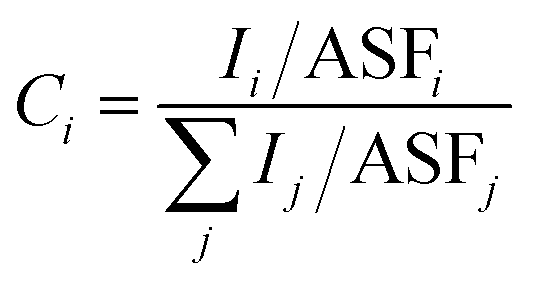DOI:
10.1039/C6RA26409C
(Paper)
RSC Adv., 2017,
7, 3179-3189
Synthesis and antibiofouling properties of crosslinkable copolymers grafted with fluorinated aromatic side chains†
Received
7th November 2016
, Accepted 11th December 2016
First published on 13th January 2017
Abstract
To obtain highly effective antifouling coatings, a series of functional ternary copolymers grafted with short fluoroalkyl or perfluoropolyether modified fluorinated aromatic side chains and cross-linkable functional groups were prepared via radical polymerization. Nuclear magnetic resonance (NMR), Fourier transform infrared (FT-IR), gel permeation chromatography (GPC), X-ray photoelectron spectroscopy (XPS) and thermogravimetric analysis (TGA) were used to characterize the fluoropolymers. The surface properties of the fluoropolymer coatings on cotton fabric, aluminum plates and glass slides were studied. The results indicate that the fluoropolymers show good thermal stabilities (Td > 303 °C), and the hydrophobicity of the fluoropolymer coatings was increased with the increase of fluorinated monomer content (water contact angle on cotton fabric was up to 144°). The antifouling characteristics were investigated by Escherichia coli and Staphylococcus aureus attachment studies. The fluoropolymers exhibited excellent antibacterial activity against E. coli and S. Aureus. The fluoropolymers might be used as promising environmentally benign marine antifouling coating materials due to their high thermal stability, hydrophobicity and antifouling effect.
1 Introduction
Marine biofouling arises from the adhesion of macro-organisms, bacteria and microalgae on artificial surfaces such as ship hulls, and results in many problems, including increased fuel consumption and maintenance costs due to corrosion.1 Antifouling technologies based on traditional chemical methods, biological methods, and physical methods have been developed over the last few decades. Although chemical methods, including self-polishing coatings based on tributyltin (TBT) or copper pyrithione, are the most effective approaches for prevention of marine biofouling, they have been banned since 2008 due to their serious environmental impacts.2,3 Hence, novel environmentally benign solutions to reduce biofouling are being sought with urgency.
Apart from biological methods such as the application of enzymes as antifouling agents,4,5 physical methods such as the modification of surface topography, hydrophobic properties, and charge potential to prevent biofouling have been considered the ultimate antifouling solution, because of their broad-spectrum effectiveness and zero toxicity. In this regard, the potency of fluoropolymers and silicon elastomers, which have been considered as the most promising candidates, have been explained in terms of their low surface energy and interesting mechanical properties.6–8 Poly(dimethylsiloxane) (PDMS) elastomers, as the most important nontoxic anti-biofouling coating materials, have been widely used in commercial applications owing to their low modulus and low surface energy.9 However, the antifouling efficacy of PDMS marine coatings is not as good as toxic paints, and regular mechanical cleaning (scrubbing) of the coated surface is also required, which increases the operational expense.10 In this respect, fluoropolymer, as another class of low surface energy coating materials, showed better antifouling performance compared with PDMS coatings; however, there is also a fatal drawback that these materials often undergo rapid molecular reconstruction in water, and thus lose their fouling release characteristics.11 For creating a long-term stable low energy surface, the fluorinated side chain of a polymer should be able to organize into an ordered structure on the surface and at the same time keep a high density of –CF3 groups on the coating surface.12 Ober and coworkers found that if a polymer contains semifluorinated alkyl substituted aromatic side chains (–C6H5CH2O(CH2)m(CF2)nF, m > 1, n > 3), the reconstruction process can be reduced due to the π–π stacking interactions between aromatic side chains.13 Coleen and coworkers reported that the strength of π–π stacking interactions can be further increased through introduction of fluorine atoms on the benzene ring.14 Another method found to be effective for creating a stable, non-reconstructable coating was to restrict chain mobility of the polymer through chemical cross-linking groups, such as epoxide functional groups.15
In this paper, we designed and synthesized a series of functional ternary copolymers grafted with short fluoroalkyl or perfluoropolyether modified fluorinated aromatic side chains and cross-linkable functional groups. Usually, for achieving a low energy surface, perfluoroalkyl chains (CnF2n+1, n > 8) and their derivatives are the most favorable functional groups.16,17 However, recent studies suggest that molecules containing perfluoroalkyl chains (CnF2n+1, n > 8) are relatively lipophilic and can accumulate in wildlife and the human body, which results in a potential risk to human health and environmental concerns. The introduction of fluorine atoms on the benzene ring avoids the use of long perfluoroalkyl chains (CnF2n+1, n > 8) while keeping the high fluorine content, and the stability of the low energy surface is increased via increased π–π stacking interactions between aromatic side chains. The low energy surfaces were fabricated on cotton fabric, aluminum plates and glass slides, and characterized by X-ray photoelectron spectroscopy (XPS). The antifouling properties of the crosslinked fluoropolymer coatings on aluminum plates to Escherichia coli and Staphylococcus aureus have been investigated.
2 Experimental section
2.1 Materials
2,3,4,5,6-Pentafluorostyrene was purchased from TCI (Shanghai, China). 1H,1H-Heptadecafluoro(2,5-dimethyl-3,6-dioxanonan-1-ol) and 1H,1H,2H,2H-perfluorooctanol were purchased from Qinba Chemical Scientific Ltd. Methyl methacrylate (MMA), glycidyl methacrylate (GMA) and other chemicals were purchased from Sinopharm Chemical Reagent Co. Ltd and used as received.
2.2 Measurements
NMR spectra were recorded at ambient temperature on a Bruker AV400 spectrometer at 400.0 and 377.0 MHz for 1H and 19F nuclei, respectively. Tetramethylsilane (TMS) was applied as the internal chemical shift reference for 1H NMR spectra and CFCl3 as an external standard for 19F NMR spectra, downfield shifts are designated as positive. Fourier transform infrared (FT-IR) spectra were recorded on a FT-IR spectrometer (Avatar 380) using KBr crystals in the infrared region of 4000–400 cm−1.
Molecular weights and distributions were determined by gel permeation chromatography (GPC) using a series of three linear Styragel columns. A Waters 1515 pump and Waters 2414 differential refractive index (RI) detector (set at 35 °C) were used. A commercial calibration kit of polystyrene standards was utilized to calibrate the GPC elution traces. As a result, the molecular weights (Mw, Mn) and their distributions were thus evaluated.
Thermogravimetric analysis (TGA) was carried out under a N2 atmosphere with a heating rate of 10 °C min−1, up to 600 °C, using a Netzsch TG 209 F1 analyzer.
The water contact angle (CA) was measured using automatic video contact-angle testing apparatus (DataPhysics OCA 40, DataPhysics Instruments GmbH, Germany). The probe liquid was deionized water. The average CA value was determined by measuring five different positions of the same sample with 5 μL each time. Drops were fitted with a Young–Laplace formula to calculate the static contact angle.
X-ray photoelectron spectroscopy (XPS) measurements were taken with a Kratos XSAM-800 using an AlKα X-ray source, and a pass energy of 40 eV. The X-ray gun was operated with a Mono Al anode, 15 keV voltage and 20 mA current. The analyzer chamber pressure was in the 10−9 Torr range.
The bacterial attachment was assessed using a scanning electron microscope (HITACHI/TM-1000).
Treatment of the cotton fabric with the polymers. The desized, bleached and cleaned cotton fabrics (3 cm × 3 cm) were soaked in the polymer solution (0.002 g mL−1, THF as solvent) for 2 h. The cotton fabrics were dried at 80 °C for 30 min and then cured at 160 °C for 3 min. Thereafter the cotton fabrics were cooled in a dryer.
Biofouling assays. The biofouling properties of the copolymer film were investigated using E. coli and S. aureus as the model microorganisms. Basically, E. coli and S. aureus were incubated in Luria-Bertani (LB) and Nutrient Broth (NB) liquid culture medium, respectively. Both strains were shaken at 37 °C for 24 h, and then diluted with the same liquid culture medium to a predetermined concentration (around 2.0 × 105 cfu mL−1), using the standard dilution method. All the polymer films coated on the aluminum sheets were sterilized at 121 °C for 30 min. Aluminum sheets were first put into separate culture plates containing 4 mL of E. coli suspension or S. aureus suspension, and then into a shaking incubator at 37 °C for 24 h. Then, these films were washed with sterile saline in order to remove dead cells and residuals, and then fixed with 2.5% glutaraldehyde solution at 0 °C for 2 h. All the films were dehydrated successively using 30%, 50%, 70%, 90%, 95% and 100% ethanol solution at room temperature for 10 minutes. Finally, the films were dried in a vacuum oven overnight and were scanned using an electron microscope.
2.3 Synthesis
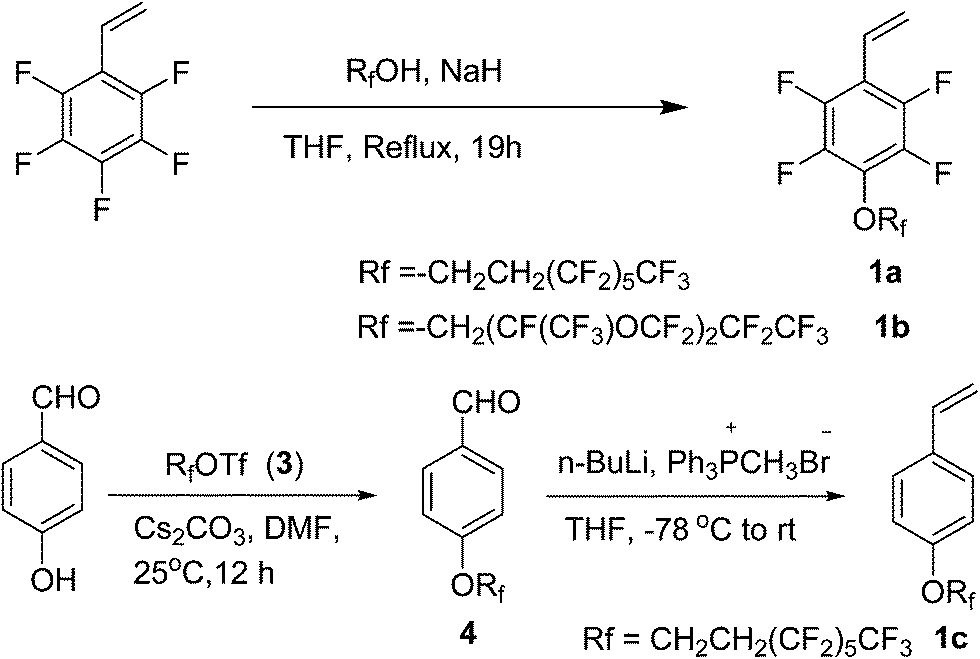
Synthesis of monomer (1a). 1H,1H,2H,2H-Perfluorooctanol (2.4 g, 6.6 mmol) was added dropwise to a solution of NaH (0.26 g, 6.6 mmol) in THF (40 mL) under a nitrogen atmosphere. The reaction mixture was stirred for 1 h at room temperature, then a solution of 2,3,4,5,6-pentafluorostyrene (1.2 g, 6 mmol) in THF (15 mL) was added slowly at 0 °C. After stirring for 1 h at 0 °C, the reaction mixture was brought to reflux for 19 h and cooled to room temperature. The reaction was quenched with ice water (40 mL), and the reaction mixture was extracted with ethyl acetate (50 mL × 3). The combined organic layer was washed with brine (20 mL), dried over anhydrous MgSO4, filtered, evaporated, and the residue was purified by column chromatography (silica gel, petroleum ether) to yield pure 1a as a colorless liquid.18 (2.16 g, 67% yield), 1H NMR (400 MHz, CDCl3) δ (ppm): 6.66 (dd, J = 18.0, 11.9 Hz, 1H), 6.08 (d, J = 18.0 Hz, 1H), 5.69 (d, J = 11.9 Hz, 1H), 4.53 (t, J = 6.7 Hz, 2H), 2.79–2.60 (m, 2H). 19F NMR (377 MHz, CDCl3) δ (ppm): −80.8 (t, J = 10.3 Hz, 3F), −113.4 (dt, J = 33.5, 18.5 Hz, 2F), −121.9 (s, 2F), −122.9 (s, 2F), −123.5 (s, 2F), −126.0 to −126.4 (m, 2F), −144.4 (dd, J = 22.1, 10.0 Hz, 2F), −158.1 (dd, J = 21.8, 9.7 Hz, 2F). IR (KBr, cm−1): 2976, 2923, 1648, 1489, 1408, 1235, 1190, 1144, 1123. MS (ESI): m/z 539 [M + H]+. HRMS (ESI-TOF): m/z [M + H]+ calcd for C16H8F17O: 539.0298; found: 539.0297.
Synthesis of monomer (1b). Colorless liquid. (90% yield), 1H NMR (400 MHz, CDCl3) δ (ppm): 6.64 (dd, J = 18.0, 11.9 Hz, 1H), 6.09 (d, J = 18.0 Hz, 1H), 5.70 (d, J = 11.9 Hz, 1H), 4.72 (d, J = 10.0 Hz, 2H). 19F NMR (377 MHz, CDCl3) δ (ppm): −80.2 to −83.3 (m, 13F), −130.0 to −130.6 (m, 2F), −134.2 to −136.2 (m, 1F), −144.0 to −144.6 (m, 2F), −145.1 to −145.9 (m, 1F), −157.7 to −160.4 (m, 2F). IR (KBr, cm−1): 2953, 2922, 1647, 1492, 1306, 1229, 1198, 1150, 1126. MS (ESI): m/z 657 [M + H]+. HRMS (ESI-TOF): m/z [M + H]+ calcd for C17H6F21O3: 656.9976; found: 656.9975.
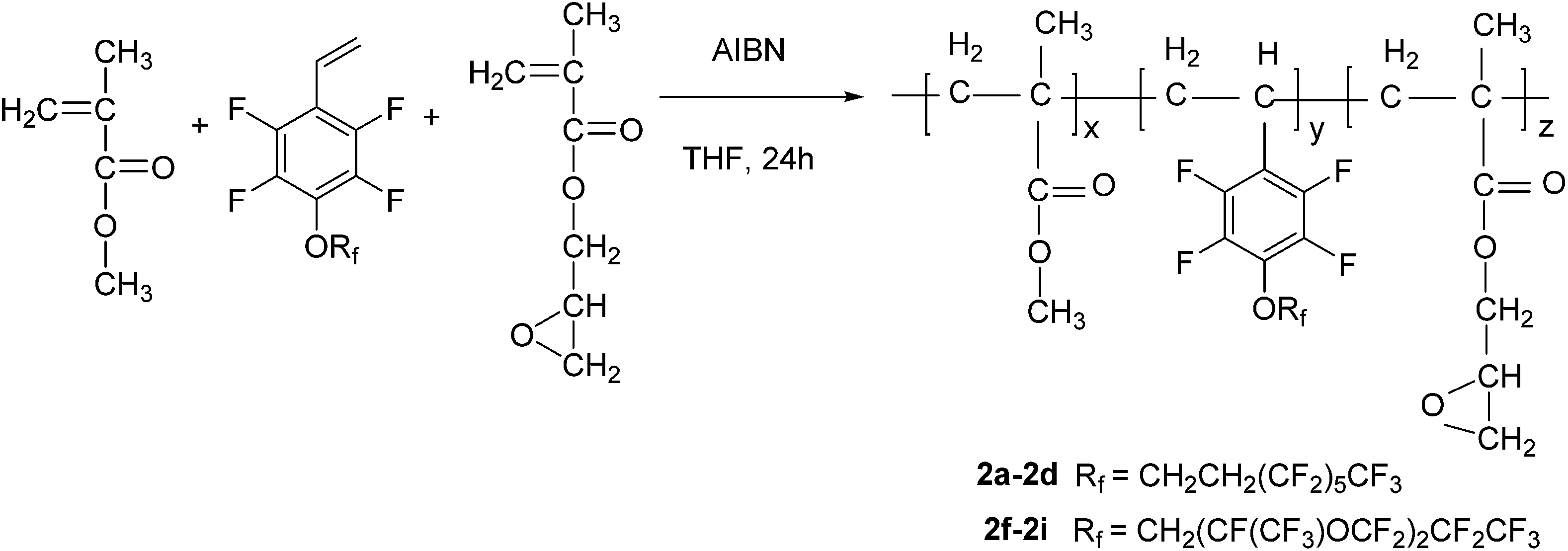
General procedure for copolymerization. To a 25 mL three-necked round-bottomed flask equipped with a reflux condenser were added MMA (1.0 g, 10.0 mmol), GMA (1.0 g, 7.7 mmol), 1a (1.0 g, 1.9 mmol, 33 wt%), AIBN (0.03 g, 0.2 mmol) and THF (10 mL) under a nitrogen atmosphere. After reflux for 24 h at 65 °C, the reaction mixture was added dropwise into ethanol (600 mL) with vigorous agitation. The precipitate was collected by filtration and re-dissolved in ethyl acetate, and re-precipitated in ethanol. This purification process was repeated three times. The product was dried in a vacuum oven overnight to afford polymer 2a as a white solid (2.52 g, 84% yield).19 1H NMR (400 MHz, CDCl3) δ (ppm): 4.31–4.46 (br), 3.78, 3.60, 3.22, 2.85, 2.64, 1.84, 1.44, 0.88–1.25 (br). 19F NMR (377 MHz, CDCl3) δ (ppm): −80.7 (s, 3F), −113.3 (s, 2F), −121.8 (s, 2F), −122.8 (s, 2F), −123.5 (s, 2F), −126.1 (s, 2F), −139.4 to −143.7 (m, 2F), −154.3 to −159.0 (m, 2F). IR (KBr, cm−1): 2996, 2955, 1728, 1652, 1492, 1239, 1196, 1145.Polymer 2b–2e were prepared following a similar procedure to that used for 2a, but the amount of MMA was kept constant, and the mass fraction of 1a was controlled as 20 wt%, 10 wt%, 5 wt%, 0 wt% to get polymer 2b (83% yield), 2c (83% yield), 2d (83% yield), 2e (87% yield), respectively. Polymers 2a–2e were all white solids.
Polymer 2f–2i were prepared following a similar procedure to that used for 2a, but the amount of MMA was kept constant, and the mass fraction of 1b was controlled as 33 wt%, 20 wt%, 10 wt%, 5 wt% to get 2f (81% yield), 2g (83% yield), 2h (82% yield), 2i (84% yield), respectively. Polymers 2f–2i were all white solids.
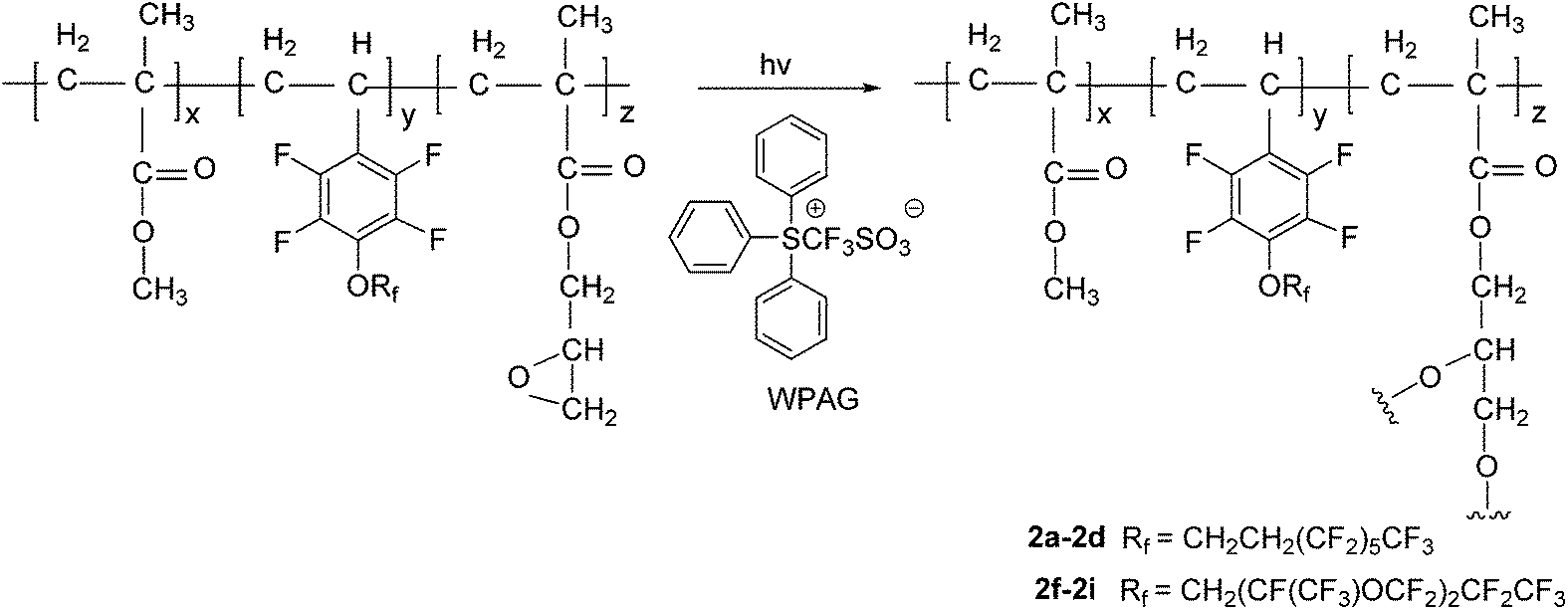
Preparation of polymer-coated surfaces. The fluoropolymer 2a (1 g) and WPAG336 (1.5 wt% relative to polymer) were dissolved in ethyl ketone (4 mL) to get a clear, homogeneous solution. The solution was evenly spread onto a piece of glass cleaned using hot piranha solution (H2SO4 (98 wt%)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O2 (30 wt%) = 7:3 v/v) using spin coating machine. The solution was allowed to evaporate for 10 min, then underwent UV irradiation using a mercury arc lamp for 20 min, followed by a post-exposure bake at 120 °C for 6 h until curing was complete. Thus the surface fluoropolymer-coated with 2a was prepared. Other polymer-coated surfaces on glass were prepared according to the same process by using fluoropolymer 2b–2i, respectively. The fluoropolymer-coated surfaces on aluminum sheets were also prepared according to a similar process.
:H2O2 (30 wt%) = 7:3 v/v) using spin coating machine. The solution was allowed to evaporate for 10 min, then underwent UV irradiation using a mercury arc lamp for 20 min, followed by a post-exposure bake at 120 °C for 6 h until curing was complete. Thus the surface fluoropolymer-coated with 2a was prepared. Other polymer-coated surfaces on glass were prepared according to the same process by using fluoropolymer 2b–2i, respectively. The fluoropolymer-coated surfaces on aluminum sheets were also prepared according to a similar process.
Synthesis of controlled monomer.
Synthesis of compound 4. 4-Hydroxybenzaldehyde (0.49 g, 4.0 mmol), triflate 3 (2.3 g, 4.8 mmol), Cs2CO3 (2.0 g, 6.0 mmol), and DMF (8 mL) were added to a 50 mL reaction tube under a nitrogen atmosphere at 25 °C. The reaction mixture was stirred for 12 h and then diluted with water, followed by extraction with ethyl acetate. The organic layer was washed with brine, and dried with anhydrous MgSO4. The combined ethyl acetate layers were concentrated under vacuum, and the residue was purified by column chromatography on silica gel (petroleum ether:ethyl acetate = 10:1) to afford 4 (1.348 g, 72% yield) as a white powder. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.93 (s, 1H), 7.89 (d, J = 7.7 Hz, 2H), 7.05 (d, J = 7.8 Hz, 2H), 4.39 (t, J = 6.6 Hz, 2H), 2.81–2.56 (m, 2H). 19F NMR (377 MHz, CDCl3) δ (ppm): −80.7 (t, J = 11.4 Hz, 3F), −113.3 (dt, J = 36.6, 18.1 Hz, 2F), −121.8 (s, 1H), −122.8 (s, 2F), −123.5 (s, 2F), −125.6 to −126.6 (m, 2F). IR (KBr, cm−1): 2738, 1693, 1504, 1239, 1200, 1146.
Synthesis of compound 1c. To a 50 mL three-necked flask were added Ph3PCH3Br (1.4 g, 4 mmol) and THF (22 mL) under a nitrogen atmosphere. Then n-BuLi (1.6 mL, 2.5 M in THF, 4 mmol) was added dropwise to the solution at −78 °C, and the mixture was stirred at −78 °C for 3 h. Following this, a solution of compound 4 (0.94 g, 2 mmol) in THF (4 mL) was added dropwise to the mixture at −78 °C. The cooling bath was removed after the addition, and the reaction mixture was stirred for 2 h, during which time the temperature increased slowly from −78 °C to room temperature. Thereafter, water was added into the reaction mixture and then extracted with diethyl ether. The organic layer was washed with brine, and dried with anhydrous MgSO4. The residue was purified by flash chromatography (petroleum ether) to give compound 1c (0.842 g, 90% yield) as a yellowish solid. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.39 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 6.69 (dd, J = 17.6, 10.9 Hz, 1H), 5.66 (d, J = 17.6 Hz, 1H), 5.18 (d, J = 10.9 Hz, 1H), 4.31 (t, J = 6.8 Hz, 2H), 2.66 (tt, J = 18.4, 6.8 Hz, 2H). 19F NMR (377 MHz, CDCl3) δ (ppm): −80.7 (t, J = 9.9 Hz, 3F), −113.2 to −113.4 (m, 2F), −121 (s, 2F), −122.8 (s, 2F), −123.5 (s, 2F), −126.0 to −126.2 (m, 2F). IR (KBr, cm−1): 2969, 2928, 1607, 1514, 1405, 1234, 1177, 1137. Analytical data are consistent with that in the literature.20
Synthesis of controlled polymer 5. The controlled polymer 5 was synthesized to explore the influence of fluorine atoms on the benzene ring. Polymer 5 was prepared as a white solid (72% yield) according to the procedure used for the preparation of 2a. 1H NMR (400 MHz, CDCl3) δ (ppm): 6.95, 6.75, 4.23–3.61 (br), 3.81, 3.62, 3.25, 2.87, 2.66, 1.85, 0.87–1.40 (br). 19F NMR (377 MHz, CDCl3) δ (ppm): −80.7 (s, 3F), −113.4 (s, 2F), −121.9 (s, 2F), −122.9 (s, 2F), −123.6 (s, 2F), −126.2 (s, 2F). IR (KBr, cm−1): 2998, 2949, 1726, 1237, 1196, 1144.
3 Results and discussion
3.1 Synthesis and characterization
In this work, we focused on developing stable and high-performance low surface energy materials as antifouling finishing agents. The strategy was to combine the advantages of perfluoroalkane or perfluoropolyether, 2,3,4,5,6-pentafluorostyrene and epoxy pendant groups to prepare cross-linkable fluoropolymers. The fluorinated monomers 1a and 1b contain fluorinated aromatic side chains grafted with short fluoroalkyl and perfluoropolyether groups, respectively, and were used to prepare the copolymers with low surface energies. The choice of methylmethacrylate (MMA) as the principal component should enable the tuning of the matrix’s mechanical behaviour by adjusting the glass transition temperature21 and GMA provides a cross-linkage via the ring-opening reaction of epoxy pendant groups. The fluorinated monomers 1a and 1b were prepared by nucleophilic substitution of 2,3,4,5,6-pentafluorostyrene with corresponding fluorinated alcohols in the presence of sodium hydride (Scheme 1). The radical copolymerization of the fluorinated monomers, MMA and GMA, was accomplished in the presence of radical initiator AIBN under a nitrogen atmosphere (Scheme 2). A series of terpolymers were synthesized with various stoichiometries of the fluorine-containing monomers (0, 5, 10, 20, 33, wt% relative to total weight of monomers), MMA and GMA (Tables 1 and 2). Antifouling surfaces with cross-linked fluroropolymers were obtained by ring-opening reaction of epoxy pendant groups, catalyzed by an organic acid under an UV lamp (Scheme 3). In order to explore the influence of fluorine atoms on the benzene ring, controlled polymer 5 was synthesized with normal perfluoroalkyl substituted styrene 1c as a fluorinated monomer (Scheme 1).
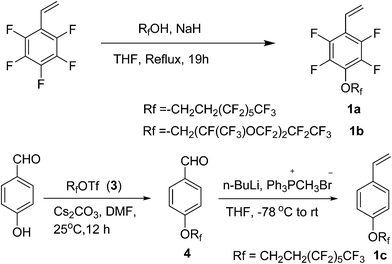 |
| | Scheme 1 Synthesis of fluorinated monomers 1a, 1b and 1c. | |
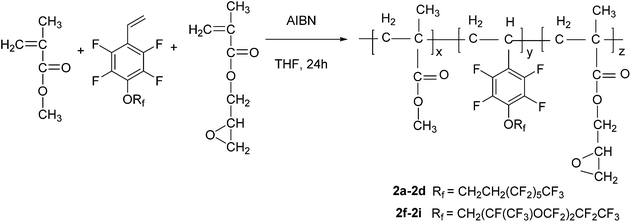 |
| | Scheme 2 Synthesis of fluoropolymers. | |
Table 1 Composition and yields of fluoropolymers 2a–2d and 5
| Polymers |
1a (g) |
MMA (g) |
GMA (g) |
1a (wt%) |
Yield (%) |
| 2a |
1 |
1 |
1 |
33 |
84 |
| 2b |
0.6 |
1 |
1.4 |
20 |
83 |
| 2c |
0.3 |
1 |
1.7 |
10 |
83 |
| 2d |
0.15 |
1 |
1.85 |
5 |
83 |
| 5 |
1 |
1 |
1 |
33 |
72 |
Table 2 Composition and yields of fluoropolymers 2e–2i
| Polymers |
MMA (g) |
1b (g) |
GMA (g) |
1b (wt%) |
Yield (%) |
| 2f |
1 |
1 |
1 |
33 |
81 |
| 2g |
1 |
0.6 |
1.4 |
20 |
83 |
| 2h |
1 |
0.3 |
1.7 |
10 |
82 |
| 2i |
1 |
0.15 |
1.85 |
5 |
84 |
| 2e |
1 |
0 |
2 |
0 |
87 |
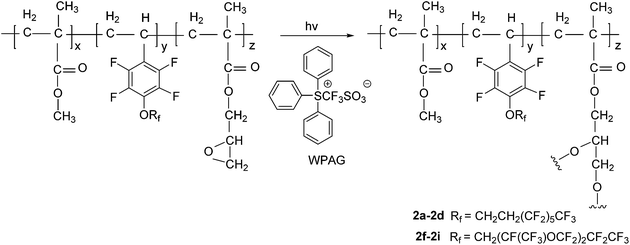 |
| | Scheme 3 The acid-catalyzed photolytic epoxide ring-opening cure chemistry. | |
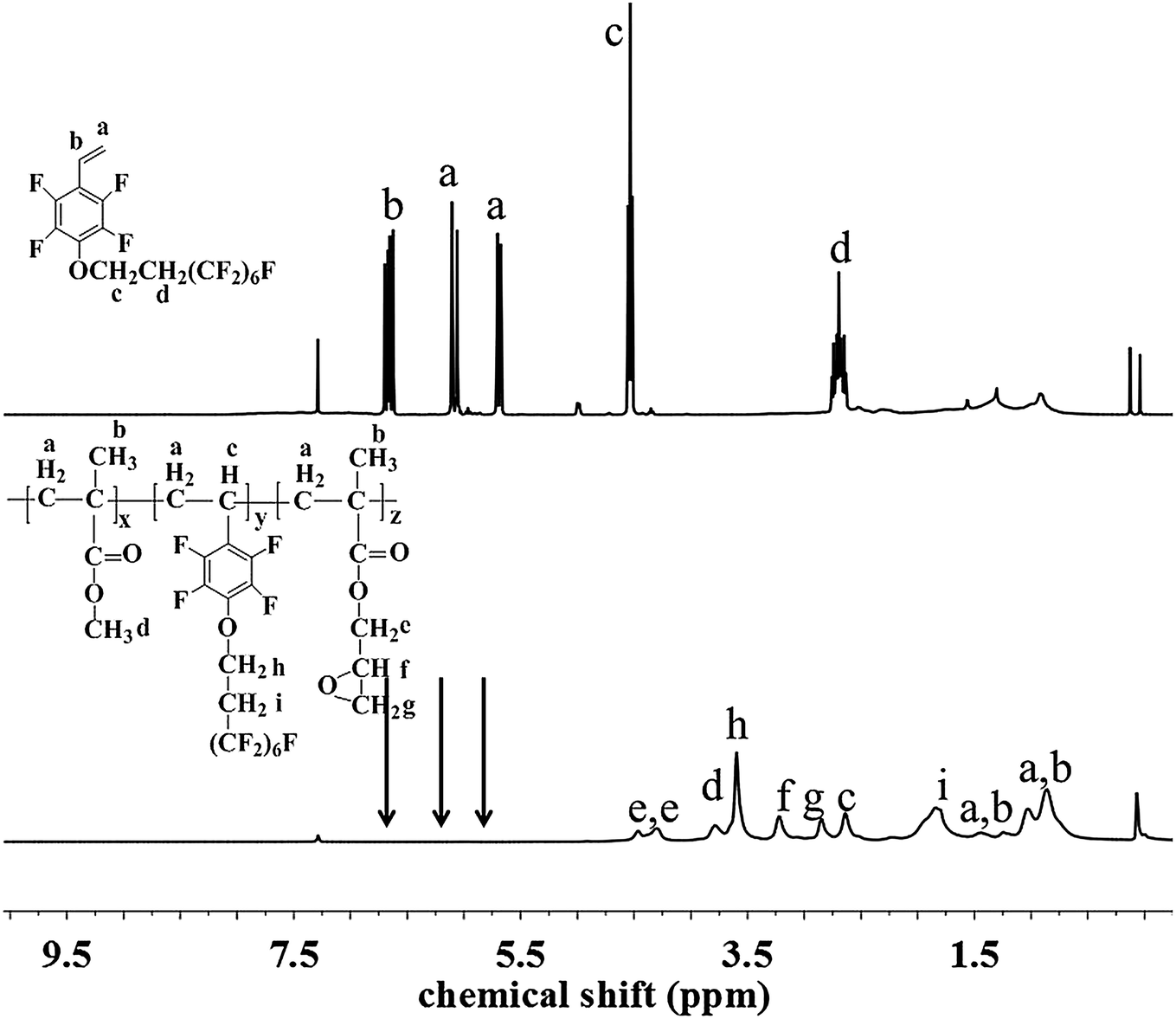
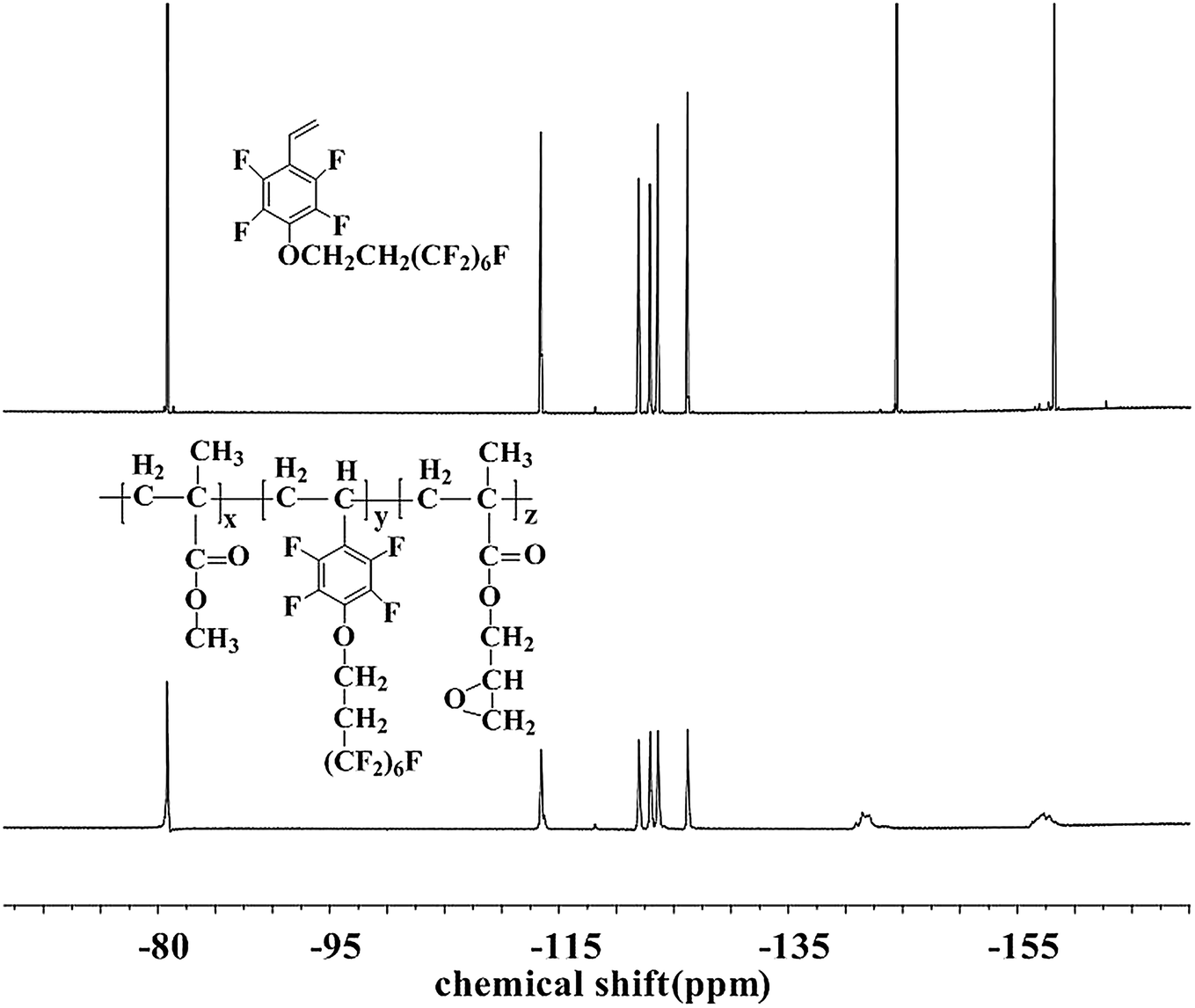
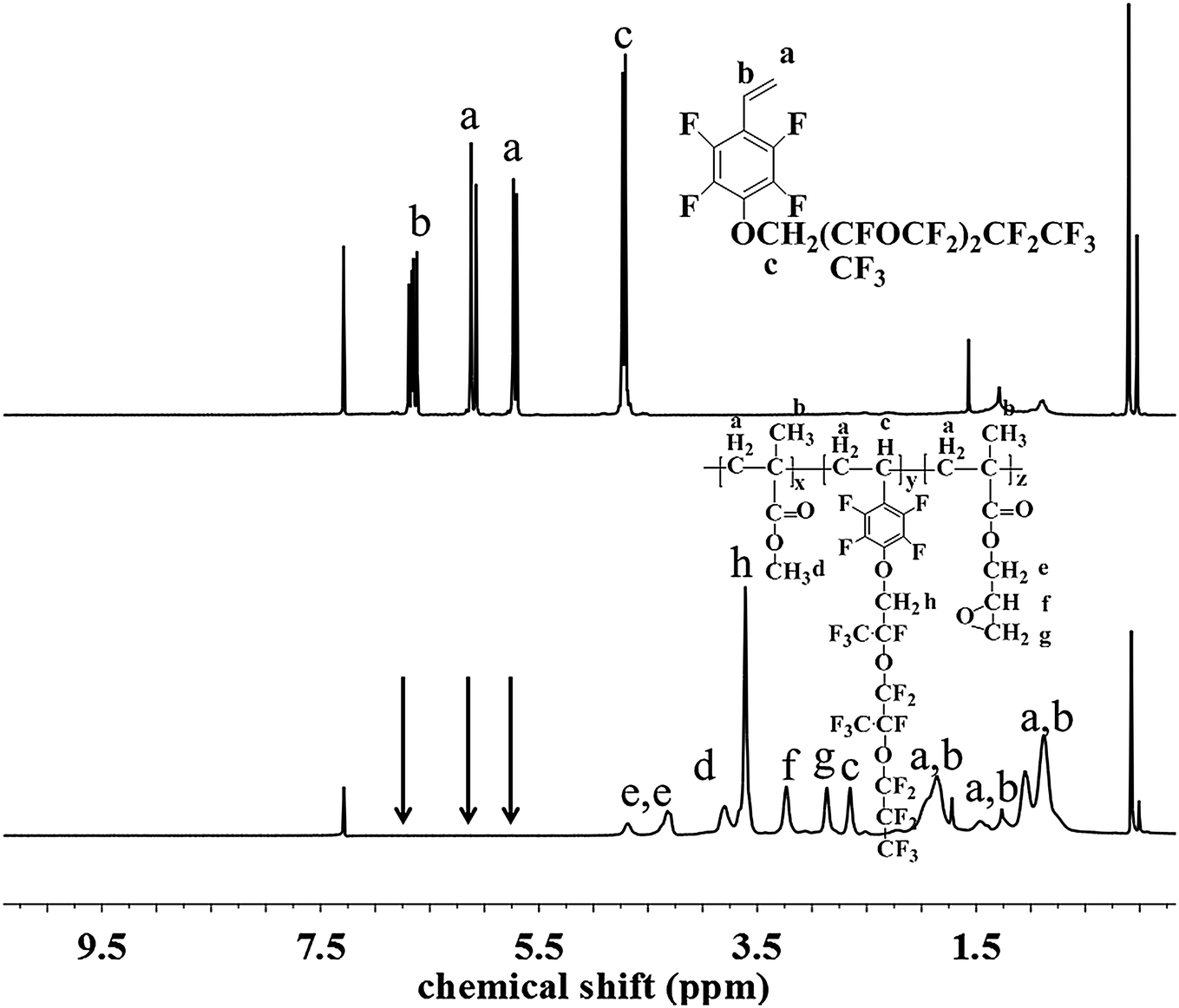
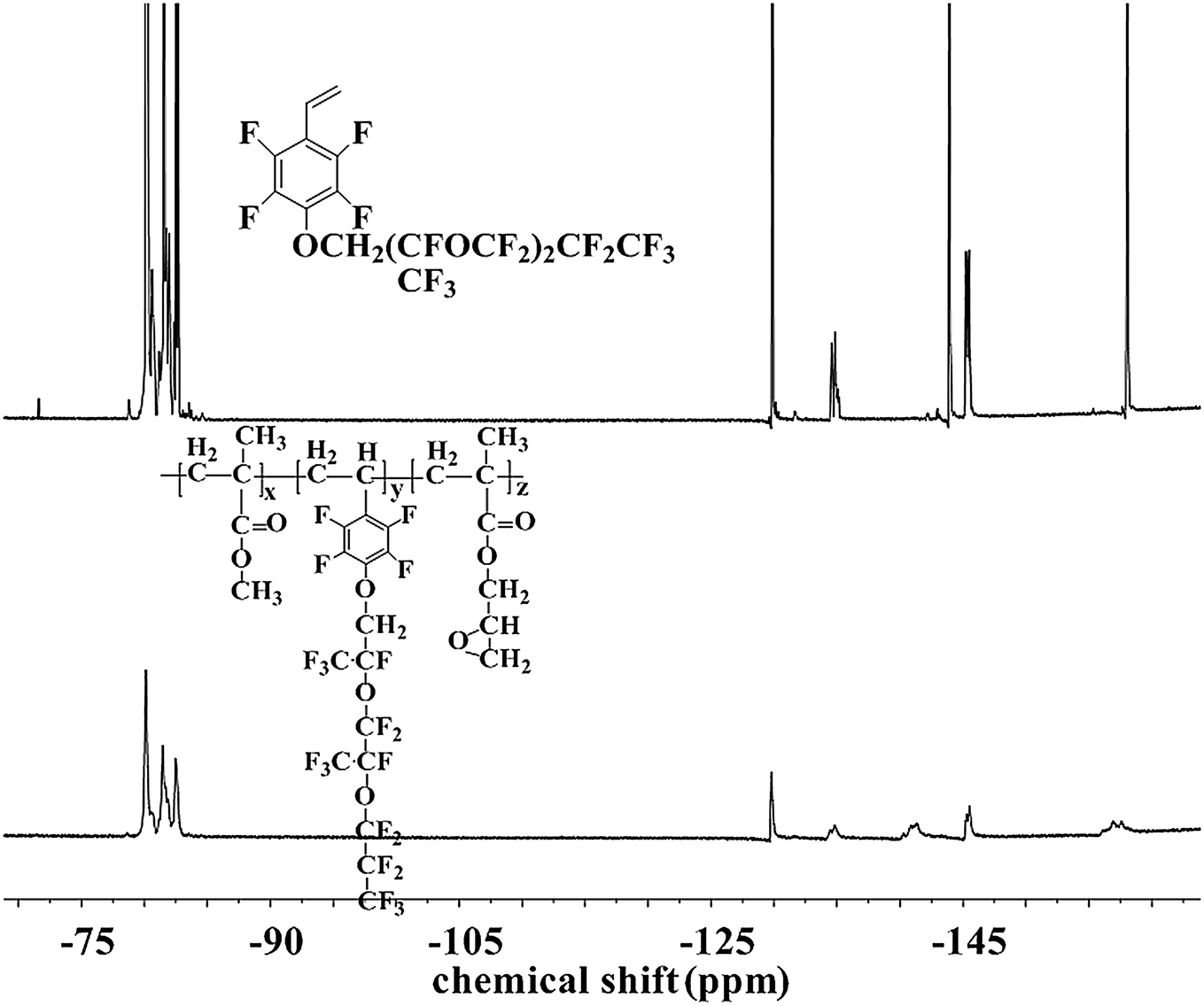
The chemical structure and composition of the fluoropolymers were characterized by 1H NMR, 19F NMR and FT-IR analyses. The 1H NMR spectra of 1a and 2a are shown in Fig. 1, in which the signals are attributed to the corresponding protons marked with letters. Proton signals at 6.66, 6.08 and 5.69 ppm in the 1H NMR spectrum of 1a can be assigned to the CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– group, and these signals disappeared and new signals are presented at 0.86–1.44 ppm in that of 2a, which demonstrated the successful incorporation of 1a in the polymerization under initiation of AIBN. Meanwhile, the 19F NMR spectra of 1a and 2a are almost the same (Fig. 2). Similar results can be seen for monomer 1b and polymer 2b, as shown in Fig. 3 and 4. The results indicated that radical polymerization reactions were successfully conducted.
CH– group, and these signals disappeared and new signals are presented at 0.86–1.44 ppm in that of 2a, which demonstrated the successful incorporation of 1a in the polymerization under initiation of AIBN. Meanwhile, the 19F NMR spectra of 1a and 2a are almost the same (Fig. 2). Similar results can be seen for monomer 1b and polymer 2b, as shown in Fig. 3 and 4. The results indicated that radical polymerization reactions were successfully conducted.
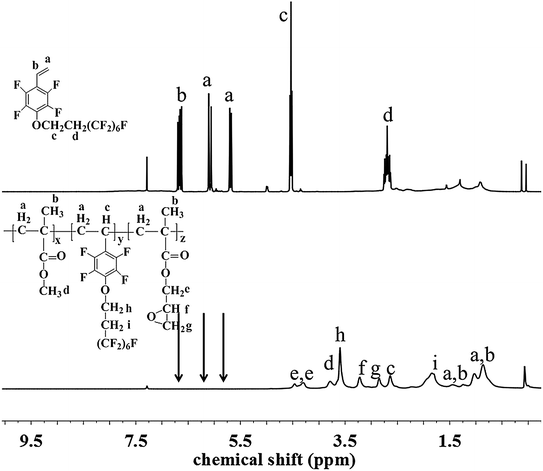 |
| | Fig. 1 1H NMR spectra of monomer 1a and polymer 2a. | |
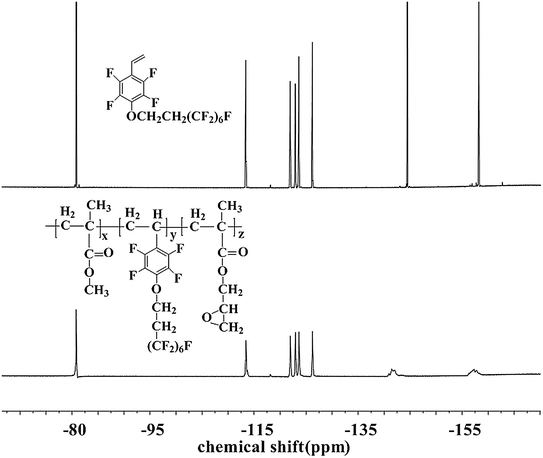 |
| | Fig. 2 19F NMR spectra of monomer 1a and polymer 2a. | |
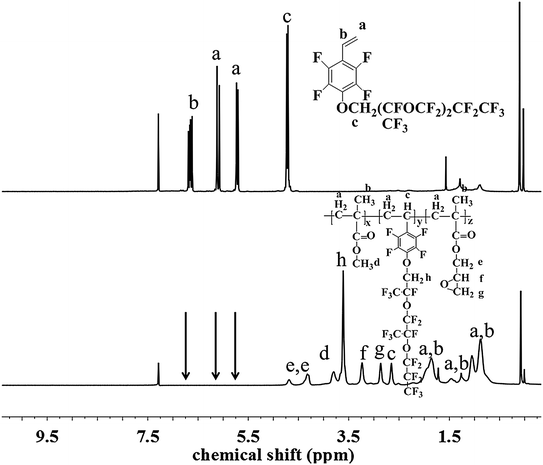 |
| | Fig. 3 1H NMR spectra of monomer 1b and polymer 2f. | |
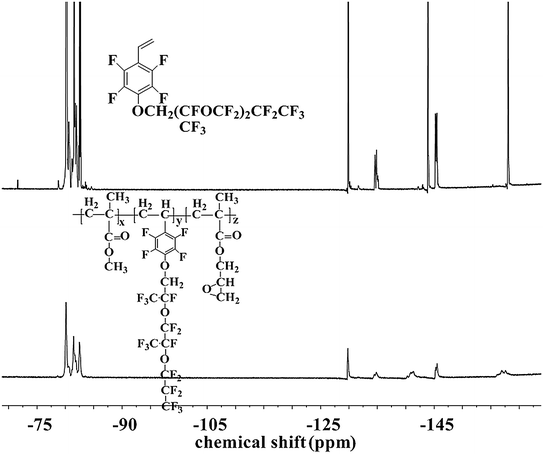 |
| | Fig. 4 19F NMR spectra of monomer 1b and polymer 2f. | |
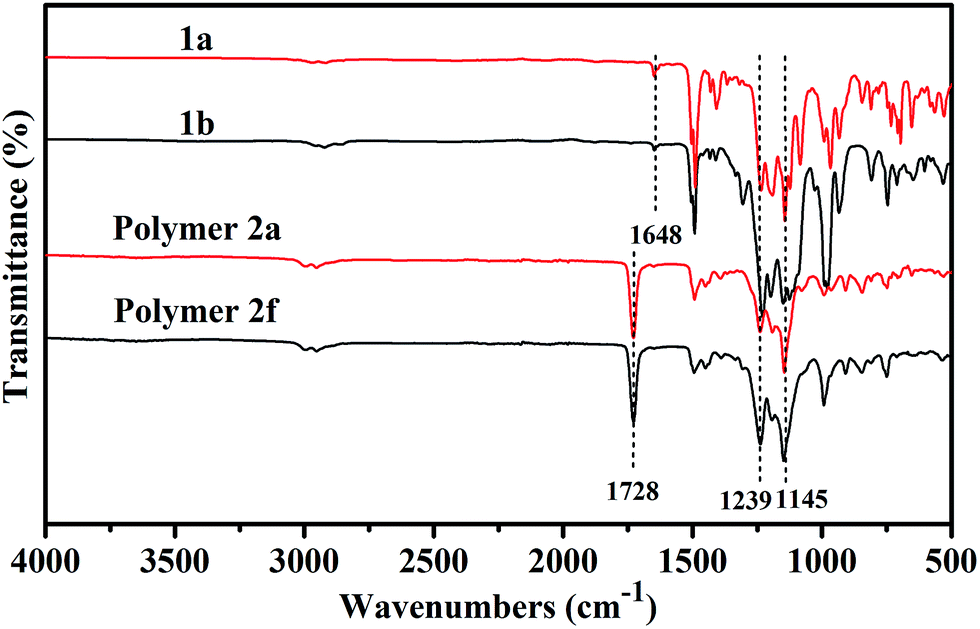
The FT-IR spectra of monomers 1a and 1b, and corresponding fluoropolymers 2a and 2f are presented in Fig. 5. In addition to the characteristic absorption bands of C–F bonds around 1145 cm−1 and 1239 cm−1, there were also characteristic absorption bands of the C–H stretch (2922–2976 cm−1), CC stretch (1648 cm−1) and aromatic bands (1493 cm−1) for monomers 1a and 1b. The disappearance of the characteristic absorption of CC (1648 cm−1) from the spectra of fluoropolymers 2a and 2f indicates that the fluorinated monomers were transferred into the polymers. In the FT-IR spectra of fluoropolymers 2a and 2f, the strong absorption band around 1728 cm−1 is attributed to carbonyl stretching vibrations. There were also characteristic absorption bands of the C–F stretch around 1145 cm−1 and 1239 cm−1. So we can conclude that the polymers are successfully synthesized with fluorinated styrene 1a or 1b, MMA and GMA as monomers via free radical polymerization.
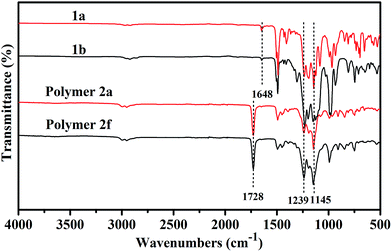 |
| | Fig. 5 FT-IR spectra of monomers 1a and 1b and polymers 2a and 2f. | |
3.2 Thermal properties
All polymers exhibit good solubility in general solvents such as CH2Cl2, THF, ethyl acetate, acetone, and so on. As shown in Tables 3 and 4, the molecular weights (Mw) of the polymers measured by GPC are higher than 3.0 × 104 g mol−1.
Table 3 Characterization of polymers 2a–2d and 5
| Polymers |
1a (wt%) |
Mw (×104 g mol−1) |
PDI (Mw/Mn) |
T10%a (°C) |
Tdb (°C) |
Fluorine ratioc (wt%) |
| Temperature at a weight loss of 10%. The onset decomposition temperature. Fluorine ratio was obtained from elemental analyses. |
| 2a |
33 |
5.16 |
1.93 |
308 |
303 |
19.91 |
| 2b |
20 |
3.98 |
2.05 |
308 |
304 |
11.92 |
| 2c |
10 |
3.58 |
2.05 |
296 |
304 |
5.27 |
| 2d |
5 |
3.98 |
2.05 |
298 |
315 |
1.96 |
| 5 |
33 |
2.95 |
1.95 |
296 |
250 |
18.3 |
Table 4 Characterization of polymers 2e–2i
| Polymers |
1b (wt%) |
Mw (×104 g mol−1) |
PDI (Mw/Mn) |
T10%a (°C) |
Tdb (°C) |
Fluorine ratioc (wt%) |
| Temperature at a weight loss of 10%. The onset decomposition temperature. Fluorine ratio was obtained from elemental analyses. |
| 2f |
33 |
4.42 |
2.01 |
314 |
332 |
22.49 |
| 2g |
20 |
3.74 |
2.08 |
298 |
282 |
12.55 |
| 2h |
10 |
3.55 |
2.02 |
302 |
260 |
4.75 |
| 2i |
5 |
3.06 |
2.01 |
296 |
281 |
3.38 |
| 2e |
0 |
1.75 |
1.67 |
292 |
282 |
0 |
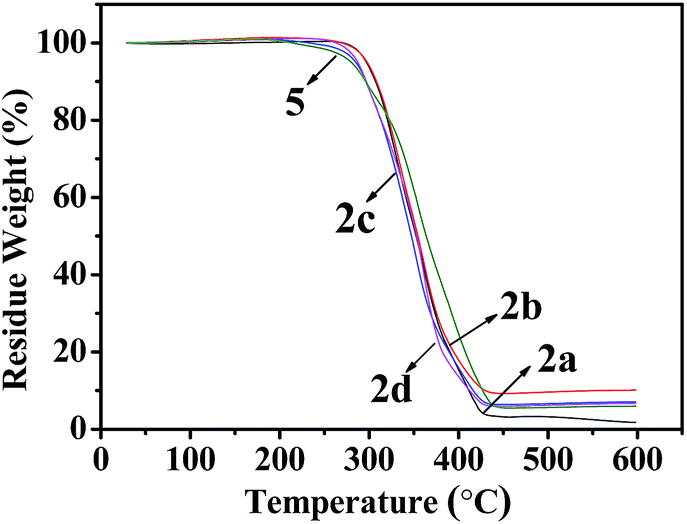
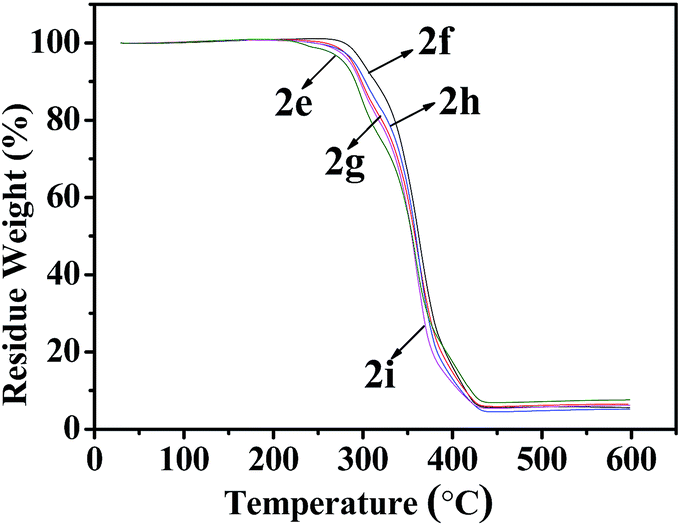
As shown in Fig. 6 and 7, Tables 3 and 4, the temperatures of the 10% weight loss of perfluoroalkyl-substituted polymers 2a–2d and perfluoropolyether-substituted polymers 2f–2i are similar and in the range of 296 to 314 °C. The onset thermal decomposition temperatures (Td) of perfluoroalkyl-substituted polymers 2a–2d were 303 °C to 315 °C, at the same time those of perfluoropolyether-substituted polymers 2f–2i were 260 °C to 332 °C. Surprisingly, the lowest Td was found for polymer 5 (250 °C), which contains a nonfluorinated benzene ring, and the highest Td was achieved for polymer 2f, which contains perfluoropolyether substituted monomer 1b in a 33 wt% feeding ratio. The results of thermal analyses demonstrated that these cross-linkable fluoropolymers exhibit good thermal stabilities, which were slightly improved by the incorporation of fluorine atoms on the benzene rings.
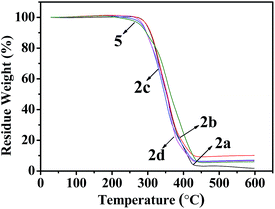 |
| | Fig. 6 TGA curves of fluoropolymers 2a–d and 5. | |
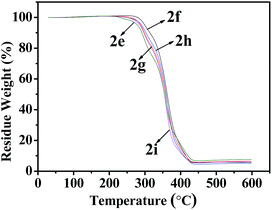 |
| | Fig. 7 TGA curves of fluoropolymers 2e–2i. | |
It is well known that hydrophobic properties are very important for materials used in marine antifouling. Fluoropolymers 2a and 2f, with a relatively high fluorine content, were coated on aluminum sheets and glass slides by spin-coating, and then the ring-opening reaction of the epoxy pendant groups was catalyzed by an organic acid under an UV lamp. The water contact angles (CAs) of the resulting coatings were measured, and the average CA of the coatings of 2a on aluminum sheets and glass slides was 112° and 109°, respectively, and that for coatings of 2f was 117° and 112°, respectively. However, the average CAs for non-fluorinated coatings of 2e on aluminum sheets and glass slides were only 88° and 79°, respectively (Table 5). The water contact angles of the polymers on different matrices demonstrated that the surface hydrophobicities of the coatings resulting from the fluorinated polymers are higher than those from the non-fluorinated polymers. Polymers with perfluoropolyether side chains show better surface hydrophobicities than the polymers with perfluoroalkyl side chains.
Table 5 Water contact angles of coatings of fluoropolymers on different matrices
| Polymers |
Cotton fabric |
Aluminum sheet |
Glass slide |
| 2a |
144° |
112° |
109° |
| 2f |
147° |
117° |
112° |
| 2e |
75° |
88° |
79° |
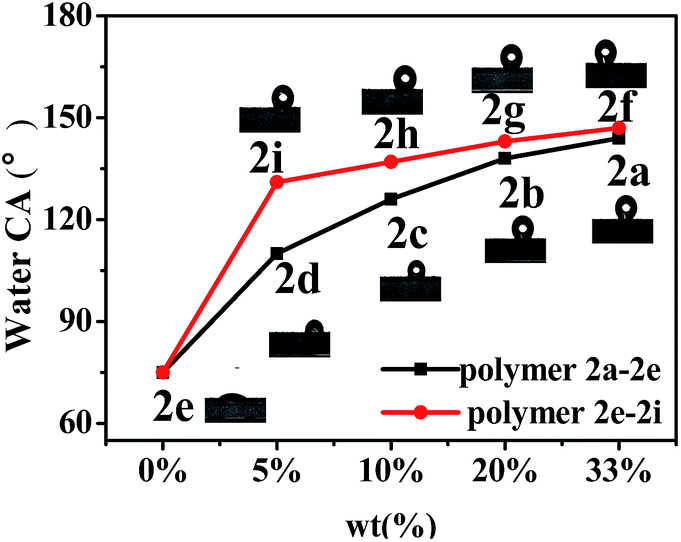
As is known, if the water CA of a polymer-coated surface is higher than 98° (surface tension < 25 mN m−1), the surface can be classified as an antifouling coating.22 In order to study the hydrophobic effect of the synthesized fluoropolymers on other matrices, 2a–2i were coated on cotton fabrics, and then the water contact angles were measured. As shown in Table 5 and Fig. 8, the highest CA on cotton fabric reached 147° for the perfluoropolyether-substituted polymer 2f coating, while for the perfluoroalkyl-substituted polymer coating it is 144°. The water contact angles of the polymers increased with the increase of the fluorine content. The average water CA of the perfluoropolyether-substituted polymer is higher than that of the perfluoroalkyl-substituted polymer, indicating that the former exhibits a lower surface energy than the latter. The water CA of the controlled polymer 5 coating on cotton fabrics is 136°, which is 8° lower than that of polymer 2a, indicating that the fluorine atom on the benzene ring improves the hydrophobicity of the polymers.
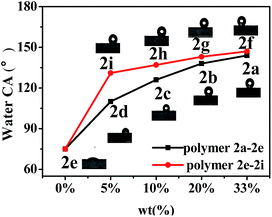 |
| | Fig. 8 Water contact angles of coatings of fluoropolymers 2a–2i on cotton fabrics. | |
3.3 XPS analysis
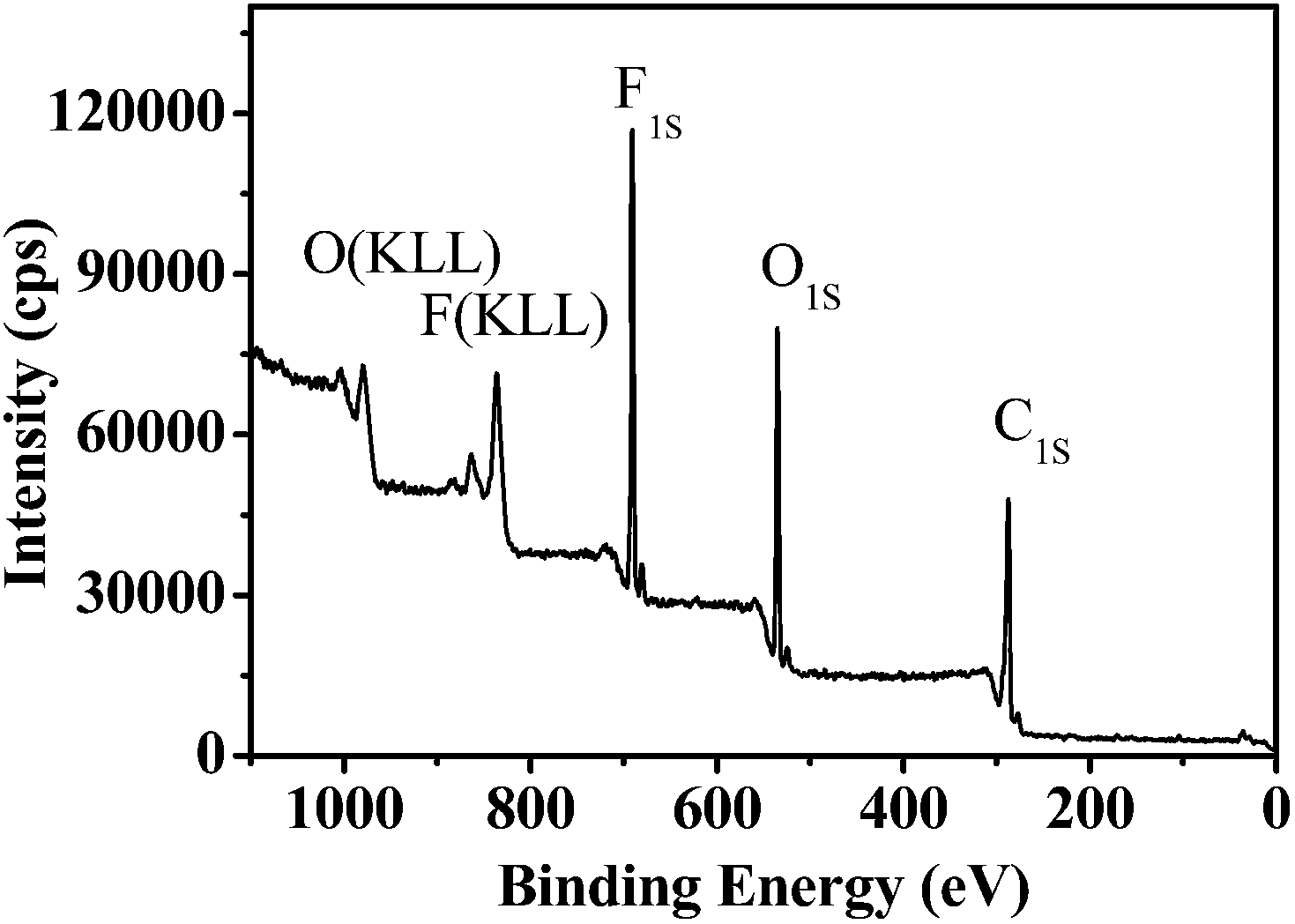
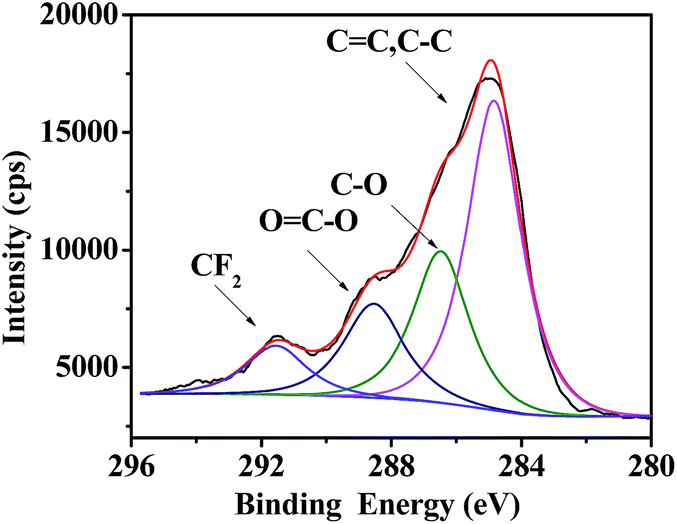
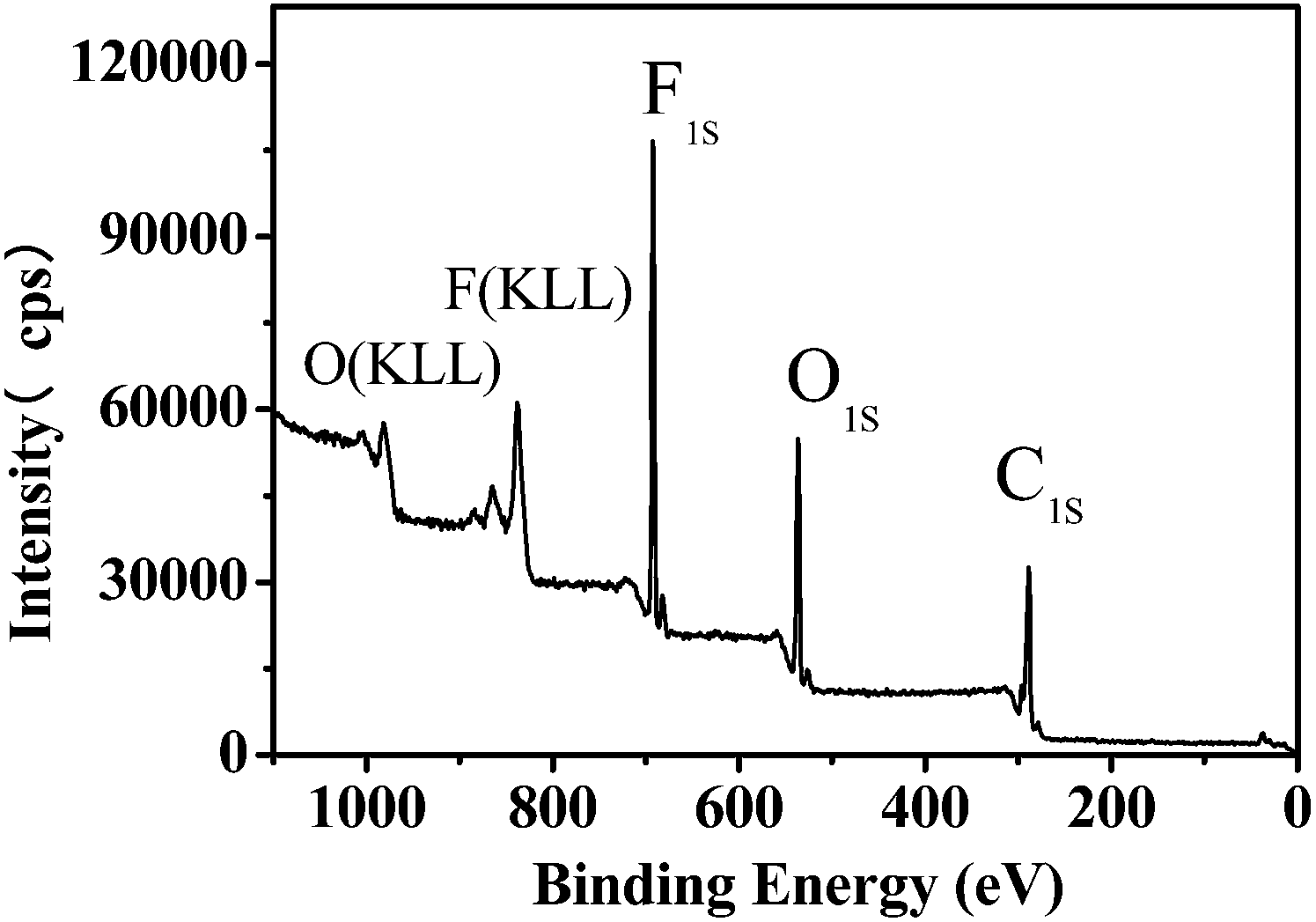
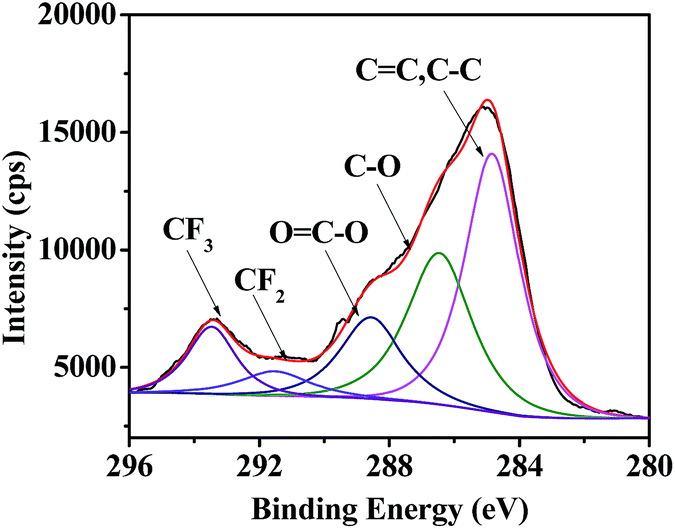
Besides hydrophobicity, the antifouling ability of a coating is strongly dependent on other surface characteristics, such as the contribution of different monomers to the surface, morphology, and so on. Therefore, the surface compositions of fluoropolymer coatings were analyzed qualitatively and quantitatively by XPS measurements. As shown in Fig. 9 and 11, the photoemission peaks of F1s, O1s, and C1s are observed at bonding energies of 691 eV, 535 eV and 288 eV, respectively. Fig. 10 and 12 exhibit the XPS of C1s for the fluoropolymer 2a and 2f coating surface, the binding energies at 284.8 eV, 286.4 eV, 288.5 eV, 291.5 eV and 293.4 eV are assigned as CC, C–C, C–O, OC–O, CF2 and CF3, respectively. The results proved that the polymers have been successfully coated on the slide surface (Fig. 12).
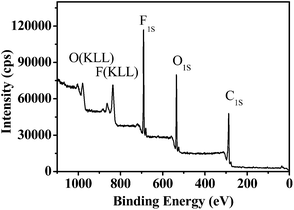 |
| | Fig. 9 XPS for the coating surface of fluoropolymer 2a on a glass slide. | |
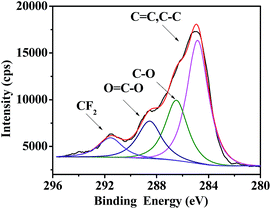 |
| | Fig. 10 XPS of C1s for the coating surface of fluoropolymer 2a on a glass slide. | |
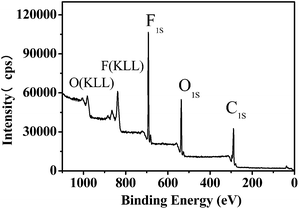 |
| | Fig. 11 XPS for the coating surface of fluoropolymer 2f on a glass slide. | |
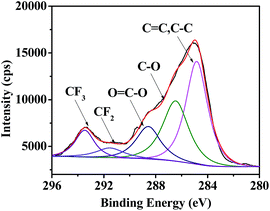 |
| | Fig. 12 XPS of C1s for the coating surface of fluoropolymer 2f on a glass slide. | |
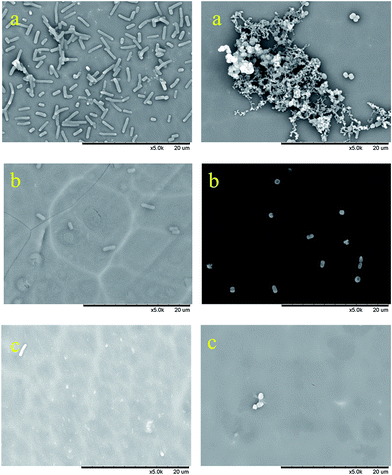 |
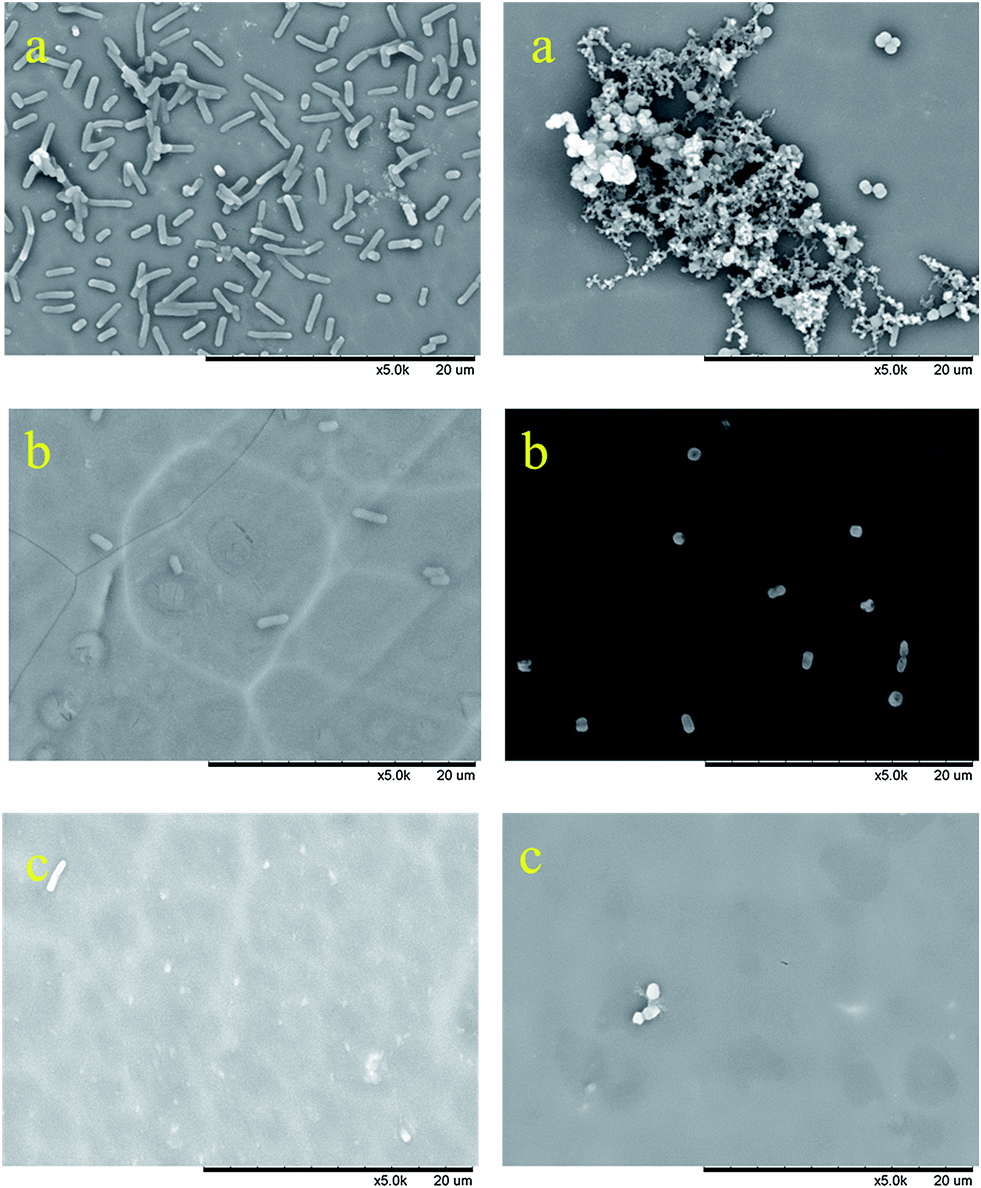
| | Fig. 13 SEM images of biofouling assay of coatings with E. coli (left) and S. aureus (right), (a) blank sample, (b) polymer 2i, and (c) polymer 2f. | |
To better explore the influence of fluorine content on the system, the coating surfaces were further studied by quantitative analysis, using the following equation:23
where
Ci is the percentage of an element,
Ii is the spectral intensity of an element which is usually expressed by peak area, ASF
i is the sensitive factor of an element (in a Kratos XSAM800 XPS spectrometer, the F
1S, C
1S and O
1S sensitivities were 1, 0.25 and 0.66 respectively). As listed in
Table 6, the fluorine content of coating surfaces with fluoropolymer
2a and
2f was 1.76% and 5.94%, higher than that of polymer
2a and
2f, indicating that the fluorine-containing segments enrich the coating surface.
Table 6 Elemental comparison of coating surfaces with fluoropolymer 2a and 2f
| Coating surface |
Elements (%, XPS) |
Fluorine ratioa (%) |
| C (%) |
O (%) |
F (%) |
| Fluorine ratio was obtained from elemental analyses. |
| 2a |
55.81 |
22.52 |
21.67 |
19.91 |
| 2f |
50.02 |
21.55 |
28.43 |
22.49 |
3.4 Biofouling assays
In this study, adhesion assays on Gram-negative (E. coli) and Gram-positive (S. aureus) bacteria were carried out to test whether the fluoropolymer coatings exhibited any antibiofouling effects.24,25 Fig. 13a–c represent the biofouling assay with E. coli (left) and S. aureus (right) on coatings with fluoropolymers containing 0% of fluorinated monomer (Fig. 13a), 5 wt% of monomer 1b (Fig. 13b), 33 wt% of monomer 1b (Fig. 13c). As shown in Fig. 13a, a large number of E. coli and S. aureus were adhered to the uncoated aluminum plate, while the number of bacteria on the fluoropolymer coated surface is dramatically decreased, as can be seen from Fig. 13b and c. These results indicated that the surface free energy and the adhesion of bacteria on the coating surface are both decreased significantly with the increase of fluorine content in the polymers. Meanwhile, the coatings containing perfluoropolyether chains shows higher antifouling activity than those containing short perfluoroalkyl chains (for the SEM images of biofouling assay of coatings of polymer 2a and 2d containing short perfluoroalkyl chains see the ESI, Fig. 39 and 40†). The antifouling ability of the hydrophobic surfaces constructed by the fluoropolymers containing perfluoroalkyl and perfluoropolyether chains can thus be verified.
4 Conclusions
Functional cross-linkable fluoropolymers composed of short fluoroalkyl groups or perfluoropolyether substituted fluorinated styrene, MMA and GMA were designed and synthesized through a free radical polymerization. XPS, TGA, contact angle measurements and germ adhesion assays were carried out to explore the surface compositions, thermal stability, hydrophobicity and antifouling ability. The fluoropolymers show good thermal stability. The hydrophobicity of the fluoropolymer coatings is enhanced with the increase of fluorine-containing monomer content in the copolymers. When a fluorine atom is introduced into the benzene ring, the stability and hydrophobicity of the fluoropolymers are slightly improved. In the germ adhesion test, the obtained fluoropolymer coatings exhibit an excellent antifouling ability. The incorporation of different amounts of fluorine-containing monomer into the copolymers reveals the excellent antibacterial activity against E. coli and S. aureus. Comparing the perfluoropolyether substituted copolymer with the short fluoroalkyl chain substituted copolymer, the former exhibits better thermal stability, hydrophobicity and antifouling effects than the latter. Therefore, in view of environmental concerns, the properties of the coatings containing perfluoropolyether chains meet the standards of green chemistry, and show far-reaching environmentally benign significance.
Acknowledgements
This work was supported by the Shanghai Municipal Education Commission (No. 13ZZ047) and Fundamental Research Funds for the Central Universities (2232015D3-13).
References
- E. Martinelli, D. Gunes, B. M. Wenning, C. K. Ober, J. A. Finlay, M. E. Callow, J. A. Callow, A. D. Fino, A. S. Clare and G. Galli, Biofouling, 2016, 32, 81–93 CrossRef PubMed.
- Y. Zhang, Y. Qi, Z. Zhang and G. Sun, J. Coat. Technol. Res., 2015, 12, 215–223 CrossRef CAS.
- Q. H. Sun, H. Q. Li, C. Y. Xian, Y. H. Yang, Y. X. Song and P. H. Cong, Appl. Surf. Sci., 2015, 344, 17–26 CrossRef CAS.
- A. Vaterrodt, B. Thallinger, K. Daumann, D. Koch, G. M. Guebitz and M. Ulbricht, Langmuir, 2016, 32, 1347–1359 CrossRef CAS PubMed.
- M. Zanoni, O. Habimana, J. Amadio and E. Casey, Biotechnol. Bioeng., 2016, 113, 501–512 CrossRef CAS PubMed.
- K. A. Pollack, P. M. Imbesi, J. E. Raymond and K. L. Wooley, ACS Appl. Mater. Interfaces, 2014, 6, 19265–19274 CAS.
- D. Pavlović, S. Lafond, A. Margaillan and C. Bressy, Polym. Chem., 2016, 7, 2652–2664 RSC.
- J. Tan, W. Liu, H. Wang, Y. Sun and S. Wang, J. Appl. Polym. Sci., 2016, 133, 43116 Search PubMed.
- K. S. Kim, N. Gunari, D. MacNeil, J. Finlay, M. Callow, J. Callow and G. C. Walker, ACS Appl. Mater. Interfaces, 2016, 8, 20342–20351 CAS.
- C. J. Kavanagh, R. D. Quinn and G. W. Swain, J. Adhes., 2005, 81, 843–868 CrossRef CAS.
- J. Zhong, H. Ji, J. Duan, H. Tu and A. Zhang, Colloids Surf., B, 2016, 140, 254–261 CrossRef CAS PubMed.
- J. Tan, W. Liu, H. Wang, Y. Sun and S. Wang, Prog. Org. Coat., 2016, 94, 62–72 CrossRef CAS.
- X. Li, L. Andruzzi, E. Chiellini, G. Galli, C. K. Ober, A. Hexemer, E. J. Kramer and D. A. Fischer, Macromolecules, 2002, 35, 8078–8087 CrossRef CAS.
- C. Pugh, C. N. Tang, M. Paz-Pazos, O. Samtani and A. H. Dao, Macromolecules, 2007, 40, 8178–8188 CrossRef CAS.
- J. C. Yarbrough, J. P. Rolland, J. M. DeSimone, M. E. Callow, J. A. Finlay and J. A. Callow, Macromolecules, 2006, 39, 2521–2528 CrossRef CAS.
- G. Galli, D. Barsi, E. Martinelli, A. Glisenti, J. A. Finlay, M. E. Callow and J. A. Callow, RSC Adv., 2016, 6, 67127–67135 RSC.
- Q. Zhang, Q. Wang, J. Jiang, X. Zhan and F. Chen, Langmuir, 2015, 31, 4752–4760 CrossRef CAS PubMed.
- S. Borkar, K. Jankova, H. W. Siesler and S. Hvilsted, Macromolecules, 2004, 37, 788–794 CrossRef CAS.
- S. J. Feng, Y. G. Huang, Q. Wang, Y. Gao and F. L. Qing, Polym. Eng. Sci., 2010, 50, 944–951 CAS.
- R. Dorigo, A. M. Garnault, D. Teyssie and S. Boileu, Integration of Fundamental Polymer Science and Technology-2, Springer, Netherlands, 1988. pp. 90–93 Search PubMed.
- L. Ruiz, J. G. Hilborn, D. Leonard and H. J. Mathieu, Biomaterials, 1998, 19, 987–998 CrossRef CAS PubMed.
- Z. K. Hu, J. A. Finlay, L. Chen, D. E. Betts, M. A. Hillmyer, M. E. Callow, J. A. Callow and J. M. DeSimone, Macromolecules, 2009, 42, 6999–7007 CrossRef CAS.
- Y. Gao, C. L. He, Y. H. Huang and F. L. Qing, Polymer, 2010, 51, 5997–6004 CrossRef CAS.
- P. Hu, T. Jiang, H. Ni, P. Ye, Z. Han, Z. Zhao, C. Zhu and X. Lu, Polym. Bull., 2016, 73, 1405–1426 CrossRef CAS.
- X. Chen, G. F. Zhang, Q. H. Zhang, X. L. Zhan and F. Q. Chen, Ind. Eng. Chem. Res., 2015, 54, 3813–3820 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26409c |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*