
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis, and antiproliferative activities of stapled melittin peptides†
Ye Wu‡
a,
Meng-fei Han‡ b,
Chao Liuc,
Tai-yu Liuc,
Yu-fei Fengc,
Yan Zouc,
Bai Li*b and
Hong-li Liao*a
b,
Chao Liuc,
Tai-yu Liuc,
Yu-fei Fengc,
Yan Zouc,
Bai Li*b and
Hong-li Liao*a
aSchool of Pharmacy, Chengdu Medical College, Chengdu 610083, China. E-mail: liaohl213@126.com
bDepartment of Traditional Chinese Medicine, Changhai Hospital, Second Military Medical University, Shanghai 200433, China. E-mail: libai9@126.com
cDepartment of Organic Chemistry, School of Pharmacy, Second Military Medical University, Shanghai 200433, China
First published on 20th March 2017
Abstract
Melittin is a 26-residue, amphipathic, cell-penetrating, α-helical anti-hepatoma peptide isolated from bee venom. However, the application of melittin as a drug is limited owing to its original conformational flexibility and low stability. In this study, we designed, synthesized, and tested a series of hydrocarbon-stapled analogs of melittin, of which, some analogs showed remarkable enhancement not only in anti-hepatoma activity, but also in α-helicity and protease resistance when compared to the parent melittin. These results disclosed the important impact of all-hydrocarbon crosslinking on the biological activity, stability, and hemolytic activity of melittin.
Hepatocellular carcinoma (HCC) is a highly malignant disease worldwide and the third and seventh most common cause of cancer deaths in men and women, respectively.1 At present, the most effective treatment of HCC is hepatic resection, liver transplantation, and traditional chemotherapy, but the overall 5 year survival rate of HCC patients still remains low (33%).2–4 Therefore, it is urgent to discover more effective agents that either inhibit the growth or induce the apoptosis of HCC cells.
As an amphipathic, cell-penetrating, α-helical 26-residue peptide, melittin is a predominant pharmacological active component of bee venom,5 which exhibits a wide range of biological activities including anti-inflammation, anti-bacterial, anti-platelet-aggregation, and anti-tumor activities.6–10 Among these, the anti-tumor effect of melittin has recently attracted significant attention.11–18 Melittin has been proven to show not only cytolytic toxicities on many types of tumor cells through direct interaction with cell transduction mediators and influencing cellular signaling pathways in vitro, but also direct inhibition of tumor growth, metastasis, invasion, and proliferation in vivo.19
Previous studies demonstrated that melittin shows a strong inhibitory effect on HCC cells. The intracellular targets of melittin for HCC cells have been reported by several groups: Rac1,20 Ca2+/calmodulin-dependent protein kinase (CaMKII)-transforming growth factor-β-activated kinase 1 (TAK1)-JNK/p38,21 histone deacetylase 2 (HDAC2),22 methyl-CpG-binding protein 2 (MeCP2),23 and cathepsin S (Cat S).24 Despite its promising potential as a new anti-HCC therapeutic,20,25–28 the application of melittin as an α-helical L peptide has been severely limited owing to the inherent drawbacks of normal peptides. For example, its original conformational flexibility could allow it to interact with various molecules besides the intended target, leading to a relatively low selectivity of binding with HCC cells.29 Their short in vivo half-life and bioavailability is also one of the major drawbacks of peptides due to their high proteolytic susceptibility. In addition, as melittin induces cell lysis by forming pores in the lipid bilayer of the cell membrane and destroys the cell membrane in a non-specific manner, it cannot be directly injected.30
In recent years, Verdine et al. developed a type of all-hydrocarbon crosslinking strategy that constrained peptides in the α-helical conformation and successfully applied it in many active L peptides.31 This method has been proven to efficiently enhance protease resistance and cell permeability.32,33 In light of this, we hypothesized that using this peptide-stapled system, the corresponding melittin analogs might increase cell permeability, alleviate its aggregation on cell membranes and reduce the hemolytic activity of melittin, achieving a reasonable level of protease stability and intracellular target engagement for anti-HCC activity.
To investigate the influence of all-hydrocarbon crosslinking on the biological activity, stability, and hemolytic activity of melittin, we planned to acquire the hydrocarbon-stapled analogs of melittin. Helical wheel projections indicated the amphipathic helical structure of melittin (Fig. 1A), of which, Val8, Thr11, Pro14, and Leu16 are the key residues for the biological activities of melittin (Fig. 1B).34 Therefore, our first criterion was to avoid the modification of these positions. Again, as cellular uptake is highly associated with hydrophobicity, staple crosslink placement at the amphipathic boundary reflects a critical step in designing a cell-penetrant stapled peptide.35 In the end, because melittin is composed of more than 25 amino acid residues, it represents a relatively high manufacture cost; thus, effort should be made to reduce the size of melittin and maintain its anti-HCC activity.
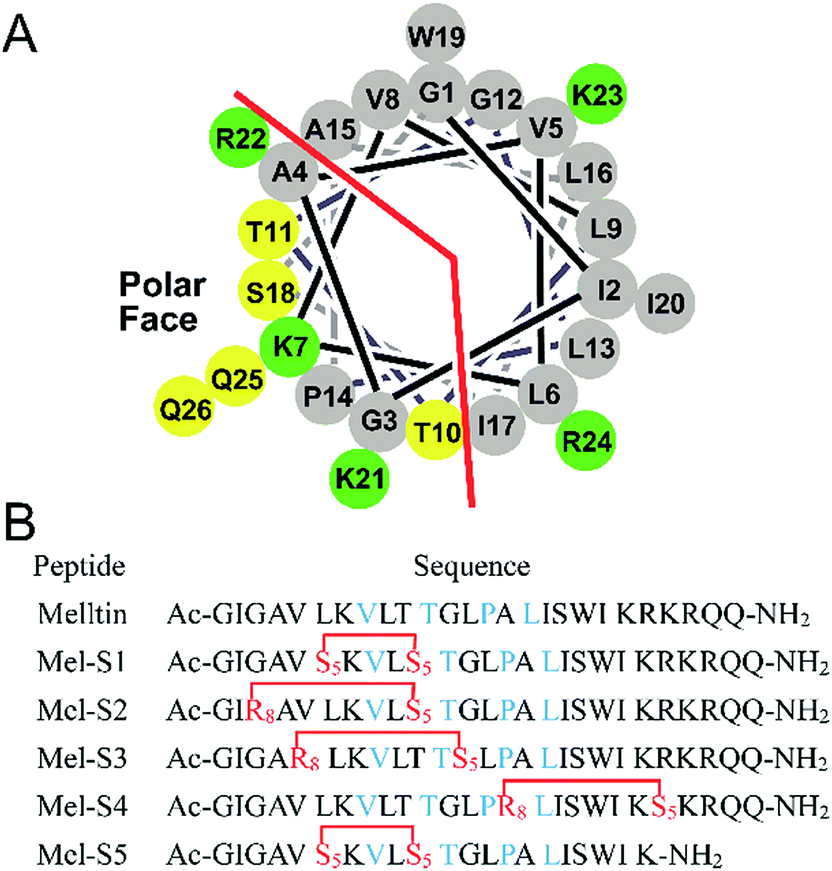 | ||
| Fig. 1 (A) Helical wheel diagram of melittin showing the polar and nonpolar faces of the helix. All residues are drawn in the helical configuration. Gray symbols represent hydrophobic residues. Yellow symbols represent polar, uncharged residues. Green symbols represent basic residues. (B) Sequences of stapled melittin peptide analogs. The color code is blue for key residues, and red for changed residues cross-linked by ring-closing metathesis (RCM). S5 = Fmoc-S5-OH; R8 = Fmoc-R8-OH; and Ac and NH2 represent N-terminal acetylation and a C-terminal primary amide, respectively. | ||
Based on the abovementioned rules, we designed a series of stapled peptide-based analogs of melittin (Fig. 1B) and incorporated two olefin-carrying amino acids (Fmoc-S5-OH) at the i and i + 4 positions (Leu6/Thr10, termed as Mel-S1 and the truncated analog, termed as Mel-S5). Additionally, we incorporated the olefin-carrying amino acids Fmoc-R8-OH and Fmoc-S5-OH at the i and i + 7 positions (Gly3/Thr10, Val5/Gly12 and Ala15/Arg22) to obtain peptides Mel-S2, Mel-S3, and Mel-S4.
For the synthesis of the stapled peptides Mel-S1 to Mel-S5 and melittin, we used the standard Fmoc solid-phase peptide synthesis (SPPS) procedures with Rink Amide MBHA resin as the solid support to assemble the linear sequence first (Fig. 2).36 After the peptide assembly was completed, the olefin-containing peptides were stapled using Grubbs' first-generation catalyst. The peptides were cleaved from the resin and globally deprotected with reagent K (80% TFA, 5% H2O, 2.5% EDT, 5% thioanisole, and 7.5% phenol). Cold ether precipitation provided crude peptides, which were purified by semi-preparative RP-HPLC.
 | ||
| Fig. 2 Synthetic route for the preparation of the stapled peptide Mel-S2. | ||
To probe structural changes induced by the conformation constraint, we determined the percent helicity of all the stapled peptides via circular dichroism (CD) measurement. The CD analysis of the peptides (Fig. 3, Table 1) indicates that the helicity of melittin is only 12%, whereas the helicity for Mel-S1 to Mel-S5 ranges from 19% to 47%, corresponding to a 1.6 to 4-fold increase. These results demonstrate that our stapled peptide strategy can improve the helicity compared to that using parent melittin. Mel-S5, the truncated analog of Mel-S1, displayed the highest degree of helicity (47%) in the aqueous solution and acquired a 1.7-fold increase compared to Mel-S1. The significant difference in their helical contents implies that the all-hydrocarbon stapling is a promising strategy for the α-helical structure stabilization of relatively short sequences. The observation that Mel-S4 shows the least level of α-helicity verifies that melittin is mostly α-helical in structure by N-terminal 20 residues.37
 | ||
| Fig. 3 CD spectra of melittin and stapled peptides. The peptides were dissolved in PBS buffer at the final concentration of 50 μM. The percent helicity was calculated based on the [θ]222 value. | ||
| Peptide | Helicity (%) | IC50 (μM) | |
|---|---|---|---|
| SMMC-7721 | HepG2 | ||
| Melittin | 12 | 3.2 | 6.1 |
| Mel-S1 | 27 | 2.5 | 1.7 |
| Mel-S2 | 29 | 2.5 | 2.7 |
| Mel-S3 | 22 | 4.0 | 4.9 |
| Mel-S4 | 19 | 1.5 | 2.0 |
| Mel-S5 | 47 | 3.8 | 4.2 |
Next, the growth inhibition of HCC cells in the presence of individual peptides was evaluated in SMMC-7721 cells and HepG2 cells using the MTT assay. Under the assay condition used in this study, all the stapled peptides enhanced or maintained anti-HCC activity compared to melittin (Table 1). Among them, the anti-HCC activity of Mel-S5 showed less promotion although its α-helicity is the highest. This observation may suggest that the highly polar, cationic C-terminal tail is indispensable for the activity of melittin. Mel-S3 also did not work very well. A reasonable explanation is that the helix-interrupting Pro14 (along with Gly12) in melittin allows for the formation of a kinked structure, creating a toroidal pore through the membrane,38 and the huge stapled group does not contribute to forming this pore. In other words, Gly12 in the sequence of melittin is critical for the interaction; therefore, it cannot be used as a site for stapling.
Unlike Mel-S3, Mel-S1 and Mel-S2 exhibit considerably enhanced activities for the growth inhibition of HCC cells. These results indicate that a higher helicity is more beneficial for the activity. However, Mel-S4 exhibits the best anti-HCC property compared to the parent peptide on SMMC-7721 cells and HepG2 cells although its helicity is only 19%. This is probably because the hydrophobic stapled group in the hydrophilic sequence contributes to improving the amphipathic property, which would effectively interact with HCC cells.39
To further research the hemolytic activity (HC50) of the stapled peptides, Mel-S4, the optimal peptide for anti-HCC activity, was evaluated by determining hemoglobin release from erythrocyte suspensions of fresh rabbit blood. As shown in Fig. 4, Mel-S4 displays a highly enhanced HC50 compared to the unmodified melittin (0.7 μM versus 2.6 μM). The cytotoxicity of compounds correlates with the capacity for cellular uptake, target binding affinity, and membrane lytic properties,35 implying that the anti-HCC activity of the stapled peptides could be attributed to both intracellular target engagement and plasma-membrane lysis.
 | ||
| Fig. 4 Dose-dependent hemolytic activity of melittin and Mel-S4. | ||
To test the protease stability of the stapled peptides, we measured their susceptibility towards α-chymotrypsin-mediated degradation at room temperature in pH 7.4 PBS buffer containing 2 mM of CaCl2, as monitored by HPLC. α-Chymotrypsin is a protease that predominantly cleaves at the carboxyl side of positively charged amino acids such as Trp and Leu. Under these conditions, the half-life (t1/2) of melittin is 4 min (Fig. 5). In contrast, more than 35% of Mel-S4 (t1/2 = 47 min) remained uncleaved even after 1 h of protease exposure. These results unambiguously demonstrate the inherent superiority of the stapled peptides over line-peptides with respect to proteolytic stability.
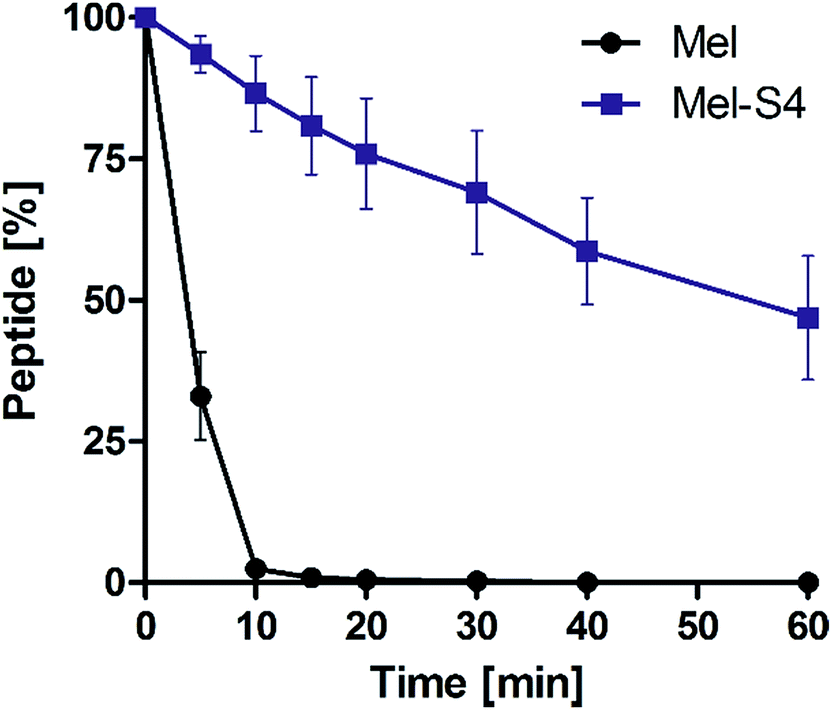 | ||
| Fig. 5 Proteolytic stability of the melittin vs. Mel-S4 in α-chymotrypsin solution (5 ng μL−1 in 50 mM PBS buffer, pH = 7.4) at the final concentration of 0.1 mM. Data points are displayed as the mean value SEM of duplicate independent experiments. The percent of residual peptide was monitored by analytic HPLC. | ||
In summary, herein, we examined the inhibitory effects of stapled peptide-based analogs of melittin on SMMC-7721 cells and HepG2 cells. We demonstrated that the stapled peptides are exceptionally stable against HCC cells and proteolytic degradation and their α-helicity can be improved via conformation constraint. Further investigation on the possibility of boosting the anti-HCC activities and reducing hemolytic activity by manipulating the sequence of Mel-S4 is currently underway. The underlying cellular and molecular mechanism remains poorly understood at present about how melittin possesses anti-HCC effects; therefore, it is a challenging work for melittin to efficiently reach biological intracellular targets without plasma membrane disruption.
Ethical statement
In the Experimental section of this manuscript, we state that all experiments with live subjects were performed in compliance with the Laboratory Animal Management Regulations of the People's Republic of China and the Three Rs (Replacement, Reduction, and Refinement) strategy of William Russell and Rex Burch, and the Animal Ethical and Welfare Committee of Chengdu Medical College has approved the experiments. Informed consent was obtained for any experimentation with human subjects.Acknowledgements
This work was financially supported by the young scholar scientific and technological innovative research team in Sichuan province (No. 2014TD0021) and provincial universities innovative research team in Sichuan province (No. 2014TD0023).Notes and references
- S. A. Hussain, D. R. Ferry, G. El-Gazzaz, D. F. Mirza, N. D. James, P. McMaster and D. J. Kerr, Ann. Oncol., 2001, 12, 161–172 CrossRef CAS PubMed.
- J. W. Sturm and M. Keese, Onkologie, 2004, 27, 294–303 CAS.
- S. F. Altekruse, K. A. McGlynn and M. E. Reichman, J. Clin. Oncol., 2009, 27, 1485–1491 CrossRef PubMed.
- M. Sato, R. Tateishi, H. Yasunaga, H. Horiguchi, H. Yoshida, S. Matsuda and K. Koike, J. Gastroenterol., 2012, 47, 1125–1133 CrossRef PubMed.
- T. C. Terwilliger and D. Eisenberg, J. Biol. Chem., 1982, 257, 6016–6022 CAS.
- W. R. Lee, K. H. Kim, H. J. An, J. Y. Kim, S. M. Han, K. G. Lee and K. K. Park, Eur. J. Pharmacol., 2014, 740, 218–226 CrossRef CAS PubMed.
- W. R. Lee, J. H. Park, K. H. Kim, Y. Y. Park, S. M. Han and K. K. Park, Toxicol. Appl. Pharmacol., 2011, 256, 209–215 CrossRef CAS PubMed.
- S. H. Park, H. J. Cho, Y. J. Jeong, J. M. Shin, J. H. Kang, K. K. Park, J. Y. Choe, Y. Y. Park, Y. S. Bae, S. M. Han, S. K. Moon, W. J. Kim, Y. H. Choi and Y. C. Chang, Am. J. Chin. Med., 2014, 42, 1139–1152 CrossRef CAS PubMed.
- J. F. Fennell, W. H. Shipman and L. J. Cole, Proc. Soc. Exp. Biol. Med., 1968, 127, 707–710 CrossRef CAS PubMed.
- H. Liu, Y. Han, H. Fu, M. Liu, J. Wu, X. Chen, S. Zhang and Y. Chen, Appl. Microbiol. Biotechnol., 2013, 97, 2877–2884 CrossRef CAS PubMed.
- M. H. Park, M. S. Choi, D. H. Kwak, K. W. Oh, D. Y. Yoon, S. B. Han, H. S. Song, M. J. Song and J. T. Hong, Prostate, 2011, 71, 801–812 CrossRef CAS PubMed.
- M. Jo, M. H. Park, P. S. Kollipara, B. J. An, H. S. Song, S. B. Han, J. H. Kim, M. J. Song and J. T. Hong, Toxicol. Appl. Pharmacol., 2012, 258, 72–81 CrossRef CAS PubMed.
- M. H. Jang, M. C. Shin, S. Lim, S. M. Han, H. J. Park, I. Shin, J. S. Lee, K. A. Kim, E. H. Kim and C. J. Kim, J. Pharmacol. Sci., 2003, 91, 95–104 CrossRef CAS.
- S. W. Ip, Y. L. Chu, C. S. Yu, P. Y. Chen, H. C. Ho, J. S. Yang, H. Y. Huang, F. S. Chueh, T. Y. Lai and J. G. Chung, Int. J. Urol., 2012, 19, 61–70 CrossRef CAS PubMed.
- H. Hu, D. Chen, Y. Li and X. Zhang, J. Pharm. Pharmacol., 2006, 58, 83–89 CrossRef CAS PubMed.
- W. C. Tu, C. C. Wu, H. L. Hsieh, C. Y. Chen and S. L. Hsu, Toxicon, 2008, 52, 318–329 CrossRef CAS PubMed.
- D. O. Moon, S. Y. Park, M. S. Heo, K. C. Kim, C. Park, W. S. Ko, Y. H. Choi and G. Y. Kim, Int. Immunopharmacol., 2006, 6, 1796–1807 CrossRef CAS PubMed.
- S. W. Ip, S. S. Liao, S. Y. Lin, J. P. Lin, J. S. Yang, M. L. Lin, G. W. Chen, H. F. Lu, M. W. Lin, S. M. Han and J. G. Chung, In Vivo, 2008, 22, 237–245 Search PubMed.
- C. C. Liu, D. J. Hao, Q. Zhang, J. An, J. J. Zhao, B. Chen, L. L. Zhang and H. Yang, Cancer Chemother. Pharmacol., 2016, 78, 1113–1130 CrossRef CAS PubMed.
- S. Liu, M. Yu, Y. He, L. Xiao, F. Wang, C. Song, S. Sun, C. Ling and Z. Xu, Hepatology, 2008, 47, 1964–1973 CrossRef CAS PubMed.
- C. Wang, T. Chen, N. Zhang, M. Yang, B. Li, X. Lu, X. Cao and C. Ling, J. Biol. Chem., 2009, 284, 3804–3813 CrossRef CAS PubMed.
- H. Zhang, B. Zhao, C. Huang, X. M. Meng, E. B. Bian and J. Li, PLoS One, 2014, 9, e95520 Search PubMed.
- X. Wu, B. Zhao, Y. Cheng, Y. Yang, C. Huang, X. Meng, B. Wu, L. Zhang, X. Lv and J. Li, Toxicol. Appl. Pharmacol., 2015, 288, 74–83 CrossRef CAS PubMed.
- Z. Zhang, H. Zhang, T. Peng, D. Li and J. Xu, Oncol. Lett., 2016, 11, 610–618 Search PubMed.
- B. Li, W. Gu, C. Zhang, X. Q. Huang, K. Q. Han and C. Q. Ling, Onkologie, 2006, 29, 367–371 CAS.
- C. Y. Qian, K. L. Wang, F. F. Fang, W. Gu, F. Huang, F. Z. Wang, B. Li and L. N. Wang, Int. J. Clin. Exp. Pathol., 2015, 8, 10403–10411 Search PubMed.
- Z. Zhang, H. Zhang, T. Peng, D. Li and J. Xu, Oncol. Lett., 2016, 11, 610–618 Search PubMed.
- L. Qu, M. Jiang, Z. Li, F. Pu, L. Gong, L. Sun, R. Gong, G. Ji and J. Si, J. Biomed. Nanotechnol., 2014, 10, 695–706 CrossRef CAS PubMed.
- H. Zhao, X. Feng, W. Han, Y. Diao, D. Han, X. Tian, Y. Gao, S. Liu, S. Zhu, C. Yao, J. Gu, C. Sun and L. Lei, J. Pept. Sci., 2013, 19, 639–650 CrossRef CAS PubMed.
- E. Perez-Paya, J. Dufourcq, L. Braco and C. Abad, Biochim. Biophys. Acta, Biomembr., 1997, 1329, 223–236 CrossRef CAS.
- C. E. Schafmeister, A. Julia Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- Q. Chu, R. E. Moellering, G. J. Hilinski, Y. W. Kim, T. N. Grossmann, T. H. Yeh and G. L. Verdine, MedChemComm, 2014, 6, 111–119 RSC.
- A. J. Krauson, J. He and W. C. Wimley, J. Am. Chem. Soc., 2012, 134, 12732–12741 CrossRef CAS PubMed.
- G. H. Bird, E. Mazzola, K. Opoku-Nsiah, M. A. Lammert, M. Godes, D. S. Neuberg and L. D. Walensky, Nat. Chem. Biol., 2016, 12, 845–852 CrossRef CAS PubMed.
- G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3–33 CAS.
- Y. X. Tan, C. Chen, Y. L. Wang, S. Lin, Y. Wang, S. B. Li, X. P. Jin, H. W. Gao, F. S. Du, F. Gong and S. P. Ji, J. Gene Med., 2012, 14, 241–250 CrossRef CAS PubMed.
- D. Sengupta, H. Leontiadou, A. E. Mark and S. J. Marrink, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 2308–2317 CrossRef CAS PubMed.
- T. T. Dinh, D. H. Kim, H. X. Luong, B. J. Lee and Y. W. Kim, Bioorg. Med. Chem. Lett., 2015, 25, 4016–4019 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental methods, analytical data, and spectra of the corresponding compounds. See DOI: 10.1039/c6ra26427a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |