
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePt/MnO2 nanosheets: facile synthesis and highly efficient catalyst for ethylene oxidation at low temperature†
Min Wangab,
Lingxia Zhang *a,
Weimin Huanga,
Yajun Zhouab,
Han Zhaoab,
Jian Lvab,
Jianjian Tianac,
Xiaotian Kanab and
Jianlin Shi*a
*a,
Weimin Huanga,
Yajun Zhouab,
Han Zhaoab,
Jian Lvab,
Jianjian Tianac,
Xiaotian Kanab and
Jianlin Shi*a
aState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, 1295 Dingxi Road, Shanghai 200050, P. R. China. E-mail: zhlingxia@mail.sic.ac.cn; jlshi@mail.sic.ac.cn
bUniversity of Chinese Academy of Sciences, 19 Yuquan Road, Beijing 100049, P. R. China
cSchool of Materials Science and Engineering, Shanghai University, 99 Shangda Road, Shanghai 200444, P. R. China
First published on 6th March 2017
Abstract
Transition metal oxides (TMOs) have been playing an indispensible role in the catalysis of redox reactions. In particular, two dimensional TMOs expose their surface/edge sites to a large extent, which brings unique catalytic features such as greatly enhanced catalytic activities. Here a series of MnO2 nanosheets have been facilely synthesized by a simple redox reaction between KMnO4 and 2-(N-morpholino)ethane sulfonic acid (MES) at room temperature. Among the obtained MnO2 samples, MnO2-48 h showed the highest performance in removing C2H4 resulting from its highest concentration of surface active oxygen species. To further improve the oxidation activity of the catalyst, a small amount of Pt nanoparticles (NPs) was subsequently loaded on MnO2 nanosheets (Pt/MnO2) by a colloidal deposition method. The Pt/MnO2 demonstrated enhanced catalytic performance and maintained complete removal of 20 ppm C2H4 at 50 °C for at least 12 h, which can be attributed to the large amount of adsorbed oxygen species and synergetic catalytic effect between Pt and the MnO2 support.
1 Introduction
Two-dimensional (2D) nanostructured materials have been attracting increasing attention in catalysis, optoelectronics, clean energy and biomedicine,1–3 thanks to their large surface-to-volume ratio and confined thickness on the atomic scale.3 Graphene,4,5MoS2,6 g-C3N4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7,8 and their derivatives are the most widely investigated 2D materials due to their easy preparation and attractive functionalities. Transition metal oxides (TMOs), which show great potential in applications such as catalysis, energy storage, and so on, have been usually obtained in 3D bulk and/or porous nanostructures, nevertheless, low-dimensional nanoclusters/nanodots (0D), nanowires/nanorods (1D) and nanosheets (2D) are arousing rapidly increasing interest. The top-down strategy of chemical or physical exfoliation9 has made it possible to obtain 2D TMOs. Recently, other strategies have also been developed to fabricate 2D TMOs. For example, by a bottom-up molecular self-assembly process, Sun et al.10 prepared a serious of TMOs such as Co3O4, ZnO, WO3, Fe3O4 and MnO2. However, the massive and low-cost synthesis of 2D nanostructured TMOs is still a great challenge.
7,8 and their derivatives are the most widely investigated 2D materials due to their easy preparation and attractive functionalities. Transition metal oxides (TMOs), which show great potential in applications such as catalysis, energy storage, and so on, have been usually obtained in 3D bulk and/or porous nanostructures, nevertheless, low-dimensional nanoclusters/nanodots (0D), nanowires/nanorods (1D) and nanosheets (2D) are arousing rapidly increasing interest. The top-down strategy of chemical or physical exfoliation9 has made it possible to obtain 2D TMOs. Recently, other strategies have also been developed to fabricate 2D TMOs. For example, by a bottom-up molecular self-assembly process, Sun et al.10 prepared a serious of TMOs such as Co3O4, ZnO, WO3, Fe3O4 and MnO2. However, the massive and low-cost synthesis of 2D nanostructured TMOs is still a great challenge.
As a typical kind of TMOs, manganese oxide is particularly promising thanks to its abundance, environmental friendliness and high catalytic performances in environmental purification and clean energy production, which are active in many reactions like NO oxidation,11,12 CO oxidation,13 water oxidation,14,15 etc. Manganese oxides are capable of mobilizing electrons, and thus the necessary mobile-electron environment for catalysis can be built.16 Birnessite, i.e. δ-MnO2, has a layered crystal structure, which is stacked by layers of edge-sharing MnO6 octahedra with a certain number of water molecules and different cations (e.g. Li+, Na+, K+, Ca2+) between the layers to balance the charge. The unique layered structure of birnessite makes it an efficient catalyst to remove HCHO, CO and volatile organic compounds (VOCs). Zhang et al.17 found that δ-MnO2 catalyst exhibited better activity in removing 170 ppm formaldehyde at 80 °C than α-, β-, and γ-MnO2 due to its special 2D layered structure. Despite the high cost, noble metals (e.g. Pt) have been loaded on δ-MnO2 to enhance its catalytic activity and made it possible to remove pollutants at low temperature even at room temperature18,19 thanks to the strong metal–support interaction (SMSI), or, synergetic catalytic effect,20 between noble metals and the MnO2 support.
Ethylene (C2H4), a common gaseous organic compound, is widely used as a raw material in chemical industry and is also an important natural plant hormone used in agriculture to force the ripening of fruits. However, it is also a typical harmful VOC which will lead to photochemical pollution of the atmosphere and cause anesthetic illness.21 Consequently, the removal of ethylene contaminant from air under mild conditions is highly significant. C2H4 is a typical thermally stable hydrocarbon compound with C–H, C–C σ bonds and C–C π bonds. The high bond energy of C–H bond (413 kJ mol−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (615 kJ mol−1) make it more difficult to be removed by catalytic oxidation than formaldehyde and CO. Supported noble metals have been proved to be highly effective in catalyzing the oxidation of ethylene at relatively low temperatures.22,23 Jiang et al.24 found that Pt loaded on MCM-41 showed higher performance to catalytically remove C2H4 than other noble metals on the same support.
C bond (615 kJ mol−1) make it more difficult to be removed by catalytic oxidation than formaldehyde and CO. Supported noble metals have been proved to be highly effective in catalyzing the oxidation of ethylene at relatively low temperatures.22,23 Jiang et al.24 found that Pt loaded on MCM-41 showed higher performance to catalytically remove C2H4 than other noble metals on the same support.
Herein, we synthesized 2D MnO2 nanosheets by a simple redox reaction between potassium permanganate (KMnO4) and 2-(N-Morpholino)ethane sulfonic acid (MES) at room temperature and subsequently loaded Pt nanoparticles (NPs) via colloidal deposition on the obtained MnO2 nanosheets. The as-prepared Pt/MnO2 catalyst showed extraordinarily high activity and durability in the catalytic oxidation of ethylene, and the complete ethylene removal has been achieved at around 50 °C for at least 12 h.
2 Experimental section
2.1 Preparation of the catalyst
2.1.2.1. The synthesis of MnO2 nanosheets. MnO2 nanosheets were prepared according to the previous report25 with little modification. 2.10 g KMnO4 was dissolved in 120 mL deionized water and 4.26 g 2-(N-morpholino)ethane sulfonic acid (MES) was dissolved in 120 mL deionized water, respectively. Then the KMnO4 solution was added dropwise into the MES solution under stirring. The resultant mixture was kept stirring for different time at room temperature. Then the brown slurry was filtered and washed with deionized water and alcohol and dried at 80 °C overnight. The obtained samples were denoted as MnO2-t, where t represents the reaction time between KMnO4 and MES.
2.1.2.2. The synthesis of Pt/MnO2. Pt/MnO2 catalyst with 2 wt% Pt was prepared by an ascorbic acid reduction method.26 In a typical procedure, 68 mg of PVP (PVP
:H2PtCl6 = 20:1, molar ratio) and 0.18 g of sodium citrate (sodium citrate:H2PtCl6 = 20:1, molar ratio) were dissolved in 100 mL deionized water. 0.8 mL aqueous chloroplatinic acid (H2PtCl6·6H2O) (20 mg mL−1) solution was added dropwise to the above solution under vigorous stirring at 80 °C. After stirring for an additional 5 min, 10 mL aqueous solution with 108 mg of ascorbic acid (ascorbic acid:H2PtCl6 = 20:1, molar ratio) was added dropwise to the mixture solution and after continual stirring for 1 h, the Pt colloid solution was obtained. Subsequently, 0.3 g MnO2-48 h powder (dispersed in 50 mL deionized water by ultrasound) was added into the Pt colloid solution. After stirring at 80 °C for 4 h, the obtained precipitate was filtered and washed several times with deionized water and absolute alcohol to remove any possible residual reactants. Finally, the product was dried at 80 °C for overnight and denoted as Pt/MnO2-48 h.
2.2 Characterizations
Field Emission Transmission Electron Microscopy (FETEM) analysis was conducted with a JEOL 200CX electron microscope operated at 200 kV. Energy dispersive spectrum (EDS) was obtained from an attached Oxford Link ISIS energy-dispersive spectrometer fixed on a JEM-2010 electron microscope. Field Emission Scanning Electron Microscopy (FESEM) analysis was conducted with a Su 8220 electron microscope. Powder X-ray diffraction (XRD) patterns were recorded on a Rigaku D/Max 2200PC diffractometer using Cu-Kalpha radiation (40 kV and 40 mA). The scanning rate was 4° min−1. The content of Pt was measured with Inductively Coupled Plasma atomic emission spectrometry (ICP-OES) (Vista AX). Nitrogen adsorption/desorption isotherms at 77 K were measured on a Micromeritics TriStar 3000 instrument. All samples were outgassed at 150 °C for 12 h under flowing N2 before the measurement. The specific surface areas were calculated by the Brunauer–Emmet–Teller (BET) methods using the desorption branches. X-ray photoelectron spectroscopy (XPS) signals were collected on VG Micro MKII instrument using monochromatic Mg Kalpha X-ray at 1253.6 eV operated at 120 W. All the element binding energies were referenced to the C (1s) line situated at 284.6 eV. The H2 temperature-programmed reduction (H2-TPR) and O2 temperature programmed desorption (O2-TPD) measurements were conducted on a pulse chemisorption system (Chemisorb 2750, Micromeritics Inc.) equipped with a TCD detector. As for H2-TPR, 50 mg sample was loaded into a quartz U-type tube. The sample was first pretreated with He (30 mL min−1) at 80 °C for 1 h and cooled down to the room temperature, then 5% H2/Ar was introduced with a rate of 25 mL min−1 and at the same time the temperature was increased to 800 °C at the rate of 10 °C min−1. For O2-TPD, the sample was first pretreated with He (30 mL min−1) at 80 °C for 1 h to remove physisorbed and interlayered H2O as well as surface oxygen. Then it was cooled to room temperature and flowed with O2 gas for 1 h. After that, it was purged with He for 30 min to remove weakly adsorbed O2. Then the temperature was increased from room temperature to 800 °C at the rate of 10 °C min−1 under the flow of He gas (30 mL min−1).2.3 Catalytic activity in C2H4 oxidation removal
The catalytic oxidation of ethylene was performed in a fixed-bed reactor with continuous flow under atmospheric pressure. 100 mg catalyst was placed in a quartz tube reactor. Typically, the as-synthesized MnO2 and Pt/MnO2 catalysts were pretreated with air at 373 K for 1 h. The typical composition of reactant gas was 20 ppm C2H4, 21 vol% O2, and balance N2. The hourly space velocity (GHSV) was 60000 mL g−1 h−1. The ethylene concentrations were analyzed by an on-line GC 2060 gas chromatograph equipped with Flame Ionization Detector (FID), and the ethylene removal efficiency was calculated by the following formula:| Removal efficiency = (Cinlet − Coutlet)/Cinlet × 100% | (1) |
3 Results and discussion
Fig. 1 shows the X-ray diffraction (XRD) patterns of the MnO2 samples obtained by the facile redox reaction approach between KMnO4 and MES at room temperature. All of the materials exhibit diffraction peaks at 12.5°, 25°, 36.5° and 65.5°, which are in good agreement with the (001), (002), (−111) and (−321) crystal planes of δ-MnO2 (JCPDS 80-1098). In the case of Pt/MnO2-48 h catalyst, the loading of Pt has not changed the crystal form of MnO2 and no reflections belonging to Pt can be observed, indicating that Pt NPs are small enough and finely dispersed on MnO2 matrix.27 Notably, the intensities of the (001) and (002) reflections of MnO2 remarkably decreases after Pt loading, suggesting that the Pt incorporation may have deteriorated the layered structure of δ-MnO2 or exfoliated δ-MnO2 into discrete nanosheets to a large extent.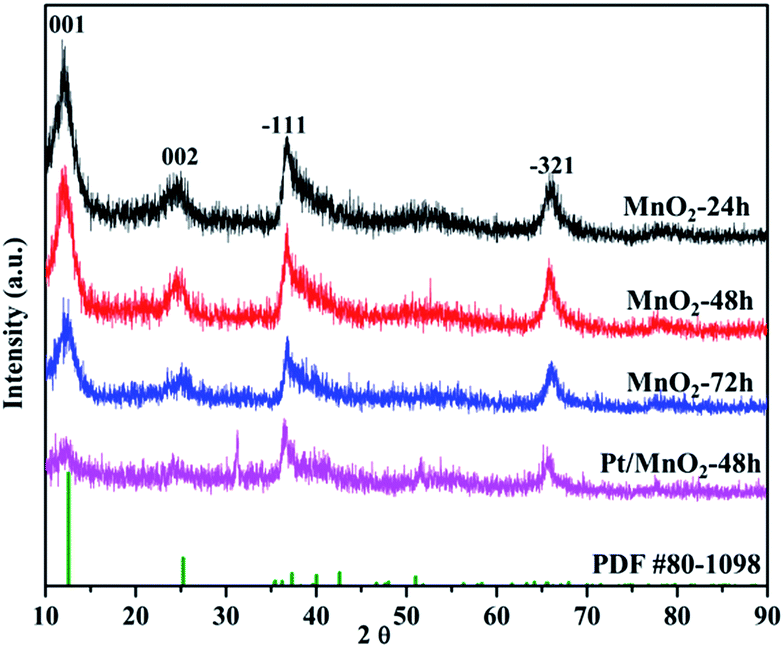 | ||
| Fig. 1 The XRD patterns of the as-synthesized MnO2 and Pt/MnO2-48 h. | ||
The morphologies of MnO2-48 h and Pt/MnO2-48 h were characterized by Field Emission Scanning Electron Microscopy (FESEM) and Transmission Electron Microscopy (TEM). As can be seen from Fig. 2A, the obtained MnO2 presents sheet-like morphology and all nanosheets are curled or crumpled due to surface tension, similar to most 2D nanosheets such as graphene. Fig. 2B shows that the Pt NPs are well decorated on the MnO2 sheets. The TEM, high resolution TEM (HRTEM), high angle annular dark-field (HAADF) TEM images and EDS spectra of the as-prepared MnO2-48 h and Pt/MnO2-48 h samples are given in Fig. 3 and 4. As can be seen in Fig. 3A, the obtained MnO2-48 h nanosheets are about 10 nm in thickness and up to hundreds of nanometers in their in-planar dimension, implying that MnO2 with 2D layered structure has been successfully prepared through the simple redox reaction between KMnO4 and MES without the complicated “up-bottom” process.9 Furthermore, the EDS spectroscopy (Fig. 3B) shows that the obtained MnO2 is composed of Mn, O and a small amount of K. Fig. 3C and D are the HRTEM images of MnO2-48 h, which clearly demonstrates a d-spacing of 0.7 nm, corresponding to the interlayer distance of (001) facet of 2D layered structure δ-MnO2. In addition, we can see that the as-synthesized MnO2 nanosheets are polycrystallized and the crystallite size is around several nanometers (Fig. 3D). Clearly, two sets of lattice fringes can be observed, that can be ascribed to (002) and (−111) planes with d-spacing of 0.35 and 0.24 nm, respectively. After the loading of 2 wt% Pt, the nanosheet morphology of MnO2 is retained, and the ∼2 nm Pt NPs are highly dispersed on the MnO2 nanosheets (Fig. 4A). EDS (Fig. 4B) spectrum of the selected area gives a Pt content of 1.99 wt%, which was further verified to be 1.90 wt% by inductively coupled plasma-optical emission spectroscopy (ICP-OES). As shown in the HRTEM image of Pt/MnO2-48 h (Fig. 4C), a lattice spacing of ca. 0.23 nm is corresponding to the (111) lattice plane of Pt. The HAADF TEM image clearly reveals the uniform dispersion of Pt NPs on the planar surface of MnO2 nanosheets.
 | ||
| Fig. 2 The FE-SEM images of (A) MnO2-48 h and (B) Pt/MnO2-48 h catalysts. | ||
 | ||
| Fig. 3 The TEM image (A), EDS spectra (B) and HRTEM images of MnO2-48 h. | ||
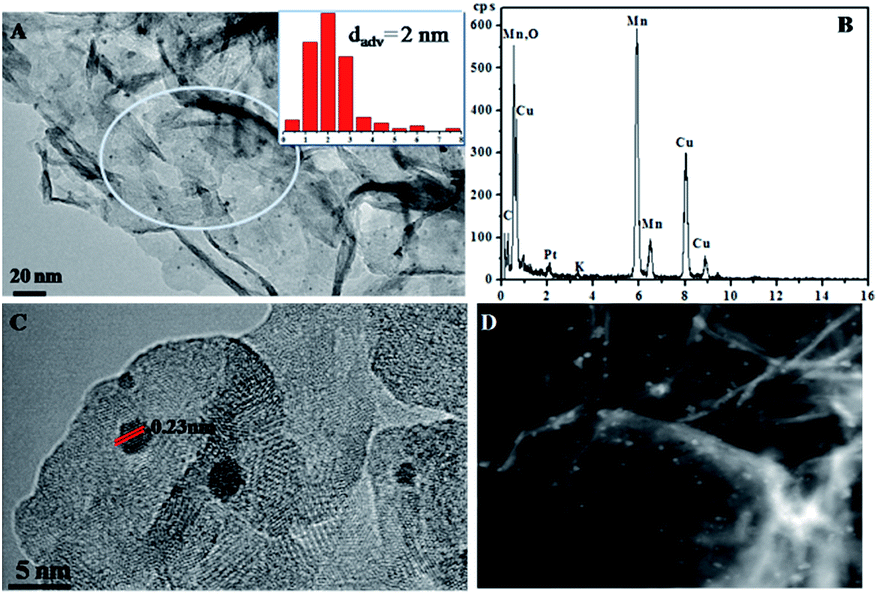 | ||
| Fig. 4 The TEM image (A) (insert: the size distribution of Pt), EDS spectra (B), HRTEM image (C) and HADDF TEM image (D) of Pt/MnO2-48 h. | ||
N2 adsorption–desorption isotherms were recorded (Fig. S1†) and the corresponding pore structure parameters are summarized in Table S1.† As shown in Fig. S1,† the specific surface area of MnO2-48 h is the highest among these three catalysts. The specific surface area of Pt/MnO2-48 h is lower than that of MnO2-48 h due to the loading of Pt NPs.
The surface chemical composition, chemical state and surface-defect of the as-synthesized MnO2 samples were analyzed by XPS. Fig. 5 shows the Mn 2p spectra and O 1s spectra of the as-synthesized MnO2 samples. As can be seen from Fig. 5A, there are two bands centred at 642.2 eV and 653.6 eV, corresponding to the binding energy of Mn 2p3/2 and Mn 2p1/2, respectively. The Mn 2p3/2 can be deconvoluted into Mn2+, Mn3+ and Mn4+ sub-bands at 641 eV, 642 eV and 644 eV,28 respectively. The proportions of Mn2+, Mn3+ and Mn4+ in the as-synthesized MnO2 samples are listed in Table 1. It is clear that the amount of Mn2+ in all the samples is very low. The percentages of Mn4+ are 31.6%, 40.4% and 30.6% in MnO2-24 h, MnO2-48 h and MnO2-72 h, respectively. The existence of Mn3+ will facilitate electron transfer between Mn3+/Mn4+ and Mn4+/Mn2+ pairs, which benefits the redox catalytic reaction.
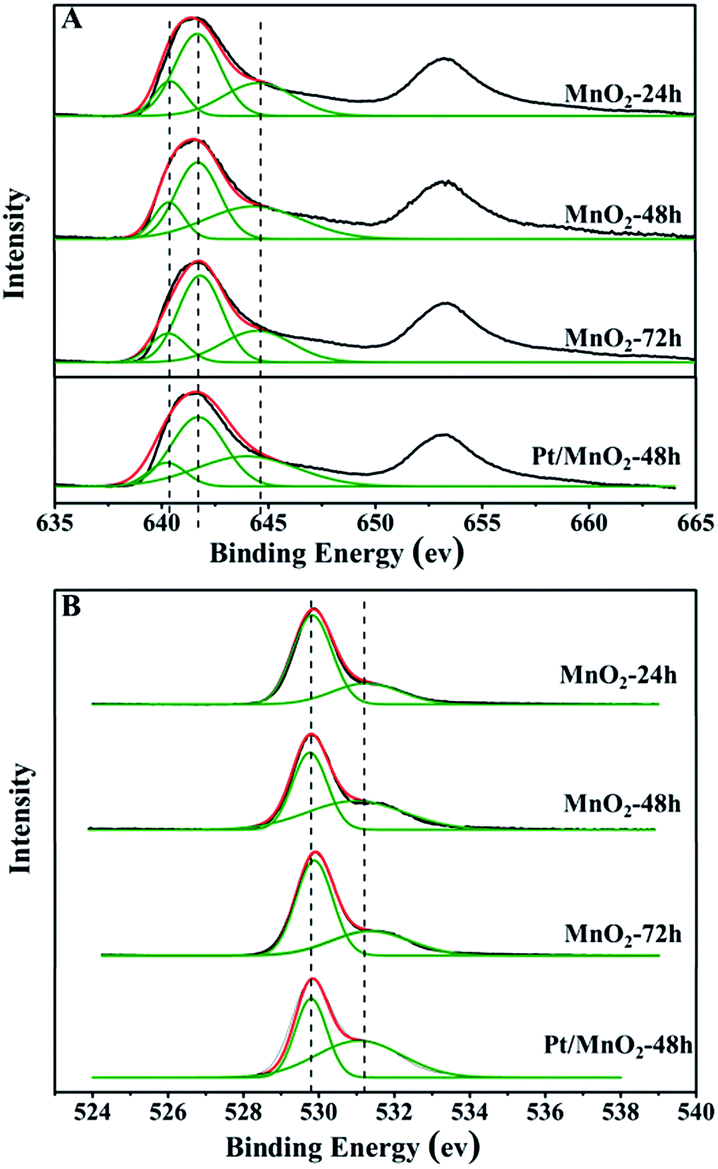 | ||
| Fig. 5 XPS spectra of Mn 2p (A) and O 1s (B) of the as-synthesized MnO2 and Pt/MnO2-48 h catalysts. | ||
| Sample | Mn2+ | Mn3+ | Mn4+ | Oads/Olat |
|---|---|---|---|---|
| MnO2-24 h | 15.7% | 52.6% | 31.6% | 0.33 |
| MnO2-48 h | 15.1% | 44.5% | 40.4% | 0.92 |
| MnO2-72 h | 15.1% | 54.3% | 30.6% | 0.53 |
| Pt/MnO2-48 h | 12.1% | 48.8% | 39.1% | 1.16 |
The O 1s spectra are shown in Fig. 5B, and all the profiles can be fitted to two sub-bands, indicating two different kinds of oxygen species in the catalysts. The one of lower binding energy corresponds to lattice oxygen (Olat), while the other at higher binding energy is for surface adsorbed oxygen (Oads).29 The surface Oads/Olat molar ratios are 0.33, 0.92 and 0.53 for MnO2-24 h, MnO2-48 h and MnO2-72 h, respectively, as listed in Table 1. Notably, the percentage of surface adsorbed oxygen increased after the loading of Pt, which may be attributed to the larger amount of surface adsorbed oxygen on the highly dispersed Pt species. It has been reported that surface active oxygen species play an important role in oxidation reactions.30 The adsorbed oxygen species result most likely from the presence of surface oxygen vacancies,31 which favours the catalytic oxidation reaction.
The Pt 4f spectrum of Pt/MnO2-48 h is shown in Fig. S2.† Two strong peaks represent the 4f7/2 and 4f5/2 electrons of Pt species, respectively.32 This further confirms the successful deposition of Pt on MnO2. The peak of 4f7/2 can be divided into two peaks located at 71 eV and 72 eV, which can be assigned to the 4f7/2 electrons of Pt0 and Pt2+, respectively. The existence of oxidized Pt (Pt2+/Pt0 ratio = 0.88) can be attributed to the formation of Pt–O and Pt–O–Mn bonds, corresponding to the increased amounts of Oads and Mn3+ species in Pt/MnO2-48 h after the Pt loading. This indicates the strong interaction between Pt and MnO2, featuring the electron transfer from Pt0 to adsorbed Oads and surface Olat in the MnO2 support. The same phenomenon has also been found in Pt/Fe3O4 system.33 This strong interaction between Pt and the MnO2 support can be named as strong metal–support interaction (SMSI), as reported in many literatures,34–36 and also recently summarized as a type of synergetic catalytic effects (type I of mutual activation of the two components).20
The reducibility of the synthesized manganese oxide catalysts, an important factor correlated with their redox activity, was investigated by H2 temperature-programmed reduction (H2-TPR). As shown in Fig. 6, there are two well-defined reduction peaks, corresponding to the stepwise reductions of MnO2 and Pt/MnO2-48 h in the temperature range of 300–500 °C. According to the previous reports, the first reduction peak centred at 350–400 °C is attributed to the reduction of MnO2 to Mn2O3, and the second one centred at 450–500 °C is assigned to the reduction of Mn2O3 to MnO.37,38 The complete reduction of MnO to metallic Mn did not happen in the reduction process of these samples. From the H2-TPR profiles of the synthesized MnO2, one can see that the reducibility of the MnO2 follows the order of MnO2-48 h ∼ MnO2-24 h > MnO2-72 h. Compared with the TPR behaviour of MnO2, the addition of Pt has dramatically altered the reduction feature of MnO2. The Pt/MnO2-48 h catalyst shows one intensive reduction peak at 137 °C. It has been reported that PtOx species can be reduced below 250 °C despite the stoichiometry of Pt4+/Pt2+ and the interaction between PtOx and support.39 Therefore, the peak at 137 °C could be attributed to the reduction of PtO to Pt. However, it should be noted that the H2 consumption (5284 μmol g−1) during the TPR of Pt/MnO2-48 h was much higher than that necessary for the reduction of PtO (<102 μmol g−1), indicating the low temperature reduction of a large part of MnO2 support due to the strong SMSI between Pt and MnO2. This is also in correspondence to the remarkably reduced band intensity (hydrogen consumption) of Pt/MnO2-48 h beyond 400 °C. This phenomenon is related to the activation of H2 on the initially reduced Pt and the spillover of the activated hydrogen to MnO2, which largely facilitates the reduction of MnO2.40,41
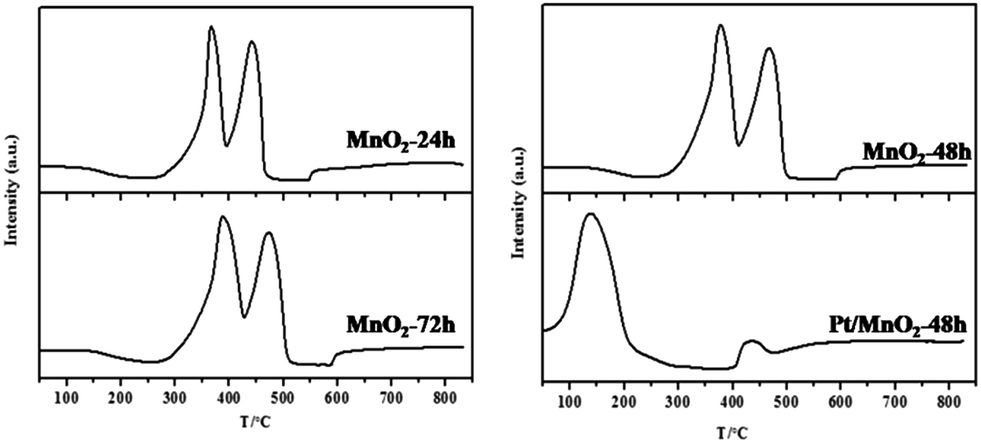 | ||
| Fig. 6 H2-TPR profiles of the as-synthesized MnO2 and Pt/MnO2-48 h. | ||
The O2 temperature programmed desorption (O2-TPD) tests were performed to further investigate the adsorbed oxygen species as well as their mobility in the synthesized MnO2 and Pt/MnO2-48 h (Fig. 7). Generally, the adsorbed oxygen will be desorbed according to the following sequence:42 O2(ad) → O2−(ad) →O−(ad) → O2−(lat). As can be seen from Fig. 7, almost all of the O2-TPD profiles of these samples show three peaks. The first peak at about 240 °C can be assigned to the desorption of chemically adsorbed O2−(ad) since the physically adsorbed O2(ad) is usually desorbed at around 50 °C.43 The second peak at about 360 °C is ascribed to desorption of chemically adsorbed O−(ad). The last desorption peak at about 600 °C can be assigned to lattice O2−(lat) desorption from MnO2 support. All MnO2 samples show the similar capacity in chemically adsorbing O2−(ad) and O2−(lat). Exceptionally, MnO2-72 h exhibits no desorption peak of O−(ad). Among the three MnO2 catalysts, the desorption temperature of the O2−(lat) in MnO2-72 h is a little higher than that of the other two MnO2 samples, indicating that the surface lattice oxygen is less mobile in MnO2-72 h. In the case of Pt/MnO2-48 h, however, the peak area between 100 °C to 400 °C is larger than that of MnO2-48 h, suggesting the enhanced chemical adsorption of O2 (O2−(ad), especially O−(ad)). Notably, Pt/MnO2-48 h adsorbs more oxygen atom O−(ad), suggesting abundant O−(ad) adsorbed on the catalyst. More importantly, the desorption temperature of lattice oxygen O2−(lat) in Pt/MnO2-48 h down-shifts substantially from 600 °C to 470 °C, indicating the dramatically enhanced mobility of lattice oxygen in Pt/MnO2-48 h. The much elevated chemical-adsorption of O2 as well as the excellent mobility of lattice oxygen in Pt/MnO2-48 h will greatly favour the enhancement on catalytic oxidation activity.
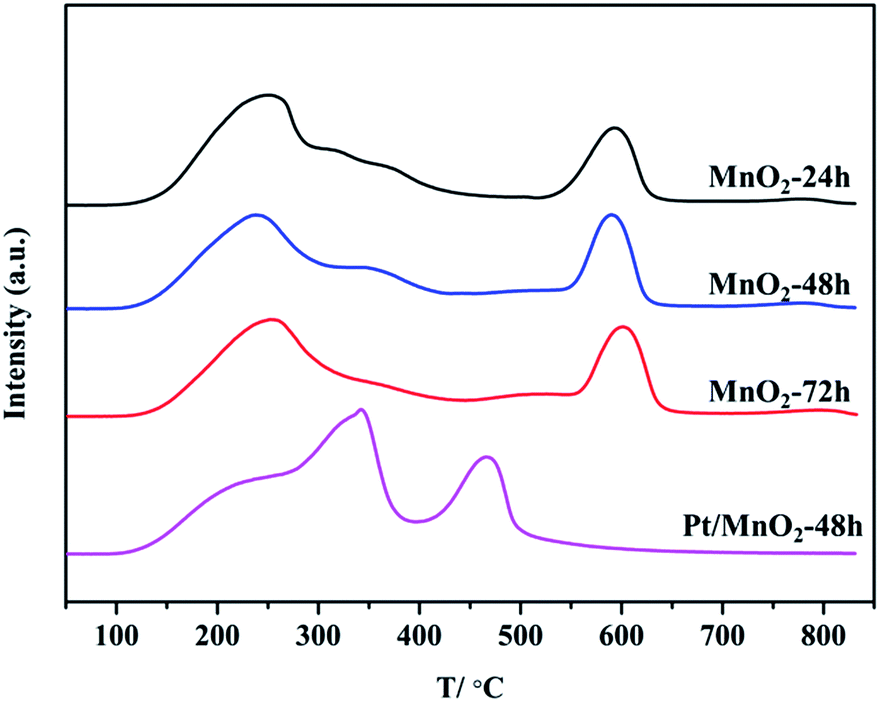 | ||
| Fig. 7 O2-TPD profiles of the as-synthesized MnO2 and Pt/MnO2-48 h. | ||
The oxidation of ethylene was used to evaluate the catalytic activity of the as-synthesized catalysts, and the results are shown in Fig. 8. The complete conversion temperatures of C2H4 are at 150 °C, 130 °C and 170 °C on MnO2-24 h, MnO2-48 h and MnO2-72 h, respectively. In particular, MnO2-48 h shows the best performance in removing 20 ppm ethylene without Pt-loading. After loading 2 wt% Pt on MnO2-48 h, the as-synthesized Pt/MnO2-48 h catalyst shows markedly enhanced catalytic activity in ethylene oxidation. The complete conversion temperature of C2H4 decreases from 130 °C to 50 °C. More importantly, the complete ethylene removal has been maintained for at least 12 h (Fig. 8B).
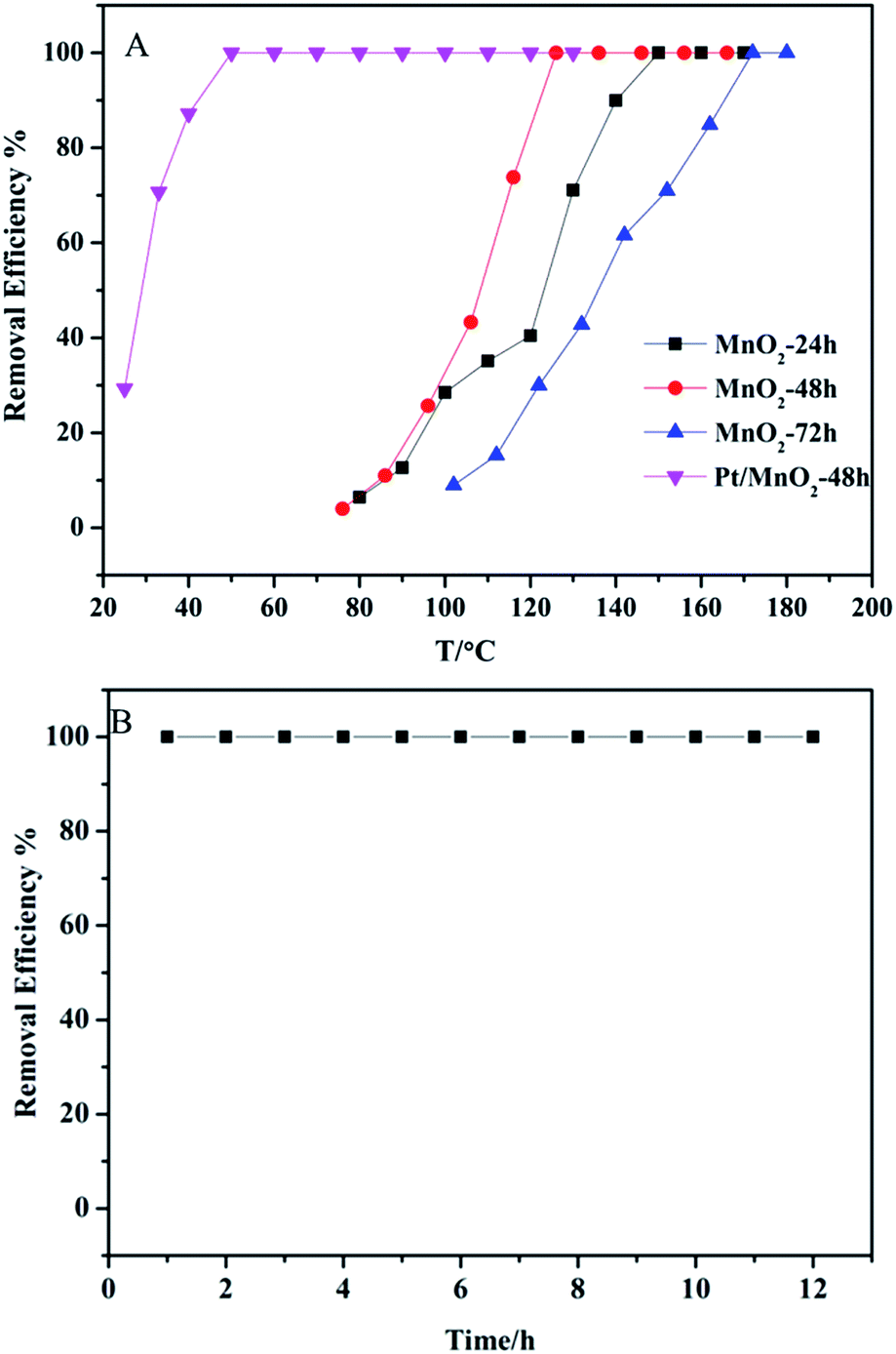 | ||
| Fig. 8 (A) C2H4 removal efficiency of by the as-synthesized MnO2 and Pt/MnO2-48 h as functions of temperature; (B) C2H4 removal efficiency by Pt/MnO2-48 h at 50 °C. Reaction condition: [C2H4] = 20 ppm, O2 = 20.0 vol%, N2 balance, GHSV = 60000 mL g−1 h−1. | ||
3.1 Discussion on the structure–activity relation
As demonstrated above, the as-prepared MnO2 materials are δ-MnO2 built up with edge-shared MnO6 octahedra with interlayer cations (K+) to balance the charge, and the sample MnO2-48 h features a 2D nanosheet morphology stacked with less than 5 layers of manganese oxide with a high surface area. Among all the MnO2 samples, MnO2-48 h processes the highest amount of surface adsorbed active oxygen species and the strongest reducibility. All these features make MnO2-48 h the most active catalyst for C2H4 oxidation removal in the present study. A small amount of Pt incorporated on MnO2-48 h remarkably enhanced the catalytic activity for ethylene oxidation. The high surface area of Pt/MnO2-48 h favours the adsorption of HCHO. Moreover, based on the XPS and O2-TPD results, the addition of Pt influences the valence distribution of Mn. The increased ratio of Mn3+/Mn4+ would favour the formation of oxygen vacancies, which greatly benefits the adsorption and dissociation of oxygen molecules. Therefore, Pt/MnO2-48 h had enhanced amount of surface adsorbed (activated) oxygen species. It is well-known that surface adsorbed oxygen species are highly active and therefore are responsible for the highly activity in catalytic oxidation of C2H4 at much lowered temperature.44 As a result, the complete conversion temperature of C2H4 on Pt/MnO2-48 h has reached down to 50 °C from 130 °C of MnO2-48 h. Another important issue which should be mentioned is that the SMSI effect, which not only changes the electron structure of Pt and MnO2 but also builds a fast channel for electron mobility and transfer between Pt and MnO2 during the catalytic reaction process, endowing the composite catalyst a significantly synergetic catalysis effect. H2-TPR results further proves the existence of the interaction between Pt and MnO2, which leads to a much strengthened reducibility of Pt/MnO2-48 h at a temperature down to 137 °C from ∼400 °C of MnO2-48 h, suggesting the surface mobility enhancement of lattice oxygen.In the process of C2H4 catalytic oxidation, as shown in Fig. 9, C2H4 molecules would be adsorbed on the active surface of Pt.45,46 The active oxygen species (O2−(ad), especially O−(ad)) adsorbed on both Pt NPs and the surface oxygen vacancies of MnO2 are primarily consumed in oxidizing C2H4 (Step I), producing HCHO according to the previous reports24 (Step II). The oxygen vacancies on Pt/MnO2 catalyst play a key role in the catalytic reaction, which enable the adsorption, dissociation, activation and migration of active oxygen species. The cycle of adsorption, activation and consumption of the active oxygen species on the oxygen vacancies of Pt/MnO2 is accompanied by the redox cycling of Mn4+/Mn3+ and Pt2+/Pt047 (Step II). The HCHO molecules will be further oxidized to HCOOH (Step III) and OHCOOH (Step IV) intermediates by the newly formed active oxygen species, and finally the OHCOOH completely decomposes into CO2 and H2O.
 | ||
| Fig. 9 The possible mechanism of C2H4 catalytic oxidation on Pt/MnO2. | ||
4 Conclusions
In summary, we have synthesized δ-MnO2 nanosheets by a simple, environment-friendly and cost-effective approach via redox precipitation reaction between potassium permanganate and 2-(N-morpholino)ethane sulfonic acid (MES) at room temperature. The obtained MnO2-48 h was able to catalyze the complete removal of low-concentration (20 ppm) ethylene at 130 °C. To further improve the catalytic ability of MnO2, Pt nanoparticles (∼2 nm in diameter) were highly dispersed on the MnO2-48 h by colloidal deposition. The obtained catalyst Pt/MnO2-48 h demonstrated markedly enhanced catalytic activity, which kept completely removing C2H4 (20 ppm) at 50 °C for at least 12 h. The high specific surface area and the enhanced amount of surface adsorbed oxygen species on Pt/MnO2-48 h contribute to its high catalytic activity in ethylene oxidation. Furthermore, the strong metal–support interaction between Pt and MnO2, is also proposed to play a role in elevating the redox catalytic activity of the Pt/MnO2 composite catalyst.Acknowledgements
The authors gratefully acknowledge financial support from National Key Basic Research Program of China (2013CB933200), National 863 plans project (2012AA062703), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Youth Innovation Promotion Association CAS (2012200).Notes and references
- Y. Chen, D. Ye, M. Y. Wu, H. R. Chen, L. X. Zhang, J. L. Shi and L. Wang, Adv. Mater., 2014, 26, 7019 CrossRef CAS PubMed.
- K. J. Koski and Y. Cui, ACS Nano, 2013, 7, 3739 CrossRef CAS PubMed.
- M. Osada and T. Sasaki, Adv. Mater., 2012, 24, 210 CrossRef CAS PubMed.
- E. Antolini, Appl. Catal., B, 2012, 123–124, 52 CrossRef CAS.
- S. H. Hur and J.-N. Park, Asia-Pac. J. Chem. Eng., 2013, 8, 218 CrossRef CAS.
- Q. Jia, X. Huang, G. Wang, J. Diao and P. Jiang, J. Phys. Chem. C, 2016, 120, 10206 CAS.
- X. Fan, L. Zhang, R. Cheng, M. Wang, M. Li, Y. Zhou and J. Shi, ACS Catal., 2015, 5, 5008 CrossRef CAS.
- Z.-A. Lan, G. Zhang and X. Wang, Appl. Catal., B, 2016, 192, 116 CrossRef CAS.
- Z. Liu, K. Ooi, H. Kanoh, W. Tang and T. Tomida, Langmuir, 2000, 16, 4154 CrossRef CAS.
- Z. Sun, T. Liao, Y. Dou, S. M. Hwang, M.-S. Park, L. Jiang, J. H. Kim and S. X. Dou, Nat. Commun., 2014, 5, 3813 CAS.
- Y. Du, Z. Hua, W. Huang, M. Wu, M. Wang, J. Wang, X. Cui, L. Zhang, H. Chen and J. Shi, Chem. Commun., 2015, 51, 5887 RSC.
- J. Wang, J. Zhu, X. Zhou, Y. Du, W. Huang, J. Liu, W. Zhang, J. Shi and H. Chen, J. Mater. Chem. A, 2015, 3, 7631 CAS.
- S. Liang, F. T. G. Bulgan, R. Zong and Y. Zhu, J. Phys. Chem. C, 2008, 112, 5307 CAS.
- P. W. Menezes, A. Indra, P. Littlewood, M. Schwarze, C. Goebel, R. Schomaecker and M. Driess, ChemSusChem, 2014, 7, 2202 CrossRef CAS PubMed.
- D. Shevchenko, M. F. Anderlund, S. Styring, H. Dau, I. Zaharieva and A. Thapper, Phys. Chem. Chem. Phys., 2014, 16, 11965 RSC.
- P. Liu, H. He, G. Wei, X. Liang, F. Qi, F. Tan, W. Tan, J. Zhu and R. Zhu, Appl. Catal., B, 2016, 182, 476 CrossRef CAS.
- J. Zhang, Y. Li, L. Wang, C. Zhang and H. He, Catal. Sci. Technol., 2015, 5, 2305 CAS.
- Y. Chen, J. He, H. Tian, D. Wang and Q. Yang, J. Colloid Interface Sci., 2014, 428, 1 CrossRef CAS PubMed.
- X. Yu, J. He, D. Wang, Y. Hu, H. Tian and Z. He, J. Phys. Chem. C, 2012, 116, 851 CAS.
- J. Shi, Chem. Rev., 2013, 113, 2139 CrossRef CAS PubMed.
- C. Y. Ma, Z. Mu, J. J. Li, Y. G. Jin, J. Cheng, G. Q. Lu, Z. P. Hao and S. Z. Qiao, J. Am. Chem. Soc., 2010, 132, 2608 CrossRef CAS PubMed.
- S. Chavadej, K. Saktrakool, P. Rangsunvigit, L. L. Lobban and T. Sreethawong, Chem. Eng. J., 2007, 132, 345 CrossRef CAS.
- U. Ackelid, L. R. Wallenberg and L. G. Petersson, Catal. Lett., 1996, 39, 129 CrossRef CAS.
- C. Jiang, K. Hara and A. Fukuoka, Angew. Chem., Int. Ed., 2013, 52, 6265 CrossRef CAS PubMed.
- W. Fan, W. Bu, B. Shen, Q. He, Z. Cui, Y. Liu, X. Zheng, K. Zhao and J. Shi, Adv. Mater., 2015, 27, 4155 CrossRef CAS PubMed.
- X. Yu, J. He, D. Wang, Y. Hu, H. Tian and Z. He, J. Phys. Chem. C, 2012, 116, 851 CAS.
- L. Nie, J. Yu, X. Li, B. Cheng, G. Liu and M. Jaroniec, Environ. Sci. Technol., 2013, 47, 2777 CrossRef CAS PubMed.
- Z. Shu, Y. Chen, W. Huang, X. Cui, L. Zhang, H. Chen, G. Zhang, X. Fan, Y. Wang, G. Tao, D. He and J. Shi, Appl. Catal., B, 2013, 140–141, 42 CrossRef CAS.
- V. P. Santos, M. F. R. Pereira, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal., B, 2010, 99, 353 CrossRef CAS.
- H. Over and A. P. Seitsonen, Science, 2002, 297, 2003 CrossRef CAS PubMed.
- B. Bai, J. Li and J. Hao, Appl. Catal., B, 2015, 164, 241 CrossRef CAS.
- Y. Nagai, H. Shinjoh and K. Yokota, Appl. Catal., B, 2002, 39, 149 CrossRef CAS.
- Z. Yan, Z. Xu, J. Yu and M. Jaroniec, Environ. Sci. Technol., 2015, 49, 6637 CrossRef CAS PubMed.
- H. Zhu, J. Catal., 2004, 225, 267 CrossRef CAS.
- M. F. Luo, Z. Y. Hou, X. X. Yuan and X. M. Zheng, Catal. Lett., 1998, 50, 205 CrossRef CAS.
- H. W. Jen, G. W. Graham, W. Chun, R. W. McCabe, J. P. Cuif, S. E. Deutsch and O. Touret, Catal. Today, 1999, 50, 309 CrossRef CAS.
- D. Delimaris and T. Ioannides, Appl. Catal., B, 2008, 84, 303 CrossRef CAS.
- X. Tang, J. Chen, X. Huang, Y. Xu and W. Shen, Appl. Catal., B, 2008, 81, 115 CrossRef CAS.
- I. D. González, R. M. Navarro, W. Wen, N. Marinkovic, J. A. Rodriguéz, F. Rosa and J. L. G. Fierro, Catal. Today, 2010, 149, 372 CrossRef.
- X. Tang, J. Chen, X. Huang, Y. Xu and W. Shen, Appl. Catal., B, 2008, 81, 115 CrossRef CAS.
- S. Hamoudi, A. Sayari, K. Belkacemi, L. Bonneviot and F. Larachi, Catal. Today, 2000, 62, 379 CrossRef CAS.
- P. Li, C. He, J. Cheng, C. Y. Ma, B. J. Dou and Z. P. Hao, Appl. Catal., B, 2011, 101, 570 CrossRef CAS.
- L. F. Liotta, M. Ousmane, G. Di Carlo, G. Pantaleo, G. Deganello, G. Marcì, L. Retailleau and A. Giroir-Fendler, Appl. Catal., A, 2008, 347, 81 CrossRef CAS.
- Y. Liu, H. Dai, J. Deng, S. Xie, H. Yang, W. Tan, W. Han, Y. Jiang and G. Guo, J. Catal., 2014, 309, 408 CrossRef CAS.
- A. F. Carlsson and R. J. Madix, J. Chem. Phys., 2001, 115, 8074 CrossRef CAS.
- R. J. Isaifan, S. Ntais and E. A. Baranova, Appl. Catal., A, 2013, 464–465, 87 CrossRef CAS.
- B. Bai, Q. Qiao, H. Arandiyan, J. Li and J. Hao, Environ. Sci. Technol., 2016, 50, 2635 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The N2 adsorption–desorption isotherms of the as-synthesized MnO2 samples and Pt/MnO2-48 h; XPS Pt 4f spectrum of Pt/MnO2-48 h; specific surface areas of the as-prepared MnO2 samples and Pt/MnO2-48 h. See DOI: 10.1039/c6ra26529d |
| This journal is © The Royal Society of Chemistry 2017 |