAS; (b) S. Zhou, S. Wang, G. Yang, X. Liu, E. Sheng, K. Zhang, L. Cheng and Z. Huang, Polyhedron, 2003, 22, 1019 CrossRef CAS.
- (a) M. Kaik and J. Gawroński, Tetrahedron: Asymmetry, 2003, 14, 1559 CrossRef CAS; (b) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS PubMed; (c) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS PubMed.
- (a) C. F. Bender and R. A. Widenhoefer, J. Am. Chem. Soc., 2005, 127, 1070 CrossRef CAS PubMed; (b) M. R. Crimmin, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data in CIF files, copies of NMR spectra and HPLC traces. CCDC 1509830, 1509831, 1509832 and 1509833. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26537e |
| This journal is © The Royal Society of Chemistry 2017 |