
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCryo scanning probe nanotomography study of the structure of alginate microcarriers†
Anton E. Efimov *ab,
Olga I. Agapovaa,
Liubov A. Safonovaac,
Maria M. Bobrovaac,
Alexey D. Volkovd,
Laura Khamkhashd and
Igor I. Agapova
*ab,
Olga I. Agapovaa,
Liubov A. Safonovaac,
Maria M. Bobrovaac,
Alexey D. Volkovd,
Laura Khamkhashd and
Igor I. Agapova
aLaboratory of Bionanotechnology, V.I.Shumakov Federal Research Center of Transplantology and Artificial Organs, Shchukinskaya str., 1, Moscow, 123182 Russia. E-mail: antefimov@gmail.com; igor_agapov@mail.ru; Tel: +7 4991906619
bSNOTRA LLC., Shcherbakovskaya str., 3, Moscow, 105318 Russia. E-mail: antefimov@gmail.com; Tel: +7 9161642475
cBioengineering Department, Biological Faculty, Lomonosov Moscow State University, GSP-1, Leninskie Gory, Moscow, 119991, Russia. E-mail: mariabobrova.msu@gmail.com; saf.lyubov.msu@gmail.com; Tel: +7 4959391000
dNational Laboratory Astana, Nazarbayev University, 53 Kabanbay Batyr Avenue, 010000 Astana, Kazakhstan. E-mail: alvolk@mail.ru; laura.khamkhash@nu.edu.kz; Tel: +7 7771809533
First published on 27th January 2017
Abstract
Tissue-specific alginate microspheres surface modified with microparticles of decellularized liver tissue were prepared. The nanostructures of these microspheres were analysed by cryo scanning probe nanotomography. Modification of the surfaces of the alginate microspheres with microparticles of decellularized liver tissue increased surface roughness, the specific effective area, and the average autocorrelation length. Up to seven times more human hepatocellular carcinoma (HepG2) cells were adhered to the modified alginate microspheres than to the unmodified microspheres. The developed alginate microspheres can be used in regenerative medicine and tissue engineering as a biodegradable cell microcarriers.
1. Introduction
Development of injectable cell carriers for non-invasive therapy is important for regeneration of damaged organs and tissues in the treatment of numerous severe diseases and injuries.1 Biodegradable carriers—polymeric microspheres, microcapsules, and microparticles—can be used as containers for drug delivery in the body, and simultaneously promote adhesion, proliferation, and differentiation of cells.2 However, the biological and mechanical properties of a microcarrier largely depend on its three-dimensional (3D) structure at the micro- and nanolevel. Therefore, appropriate methods for nanostructural investigations are required to improve the efficacy of the final products.To effectively study microcarrier structure, a method that can accurately analyse the surface nanostructure is required to evaluate the adhesion properties of the product. Preservation of the native structure of the sample during analysis should be prioritized in method selection.
The micro- and nanostructures of samples can be studied by many different methods, including scanning electron microscopy (SEM),3 and transmission electron microscopy.4 Combination of SEM and etching of the surface with a focused ion beam is a promising method. In this method, reconstruction of the 3D structure is carried out by integrating successive SEM images of the surfaces, which are obtained after etching of material layers by ion beams. This method is effective for a wide range of samples,5,6 but the impact of electrons and vacuum conditions can alter the structures of biological samples, which is problematic.7 X-ray tomography is widely used for studying polymers, cell carriers, and biomaterials on the micro level,8 but is not suitable for nanoscale investigations. In addition, to obtain the maximum resolution with this method, the sample size should not exceed 1 μm.9 These restrictions are very inconvenient for the analysis of biological and polymeric materials of light chemical elements.
Scanning probe microscopy (SPM) is a technique that can be used for direct surface characterization on the nanoscale. SPM imaging is based on physical interaction of an ultrasharp probe tip with the sample surface, and may yield results that are fundamentally different from those of electron microscopy or X-ray tomography. In addition, SPM allows analysis of several aspects of the surface, including phase imaging, the distribution of electrical conductivity,10,11 measurement of force curves, magnetic12 or electrostatic force microscopy, and chemical force microscopy.13,14
Nanoscale surface characterisations of microcarriers, including alginate microspheres, using SPM demonstrated the efficiency of this method for assessment of the surface parameters of unmodified microspheres with diameters of 850 and 425 μm and with surface roughness between 0.9 and 14.4 nm.15,16 However, direct application of SPM to microspheres with smaller diameters, much greater surface roughness, and surface modifications may be difficult because of the curvature and relief of the surface, and errors introduced into the resulting image by the SPM probe because of convolution effects.17
Cryo scanning probe nanotomography (cryoSPNT) can be used to overcome the issues discussed above. This method combines SPM and ultramicrotome at low temperatures in a single measurement system with the SPM apparatus installed inside the cryochamber of the ultramicrotome. This allows analysis of sample surfaces by SPM immediately after sectioning without the need for transfer of the sample, which means that analysis of the next ultrathin section can proceed routinely. Data obtained from successive SPM measurements of sample block surfaces obtained after ultrathin sections are removed by cryo-ultramicrotomy can be integrated for nanoscale tomographic studies of the 3D morphological and structural properties of the samples.18–20
Analysis of sample nanostructure in the volume of a sample allows studies to be conducted with nanoscale resolution,21,22 without destruction of the sample by chemical fixation or by an electron beam, and also avoids issues with weak electron microscopy contrast.23,24 However, this method is relatively new, and its applications to different kinds of bionanomaterials are still being developed. Analysis of cross-sectional images of a microcarrier and microparticles on the surface, and profiles of its borders, in successive SPM images enables reconstruction of a 3D image of the surface of a microcarrier with minimal morphology artefacts introduced by the SPM probe.
Microcarriers can be fabricated from a number of different biological (e.g. collagen,25 alginate,26 and silk fibroin27,28) and synthetic (e.g. silicone,29 and polylactide-co-glycolide30) polymers. For regenerative medicine applications, it is important to find ways to increase the tissue specificity of a microcarrier because this should improve biocompatibility and regenerative properties. Coating of a microcarrier's surface with bioactive molecules or microparticles, such as fragments of decellularized tissues, may greatly improve adhesion of cells on its surface and increase proliferation. This modification of surface structures by biomimicry may be highly effective because the fragments of the native tissue-specific extracellular matrix can greatly enhance adhesion of cells of the same tissue on the carrier's surface.31
In this work, cryoSPNT was applied for the first time to a study of microcarrier structure. The main aims of this work were to apply cryoSPNT to hydrogel microcarriers and to conduct a comparative cryoSPNT study of the nanostructures of model multiscale microcarrier systems of alginate with and without surface modification with decellularized rat liver microparticles. We also studied the effect of surface modification on the adhesion of human hepatocellular carcinoma (HepG2) cells to the microcarrier surface. HepG2 cells were chosen because of their ability to reproduce many features of hepatocytes.32 Moreover, HepG2 cells cultured in decellularized liver matrix can be used as an in vitro 3D model for pharmacological studies.33
2. Materials and methods
2.1 Preparation of alginate spherical microcarriers
A 16.5 mg mL−1 sodium alginate solution was prepared by dissolving 3.3 g of sodium alginate in 200 mL of double-distilled water in a 400 mL beaker with stirring for 2 h at room temperature. The solution was then allowed to stand at room temperature to release air bubbles. Next, the solution was diluted four times to a concentration of 4.125 mg mL−1 by adding double-distilled water with vigorous stirring. The resulting solution was degassed in a desiccator for 3 h to release all the air bubbles. Alginate microparticles were prepared from this solution using an encapsulator (B-390, BÜCHI Labortechnik AG, Flawil, Switzerland). The feed rate of the solution from the syringe was 5 mL min−1. A 100 mM calcium chloride solution was used as the receiving solution. The resulting microcarriers had diameters between 200 and 300 μm. The microcarriers were stored in a solution containing 30% ethanol (v/v) and 100 mM calcium chloride. The microcarrier cytotoxicity was determined by the MTT assay.342.2 Preparation of microparticles of decellularized rat liver
Wistar rats (200–250 g) were used to obtain liver for decellularization. The animals were maintained in conventional housing on a standard diet before sampling. All studies conformed to the Guide for the Care and Use of Laboratory Animals, published by the United States National Institutes of Health (Publication no. 85-23, revised 1996), and were approved by the Local Ethical Committee of V.I.Shumakov Federal Research Center of Transplantology and Artificial Organs (Moscow, Russia). Heparin (250 μL) was injected intraperitoneally to Wistar rats 20 min before liver sampling to reduce thrombogenesis. After 30 min, 200 μL of pentoxifylline was injected as a vasoprotective drug. After another 30 min, 0.20 mL of Zoletil was injected for parenteral general anaesthesia. Next, a ventral midline incision was made with scissors. A longitudinal section along the midline was performed to open the abdominal cavity, and the muscle flaps were turned out. The portal vein was cannulated and the blood was washed out with 200 mL of phosphate buffered saline (PBS, pH 7.4) at 150 mL h−1. The PBS contained sodium chloride 137 mM, potassium chloride 2.7 mM, sodium hydrogen phosphate 10 mM, and potassium dihydrogen phosphate 1.76 mM. The liver was removed and decellularized at a speed of 150 mL h−1 in the following solutions sequentially: (1) 500 mL of PBS containing 0.1% sodium dodecyl sulfate and 1% Triton X-100, (2) 500 mL of PBS containing 0.1% sodium dodecyl sulfate and 2% Triton X-100, and (3) 500 mL of PBS containing 0.1% SDS and 3% Triton X-100. The rat liver was cut into pieces (average size 1–3 mm) with scissors. The pieces of decellularized liver were transferred to a test tube, and the volume was made up to 15 mL with a solution of 15% glycerol in PBS, followed by incubation at room temperature for 20 min, and centrifugation for 10 min at 8500 × g. The pellet was retained, frozen in liquid nitrogen, and ground for 5 min in a mortar and pestle that had been pre-cooled in liquid nitrogen. The resulting powder was transferred to a clean pre-cooled tube, and 70% ethanol was added to reach a total volume of 25 mL. The resulting suspension was centrifuged three times for 10 min each time at 8500 × g. Seventy percent of the supernatant volume was removed to a separate tube after each centrifugation step. The resulting microparticle suspension was transferred to a 1.5 mL Eppendorf tube (Eppendorf, Hamburg, Germany). The tubes were sealed and stored at 4 °C in 70% (v/v) ethanol. The protein concentration in the suspension of liver microparticles was assessed for 100 μL of the suspension of decellularized liver microparticles, which was placed into a Petri dish and dried for 3 days at room temperature in the air. The protein concentration was calculated using the equation| Cm = (M2 − M1)/V, |
The sizes of microparticles in the decellularized extracellular matrix were investigated using an inverted light microscope (Axiovert 405 M, Carl Zeiss, Oberkochen, Germany) and a hemocytometer (Heinz Herenz, Hamburg, Germany). Ten microlitres of a dilute suspension of the microparticles was applied to the hemocytometer. The microparticle size (1–5 μm) was estimated visually using the size mesh of the hemocytometer chamber. The MTT assay was performed for cytotoxicity analysis.34
2.3 Estimation of cell adhesion of HepG2 cells on the surfaces of the microcarriers
All operations were carried out in a sterile laminar flow hood. Aliquots (300 μL) of the suspension of alginate microcarriers were added to the wells of sterile 48-well plates. Ethanol was removed from the microcarriers by washing with PBS (pH 7.4) before cell culturing. The microcarriers were left for sedimentation for 10 min, and then incubated with PBS (pH 7.4) for 30 min. These steps were repeated twice. A HepG2 culture cell suspension with a concentration of 4.2 × 106 cells per mL in incubation medium (DMEM high glucose and Ham's F12 nutrient mixture in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, 10% fetal bovine serum, and 100 μg mL−1 gentamicin) was then added to the wells at 200 μL per well. Next, the plates were incubated at 37 °C under 5% CO2 for 24 h.
:1 ratio, 10% fetal bovine serum, and 100 μg mL−1 gentamicin) was then added to the wells at 200 μL per well. Next, the plates were incubated at 37 °C under 5% CO2 for 24 h.
The MTT assay was used to estimate the number of adhered cells.34 For the assay, the microcarriers were transferred into wells filled with PBS (pH 7.4). The plates were incubated on a thermo-shaker (Eppendorf Research) for 5 min at 180 rpm. Then, the microcarriers were left for sedimentation for 10 min, and then supernatant was removed. The wells containing microcarriers were filled with PBS (pH 7.4) again. These steps were repeated three times. The dense suspension of microcarriers was then transferred to the wells of a 96-well plate at 100 μL per well, and then 50 μL of 2.5 μg mL−1 solution of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, which is a substrate of mitochondrial dehydrogenase, was added to each well. The plate was incubated at 37 °C under 5% CO2 for 4 h to allow for precipitation of dark blue formazan crystals. Then, 100 μL of dimethylsulfoxide (DMSO) was added to each well, and the plates were incubated at room temperature until the formazan crystals completely dissolved. The optical density of the solution was measured at 540 nm using a photometer (AIFR-01 Uniplan, Pikon, Moscow, Russia).
2.4 Modification of the surfaces of the alginate microcarriers using the decellularized rat liver microparticles
Surface modification of the alginate microcarriers was carried out by covalent linking with the decellularized rat liver microparticles. The alginate microcarriers were added to PBS (50 mM sodium dihydrogen phosphate, 100 mM sodium chloride, pH 6.15), followed by centrifugation for 5 min at 900 × g. N-Hydroxysuccinimide (NHS) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) were used as modifying agents because they allow covalent bond formation between carboxyl groups, of which there are many in the alginate structure, and amino groups in the components of the extracellular matrix. First, EDC was added to the suspension of alginate microcarriers, followed by incubation at room temperature with stirring for 1 h. Then, NHS was added, and the solution was incubated for another hour under the same conditions. The final concentrations of EDC and NHS in the alginate microcarrier suspension at were 3 mM and 10 mM, respectively.The modifying agents were removed by centrifugation for 5 min at 900 × g. The alginate microcarriers in the pellet were resuspended in 700 μL of PBS (pH 6.15). Decellularized rat liver microparticles were added to PBS (50 mM sodium dihydrogen phosphate, 100 mM sodium chloride, pH 6.15), followed by centrifugation for 5 min at 900 × g. The suspension of decellularized rat liver matrix microparticles was added to the suspension of alginate microcarriers to reach final concentrations of decellularized rat liver matrix microparticles of 1 mg mL−1, 3 mg mL−1, 6 mg mL−1, 9 mg mL−1, 12 mg mL−1, and 20 mg mL−1. The mixture was incubated at room temperature with stirring for 12 h. Further aliquots of the microcarrier suspension were incubated in a 100 mM glycine aqueous solution at room temperature for 1 h, followed by centrifugation for 5 min at 900 × g. The modified alginate microcarriers in the pellet were resuspended in PBS (pH 7.4) containing 100 mM calcium chloride and 30% ethanol (v/v). A microscope (Carl Zeiss Axiovert 25, Carl Zeiss, Oberkochen, Germany) with 10× objective and 10× ocular lenses was used to investigated modification of the microcarriers.
To confirm modification occurred, the alginate microcarriers were incubated at room temperature with Coomassie Brilliant Blue R-250 solution for 1 h. After centrifugation for 5 min at 900 × g, the microcarriers in the pellet were resuspended in 1 mL of double-distilled water and left for 30 min. This procedure was repeated twice more. The MTT assay was performed for cytotoxicity analysis.34
2.5 Preparation of samples of alginate microcarriers for cryoSPNT
Samples were fixed in a 2.5% glutaraldehyde solution in PBS (pH 7.4), and incubated for 2 h in the dark at 4 °C. Then, the microcarriers in the pellet were filled with PBS (pH 7.4) by added 1 mL of PBS and incubating for 15 min at room temperature. This procedure was repeated. Then, the pellet was resuspended in either a 2.3 M sucrose solution, or DMSO (5%, 10% and 20% solution), or glycerol (5% and 10% solution). An aliquot (50 μL) of the microcarriers suspension was then transferred to a microtube and incubated at 4 °C for 12 h.2.6 CryoSPNT
The 3D structure of the microcarriers was investigated using cryoSPNT with the scanning probe microscope integrated in the cryochamber of a cryo-ultramicrotome (Leica EM UC6 and FC6, Leica Microsystems GmbH, Austria). Fig. 1 shows a photograph of the SPM module installed in the cryochamber. Details of the integrated cryoSPNT system are published elsewhere.20,35 This setup allows for successive SPM of the surfaces of frozen samples immediately after cryo-ultramicrotome sectioning in the cryochamber. The maximum scanning area at room temperature was 50.0 × 50.0 μm. Successive sections (thickness 80 nm) of the sample were made using a diamond knife (35 CryoAFM, Diatome AG, Switzerland) with a cutting edge width of 2.0 mm. The SPM measurements were made in semi-contact mode at a scan rate of 0.75 Hz using cantilevers with quartz resonators (A-probe, Nanosensors, Neuchatel, Switzerland) with a resonant frequency between 44 and 48 kHz and a tip curvature radius of <15 nm. | ||
| Fig. 1 CryoAFM module installed into the ultramicrotome cryochamber of the cryoSPNT system. (1) Sample, (2) AFM probe. | ||
2.7 Analysis of the liver matrix microparticles structure by SEM
Decellularized liver samples for SEM were fixed in 2.5% glutaraldehyde in PBS (pH 7.4) for 2 h in the dark at room temperature. Then, the fixed samples were washed five times for 5 min each time in PBS. The microcarriers in the pellet were filled with PBS (pH 7.4) by adding 1 mL of PBS and incubating for 5 min at room temperature. This procedure was repeated five times, followed by dehydration with increasing concentrations of ethanol (10%, 20%, 30%, 50%, 70%, 96% v/v). After that the samples were transferred into acetone. The samples were dried in a critical point dryer (HCP-2, Hitachi Ltd., Japan). An IonCoaterIB-3 (Eiko Engineering, Japan) was used to coat the dried samples with a 20 nm layer of gold under an argon atmosphere with an ion current of 6 mA. The samples were then analysed by SEM (Camscan S2, Cambridge Instruments, Great Britain) with a resolution of 10 nm and operating voltage of 20 kV. MicroCapture software (SMA, Moscow, Russia) was used for image acquisition.3. Results
3.1 Fabrication and assessment of the microcarriers
The alginate microcarriers were spherical or close to spherical with an approximate diameter of 200 μm (Fig. 2a). This size is suitable for using the microcarriers in injectable applications.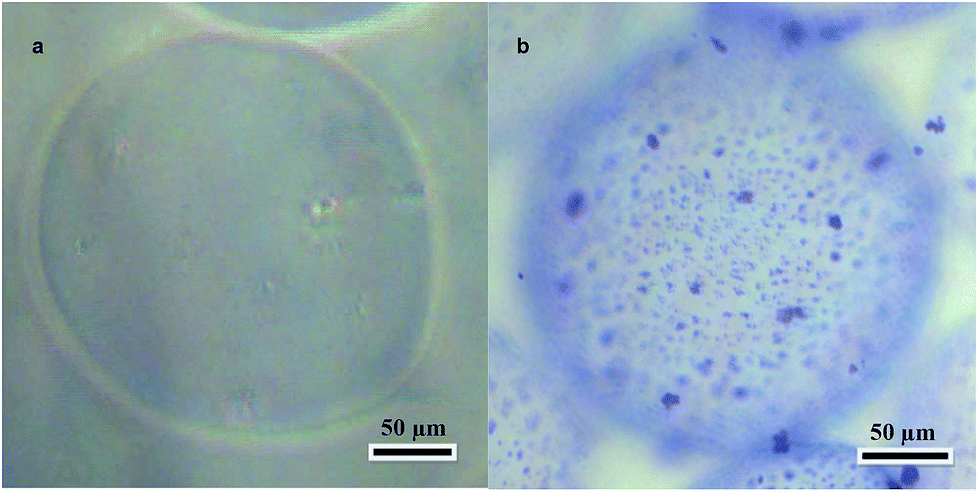 | ||
| Fig. 2 (a) Unmodified alginate microcarrier. Light microscopy, magnification 100×. (b) Alginate microcarrier modified with liver microparticles. Light microscopy, staining Coomassie Brilliant Blue R-250, magnification 100×. | ||
Extracellular matrix microparticles were prepared by mechanical grinding of decellularized rat liver frozen in liquid nitrogen. The size and structure of the decellularized rat liver microparticles were observed by optical and SEM. The microparticle size range was 1–5 μm, and the particles had a porous structure (Fig. S1, ESI†).
The alginate microcarriers were modified by covalent linking with the decellularized rat liver matrix microparticles. The maximum surface density of microparticles was achieved with a 12 mg mL−1 decellularized rat liver microparticle suspension. A decellularized rat liver microparticle suspension concentration of less than 12 mg mL−1 was effective for modification but the microparticle density was too low. A 20 mg mL−1 microparticle suspension resulted in microparticle aggregation. An image of a modified microcarrier is shown in Fig. 2b. Coomassie Brilliant Blue R-250 staining was used to visualize the microparticles because it binds to proteins in the extracellular matrix and not the alginate. Optically dense granules with sizes in range of 1–5 μm were detected on the surfaces of the modified alginate microcarriers. They were stained dark blue and corresponded to microparticles of the extracellular matrix of the liver. Unmodified samples did not show any staining. None of the samples showed cytotoxicity in the MTT assay (data not shown).
3.2 CryoSPNT studies of the microcarriers
CryoSPM studies were performed using cryoSPNT.20,35 Sectioning of samples with the ultramicrotome and SPM are possible only with frozen samples because the alginate microcarriers are hydrogels when in suspension. One of the most important aspects of the development of a sample preparation method and preliminary characterization of alginate microcarriers is the choice of cryoprotection technique. Conventional freezing, for example, immersion of samples of alginate microcarriers in liquid nitrogen, causes crystallization of water contained in the hydrogel and alginate in suspension. Formation of ice crystals leads to irreversible destruction of the structure of the microcarriers. Accordingly, to perform structural studies of hydrogel microcarriers, a cryopreparation method that does not result in ice crystal formation is required. There are various methods that can be used, including cryoprotectants and methods for freezing biological samples at high pressure.36,37To date, there have been no reports of vitrification of hydrogel microparticles with diameters between 200 and 300 μm.
Freezing at high pressure is only effective for samples up to 200 μm thick, and also requires expensive equipment.38
We investigated freezing of the suspension of alginate microcarriers with the addition of different concentrations of three commonly used cryoprotectants (sucrose, dimethylsulfoxide (DMSO), and glycerol). The samples were then sectioned with the cryo-ultramicrotome. The results showed that DMSO (5%, 10%, and 20% solutions) and glycerol (5% and 10% solutions) were not effective because the sample was still viscous at low temperatures (−80 to −140 °C) and could not be used to obtain high-quality ultrathin sections with the ultramicrotome. The results obtained with different sucrose concentrations indicated that the optimal concentration of sucrose in the suspension was 2.3 M. For this concentration, observing of the block-face surfaces of the ultrathin cryosectioned samples with optical microscopy showed that the surface quality was optimum for low temperature SPM nanostructure studies. No surface irregularities were detected over an area of 0.8 to 1.2 mm, and individual microcarriers could be identified on the section surface.
SPM measurements of the surfaces of frozen microcarriers after ultrathin sectioning were performed in semicontact mode with a raster scanning rate of 0.75 Hz.
Fig. 3 shows a cryoSPM image of the cryosectioned surface of the core of the alginate microcarrier. The results showed that the alginate microcarriers were homogeneous inside, and no cavities or other large irregularities were detected.
 | ||
| Fig. 3 CryoSPM image of the surface of the core region of ultramicrotome sections of the alginate microcarriers. Scan size 10.0 × 10.0 μm, height variation 35 nm. | ||
Fig. 4 shows a cryoSPM image of a section of decellularized rat liver microparticles on the surface of the alginate microcarrier. This image shows that the liver microparticles, which were about 5 μm wide and 1 μm thick, were distributed over the microcarrier surface. The liver microparticles had a granular microstructure with 20–50 nm nanogranules, whereas the nanostructure of the alginate microcarriers was more homogeneous. It is likely that what we observed was the distribution of proteins from the extracellular matrix inside the liver microparticles.
 | ||
| Fig. 4 CryoSPM image of ultramicrotome section of decellularized liver microparticle from Wistar rats on the surface of alginate microcarrier. Scan size 5.0 × 4.0 μm, height variation 35 nm. Inset scan size 1.25 × 1.0 μm, height variation 20 nm. (1) Microparticle of extracellular matrix, (2) alginate microcarrier. | ||
Analysis of obtained cryoSPM data allowed estimation of the surface density of tissue microparticles on the surface of microcarriers at a value of 15 ± 3%.
Successive cryoSPM measurements after ultrathin sectioning enabled us to reconstruct the 3D structures of the alginate microcarriers, both with and without modification with microparticles of decellularized rat liver. Each 3D reconstruction used 13 SPM images of the microcarrier surface topography that were obtained in series after successive cryo-ultramicrotome sectioning (thickness 80 nm) at −120 °C. The size of each image (scan) was 5.0 × 5.0 μm with a resolution of 400 × 400 pixels.
Fig. 5 shows 3D reconstructions of the surfaces of the alginate microcarriers (a) and microparticles (b) in a suspension volume of 5.0 × 5.0 × 1.1 mm. Visualization of the 3D reconstructions was performed using 3D Constructor Image Pro AMS 6.0 software (MediaCybernetics, Inc., Rockville, MD, USA) by layer-by-layer integration of all obtained SPM images in series. Physical dimensions of voxels were defined by thicknesses of sections and pixel resolutions of the initial SPM images. This method enabled reconstruction of the surfaces of the microcarriers and microparticles by combining sections of their borders in each successive SPM image. Morphological parameters of the microcarrier surfaces can greatly affect cell adhesion to microcarriers and their biological properties because cells interact primarily with the surfaces of the microcarriers.
 | ||
| Fig. 5 CryoSPNT 3D reconstructions of (a) the surface of unmodified alginate microcarrier, and (b) decellularized liver microparticle from Wistar rat liver on the alginate microcarrier. Reconstructed volume 5.0 × 5.0 × 1.1 μm. | ||
It should be noted that an adequate evaluation of surface 3D micro- and nanomorphology of microcarriers and microparticles is very difficult with other microscopic methods, such as conventional SPM, because the microparticles are adhered to the spherical surface of the hydrogel microcarrier.
Statistical analysis of the reconstructed surfaces allowed determination and analysis of the 3D nanoscale parameters of these surfaces, such as average roughness (Ra), effective surface area (σ), and the length of autocorrelation (L1). Details for these parameters and the methods used to calculate them are given in the ESI.† These surface parameters can have a large impact on the adhesion of cells to microcarriers.
The 3D reconstruction and mathematical analysis showed large differences in surface roughness between the alginate microcarriers and microparticles. The calculated average roughness of the microcarrier surface was 82.3 ± 7.4 nm, whereas that of the microparticle surface was 212.4 ± 28.2 nm.
In addition to roughness, the effective surface area is also important parameter in determining the biological properties. This is calculated as the ratio of surface area to the area of its two-dimensional projection on the plane. This parameter determines the degree of surface development. The calculated effective areas for reconstructed 3D surfaces of the alginate microcarriers and microparticles were 1.103 ± 0.01 and 1.205 ± 0.025, respectively.
Another parameter used for characterization of nanoscale 3D surfaces is the autocorrelation length, which is the distance over which the 3D relief is formed.21 This parameter can be used as an estimate of the lateral measurements of characteristic relief features. In this case, the calculated autocorrelation lengths were 115 ± 22 nm and 246 ± 38 nm for the alginate microcarriers and microparticles, respectively.
These results showed there were significant differences (p < 0.05) between the surface parameters of the microcarriers and the microparticles bound to microcarriers.
3.3 Estimation of HepG2 cell adhesion on the surfaces of the microcarriers
Estimation of the cell adhesion properties of the microcarriers was performed using HepG2 cells. Quantitative estimation was carried out with the MTT assay (Fig. 6). | ||
| Fig. 6 Comparative estimation of the adhesion of human hepatocellular carcinoma (HepG2) cells on alginate microcarriers with or without modification with decellularized liver microparticles. MTT assay data. Standard deviation values are for five independent measurements. | ||
The number of cells adhered to the modified alginate microcarrier surface was about seven times that on the unmodified microcarrier surface. This indicates that modification with microparticles greatly improves the adhesion.
4. Discussion
Surface medication by chemical or physical methods can be used to solve the problem of low cell adhesion on the surfaces of alginate microcarriers.39 One such modification uses decellularized liver matrix. Liver decellularization can be performed by cryogrinding and does not require collection of dissolved components of the extracellular matrix. The composition of the undissolved extracellular liver matrix after decellularization by a similar method was investigated in an earlier study, and the following major components were detected: structural proteins (e.g. collagen and elastin), adhesion proteins (e.g. fibronectin and laminin), glycosaminoglycans, proteoglycans, and growth factors.40,41 The biological activity of the extracellular matrix of decellularized liver has been demonstrated in many studies,42,43 and the results are promising. In agreement with previous studies, our results indicated that microspheres produced of natural materials could be used as biodegradable and biocompatible cell carriers for tissue regeneration, and injected into the body for effective delivery of cells within affected areas.25,44–48The influence of macro- and microstructure on mechanical properties and biocompatibility of 3D structures for tissue engineering and regenerative medicine has been confirmed in numerous publications,4,19,49–51 but the influence of nanostructure on these properties is not as clear. For example, in one earlier study,47 hollow nanofiber polylactide microspheres were used as carriers of injected chondrocytes to regenerate cartilaginous tissue. Compared with the degree of chondrocytes adhesion on microspheres, the character of the surface substantially affected cell adhesion.48 It should be noted that we were able to explore the nanostructure of the surfaces of modified alginate microcarriers and the microparticles on their surface in a state close to native by cryofixation using cryoSPNT.
In this study, cryoSPNT allowed reconstruction of 3D profiles of surfaces of the alginate microspheres and decellularized liver microparticles. These profiles were used to obtain surface roughness, effective surface area, which determines the degree of surface development, and autocorrelation length.
The increase in cell adhesion on the surfaces of the modified alginate microcarriers compared with the unmodified microcarriers can be explained both by an increase in surface roughness of the microcarriers after modification, and by specific features of the decellularized liver microparticles. Previously hidden receptor binding sites, such as RGD-sequence on fibronectin and collagen, may be exposed on the liver microparticles and could promote adhesion of cells.52 It is also possible that with our manipulations the soluble components of the extracellular matrix, such as growth factors, could be extracted from the decellularized liver microparticles. The cryoSPNT method allows investigation of hydrogels with maximal preservation of their native structure. CryoSPNT can be used to study the quantitative parameters of the 3D micro- and nanomorphology of microscopic objects with complex shapes made of soft and hydrated biomaterials.
5. Conclusions
Cell macro- and microcarriers, including carriers for stem cells, are widely used in regenerative medicine and tissue engineering. We prepared biodegradable alginate spherical microcarriers modified with microparticles of decellularized liver tissue on the surface. Tissue-specific microcarriers can be obtained by surface modification of alginate microparticles with any micromilled decellularized tissue. Surface modification increases cell adhesion to the microcarrier by up to seven times. CryoSPNT enables control of surface modification, 3D structure, and nanostructural parameters of microparticles and their interface with the microcarrier.Acknowledgements
This work was supported by the Science Committee of the Ministry of Education and Science of the Republic of Kazakhstan (grant number 4360/GF4).Notes and references
- M. M. Moisenovich, O. Pustovalova and J. Shackelford, et al., Biomaterials, 2012, 33(15), 3887 CrossRef CAS PubMed.
- Q. Dang, C. Liu, Y. Wang, J. Yan, H. Wan and B. Fan, Carbohydr. Polym., 2016, 136, 516 CrossRef CAS PubMed.
- J. Huang, J. Xiong and J. Liu, et al., Bio-Med. Mater. Eng., 2015, 26(suppl. 1), S197 CrossRef PubMed.
- S. Wang, T. J. Kowal, M. K. Marei, M. M. Falk and H. Jain, Tissue Eng., Part A, 2013, 19(13–14), 1632 CrossRef CAS PubMed.
- L. Holzer, F. Indutnyi, P. H. Gasser, B. Munch and M. Wegmann, J. Microsc., 2004, 216, 84 CrossRef CAS PubMed.
- A. Zankel, J. Wagner and P. Poelt, Micron, 2014, 62, 66 CrossRef CAS PubMed.
- A. Al-Abboodi, J. Fu, P. M. Doran and P. P. Chan, Biotechnol. Bioeng., 2013, 110(1), 318 CrossRef CAS PubMed.
- S. T. Ho and D. W. Hutmacher, Biomaterials, 2006, 27(8), 1362 CrossRef CAS PubMed.
- M. J. Moore, E. Jabbari and E. L. Ritman, et al., J. Biomed. Mater. Res., Part A, 2004, 71(2), 258 CrossRef PubMed.
- A. Alekseev, A. Efimov, K. Lu and J. Loos, Adv. Mater., 2009, 21(48), 4915 CrossRef CAS PubMed.
- A. Alekseev, D. Chen and E. E. Tkalya, et al., Adv. Funct. Mater., 2012, 22(6), 1311 CrossRef CAS.
- U. Hartmann, J. Appl. Phys., 1988, 64(3), 1561 CrossRef.
- A. Noy, D. V. Vezenov and C. M. Lieber, Annu. Rev. Mater. Sci., 1997, 27, 381 CrossRef CAS.
- J. Zlatanova, S. M. Lindsay and S. H. Leuba, Prog. Biophys. Mol. Biol., 2000, 74(1–2), 37 CrossRef CAS PubMed.
- M. Lekka, D. Sainz-Serp, A. J. Kulik and C. Wandrey, Langmuir, 2004, 20(23), 9968 CrossRef CAS PubMed.
- A. M. Rokstad, I. Lacik, P. de Vos and B. L. Strand, Adv. Drug Delivery Rev., 2014, 67–68, 111 CrossRef CAS PubMed.
- A. E. Efimov, A. G. Tonevitsky, M. Dittrich and N. B. Matsko, J. Microsc., 2007, 226, 207 CrossRef CAS PubMed.
- K. E. Mochalov, A. E. Efimov and A. Bobrovsky, et al., ACS Nano, 2013, 7(10), 8953 CrossRef CAS PubMed.
- A. E. Efimov, M. M. Moisenovich, V. G. Bogush and I. I. Agapov, RSC Adv., 2014, 4(105), 60943 RSC.
- A. E. Efimov, H. Gnaegi, R. Schaller, W. Grogger, F. Hofer and N. B. Matsko, Soft Matter, 2012, 8(38), 9756 RSC.
- G. Binnig, C. F. Quate and C. Gerber, Phys. Rev. Lett., 1986, 56(9), 930 CrossRef PubMed.
- S. N. Magonov and D. H. Reneker, Annu. Rev. Mater. Sci., 1997, 27(1), 175 CrossRef CAS.
- N. B. Matsko, Ultramicroscopy, 2007, 107(2–3), 95 CrossRef CAS PubMed.
- M. O. Gallyamov, Macromol. Rapid Commun., 2011, 32(16), 1210 CrossRef CAS PubMed.
- L. Yao, F. Phan and Y. Li, Stem Cell Res. Ther., 2013, 4(5), 109 CrossRef PubMed.
- Y. Peng, H. Y. Sun, Z. C. Wang, X. D. Xu, J. C. Song and Z. J. Gong, Chemotherapy, 2015, 61(1), 32 CrossRef CAS PubMed.
- N. Bhardwaj, R. Rajkhowa, X. Wang and D. Devi, Int. J. Biol. Macromol., 2015, 81, 31 CrossRef CAS PubMed.
- T. K. Mwangi, R. D. Bowles, D. M. Tainter, R. D. Bell, D. L. Kaplan and L. A. Setton, Int. J. Pharm., 2015, 485(1–2), 7 CrossRef CAS PubMed.
- E. L. Pastor, E. Reguera-Nunez, E. Matveeva and M. Garcia-Fuentes, PeerJ, 2015, 3, e1277 Search PubMed.
- S. Pascual-Gil, T. Simon-Yarza, E. Garbayo, F. Prosper and M. J. Blanco-Prieto, J. Controlled Release, 2015, 220, 388 CrossRef CAS PubMed.
- J. E. Arenas-Herrera, I. K. Ko, A. Atala and J. J. Yoo, Biomed. Mater., 2013, 8(1), 014106 CrossRef CAS PubMed.
- G. Mazza, K. Rombouts and L. Urbani, et al., Sci. Rep., 2015, 5, 13079 CrossRef CAS PubMed.
- K. H. Hussein, K. M. Park and P. K. Teotia, et al., Transplant. Proc., 2013, 45(8), 3092 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65(1–2), 55 CrossRef CAS PubMed.
- A. E. Efimov, O. I. Agapova and I. I. Agapov, Biomed. Eng., 2015, 49(3), 132 CrossRef.
- R. I. Koning and A. J. Koster, Ann Anat, 2009, 191(5), 427 CrossRef PubMed.
- K. T. Tokuyasu, J. Cell Biol., 1973, 57(2), 551 CrossRef CAS PubMed.
- D. Studer, B. M. Humbel and M. Chiquet, Histochem. Cell Biol., 2008, 130, 877 CrossRef CAS PubMed.
- B. Balakrishnan, M. Mohanty, P. R. Umashankar and A. Jayakrishnan, Biomaterials, 2005, 26(32), 6335 CrossRef CAS PubMed.
- H. Ren, X. Shi and L. Tao, et al., Liver Int., 2013, 33(3), 448 CrossRef CAS PubMed.
- P. M. Baptista, M. M. Siddiqui and G. Lozier, et al., Hepatology, 2011, 53(2), 604 CrossRef CAS PubMed.
- B. E. Uygun, A. Soto-Gutierrez and H. Yagi, et al., Nat. Med., 2010, 16(7), 814 CrossRef CAS PubMed.
- A. Soto-Gutierrez, L. Zhang and C. Medberry, Tissue Eng., 2011, 17, 677 CrossRef CAS PubMed.
- O. Khanna, J. C. Larson, M. L. Moya, E. C. Opara and E. M. Brey, J. Visualized Exp., 2012, 66, e3388 Search PubMed.
- J. S. Rasey, M. M. Cornwell and B. J. Maurer, et al., Br. J. Cancer, Suppl., 1996, 27, S78 CAS.
- A. Tonevitsky, I. Agapov, O. Chelnokova, M. Moisenovich and U. Marx, Arzneimittelforschung, 2002, 52(6), 500 CAS.
- K. Hayashi and Y. Tabata, Acta Biomater., 2011, 7(7), 2797 CrossRef CAS PubMed.
- X. Liu, X. Jin and P. X. Ma, Nat. Mater., 2011, 10(5), 398 CrossRef CAS PubMed.
- J. Gao, P. M. Crapo and Y. Wang, Tissue Eng., 2006, 12(4), 917 CrossRef CAS PubMed.
- Y. J. Lee, A. Elosegui-Artola, K. H. T. Le and G.-M. Kim, Cell. Mol. Bioeng., 2013, 6(4), 482 CrossRef CAS.
- A. Ketabchi, K. Komm, M. Miles-Rossouw, D. A. Cassani and F. Variola, PLoS One, 2014, 9(3), e92080 Search PubMed.
- P. Canning, L. Tan and K. Chu, et al., J. Mol. Biol., 2014, 426(13), 2457 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1, microparticles of the extracellular matrix of decellularized Wistar rat liver (optical microscopy image). Appendix: description of the calculation methods for 3D surface parameters. See DOI: 10.1039/c6ra26516b |
| This journal is © The Royal Society of Chemistry 2017 |