
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyethylene glycol 400 (PEG400) affects the systemic exposure of oral drugs based on multiple mechanisms: taking berberine as an example
Bing-Liang Ma†
a,
Yan Yang†a,
Yan Daia,
Qiao Lia,
Ge Lin*b and
Yue-Ming Ma*a
aDepartment of Pharmacology, School of Pharmacy, Shanghai University of Traditional Chinese Medicine, 1200 Cai Lun Road, Shanghai, 201203, China. E-mail: mayueming_117@hotmail.com; Fax: +86-21-5132-2386; Tel: +86-21-5132-2386
bSchool of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong, Lo Kwee-Seong Integrated Biomedical Sciences Building, Room 505A, Area 39, Shatin, Hong Kong, China. E-mail: linge@cuhk.edu.hk; Fax: +86-852-2603-5123; Tel: +86-852-3943-6824
First published on 12th January 2017
Abstract
Polyethylene glycol 400 (PEG400) is a commonly used co-solvent. Recent studies suggested that it might affect the systemic exposure of oral drugs based on mechanisms besides increasing their solubility. In this study, we systematically explored the dose related effects and underlying mechanisms of PEG400 on the systemic exposure of oral berberine, a new drug candidate having multiple pharmacological effects. The results showed that low concentration (5% and 15%) PEG400 decreased but high concentration (50%) PEG400 increased the absorption of berberine across mouse gut sacs by 6.5-fold. As expected, 50% PEG400 increased the solubility of berberine from 1.7 to 10.5 mg mL−1. Surprisingly, 50% PEG400 reversibly opened intestinal paracellular tight junctions, which was verified by the increased absorption of the marker compound, fluorescein isothiocyanate-labeled dextran (molecular weight [Mw]: 4400 g mol−1, FD4). However, 50% PEG400 did not influence the intestinal efflux, liver uptake, or hepatic metabolism of berberine. The systemic exposure (AUC0–4 h) of berberine in mice receiving orally administered berberine (200 mg kg−1) dissolved in water or 50% PEG400 reached 30.6 ng h mL−1 or 188.1 ng h mL−1, respectively. The enhancive effect of PEG400 was significantly inhibited by cycloheximide pretreatment. In conclusion, our data showed that high concentration of PEG400 increased the systemic exposure of berberine by increasing its solubility, prompting its intestinal paracellular absorption and lymphatic transport. Our results provide new insights into the effects and mechanisms of PEG400 on the systemic exposure of oral drugs. In addition, the results would encourage further exploration on the application of PEG400 in drug delivery.
Introduction
Solubility is one of the fundamental parameters controlling oral drug absorption.1 Poor aqueous solubility usually leads to limited systemic exposure of oral drugs. Various solubility-enhancing approaches have been developed and are routinely used to tackle this problem.2 The application of co-solvent, such as polyethylene glycol 400 (PEG400),2 is one of the commonly used techniques for improving solubility.PEG400 is usually considered as inert.3 However, recent studies have revealed that this compound has various bioactivities. For example, PEG400 inhibits drug efflux transporters such as p-glycoprotein (P-gp)4 and uptake transporters like organic anion-transporting polypeptide (OATP) 1A2.5 PEG400 also inhibits drug metabolizing enzymes, including intestinal6 and hepatic cytochrome P450 3A (CYP3A).7 In addition, PEG400 accelerates gastrointestinal transit.8,9 Other studies have shown that PEG400 affects the permeability of drugs. For example, PEG400 decreases the apparent permeability of carbamazepine by reducing its thermodynamic activity and by reversing solvent drug.10,11 In contrast, PEG400 increases the permeability of testosterone across a phospholipid vesicle-based barrier.12 Bendels et al.13 reported that PEG400 causes enhanced permeability of weak acids and neutral compounds but depressed permeability of weak bases. Thus, the diverse effects of PEG400 on solubility, permeability, drug metabolizing enzymes, transporters, and gastrointestinal transit time may have profound influences on the intestinal absorption and systemic exposure of oral drugs. However, although PEG400 is widely used, systematic studies on the influences of PEG400 on the systemic exposure of oral drugs and underlying mechanisms have not been reported.
Berberine (Fig. 1), an isoquinoline alkaloid isolated from medicinal plants including plants of the family Ranunculaceae, such as Coptis chinensis Franch,14 has been reported to have multiple pharmacological effects,15 including antidiabetic,16 lipid-reducing,17 anticancer,18 anti-inflammatory,19 antidiarrheal,20 and antimicrobial21 activities. Berberine was introduced in clinical practice in Europe in 2014 because of its beneficial effects on treating type 2 diabetes mellitus and improving lipid metabolism in patients.22 However, due to its poor aqueous solubility,23 extensive first pass metabolism,24 and efflux by intestinal25 and hepatic P-gp,26 the systemic exposure of berberine is extremely low (0.36%);24 this dramatically limits its clinical applications. Thereby, a lot of strategies have been developed to promote the intestinal absorption of oral berberine.27 PEG400 has been used with other excipients to increase the systemic exposure of oral berberine.28,29 However, the effects of PEG400 alone and underlying mechanisms have not been systematically evaluated.
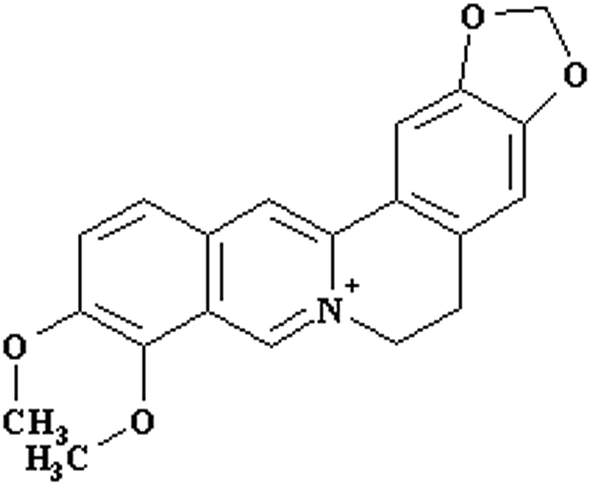 | ||
| Fig. 1 Structure of berberine. | ||
In this study, we aimed to systematically evaluate the concentration related effects and underlying mechanisms of PEG400 on the intestinal absorption and systemic exposure of oral drugs taking berberine as an example. Although the result of this study would not directly encourage the clinical applications of high concentration of PEG400, it does provide new insights into the pharmaceutical effects of PEG400.
Materials and methods
Materials
Berberine hydrochloride and palmatine hydrochloride with purities of more than 98% were obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Berberrubine and demethyleneberberine with purities of more than 98% were synthesized and identified as reported.30 Polyethylene glycol 400 (PEG400), fluorescein isothiocyanate-labeled dextran (molecular weight [Mw]: 4400 g mol−1, FD4), the QuantiPro bicinchoninic acid (BCA) assay kit, and reduced nicotinamide adenine dinucleotide phosphate (NADPH) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The lactate dehydrogenase (LDH) activity assay kit was purchased from keyGEN BioTECH (Nanjing, China). Acetonitrile was the product of Merck (Darmstadt, Germany). Pure water used in the current study was prepared using a Milli-Q system (Millipore Corporation, Billerica, MA, USA). All other materials were of analytical grade or better.Animals
Grade II male and female Kun-Ming (KM) mice (22–24 g) were purchased from Shanghai Slac Laboratory Animal Co., Ltd. (Shanghai, China). The mice were housed in an air-conditioned room at 22–24 °C under a 12 h dark/light cycle and given food and water ad libitum. The mice were fasted overnight before the experiments. All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of Shanghai University of Traditional Chinese Medicine (Approval Number: 2015007), and all experiments were performed according to the guideline of this committee.Validated high-performance liquid chromatography (HPLC) with ultraviolet (UV) detection30
The HPLC was equipped with a 2695 separation module, a 2487 dual-wavelength absorbance detector, and an Empower2 Chemstation (Waters, Milford, MA, USA). Separation and determination were performed using an XTerra RP 18 analytical column (5 μm, 3.9 × 150 mm; Waters) at 35 °C, with detection at 346 nm. The samples were eluted through the column with a gradient of water/formic acid/triethylamine (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1:0.2, v/v/v) and methanol (0 min, 75:25; 15 min, 60:40; 15.5 min, 75:25; 20 min, 75:25) at a flow rate of 0.5 mL min−1. The standard sets had good linearity within their own quantitative range [0.078–2.5 μM for berberrubine and demethyleneberberine, two major phase I metabolites of berberine30]. The accuracy, precision, recovery, and stability tests all met the requirements of the quantitative determination in biological samples.
:0.1:0.2, v/v/v) and methanol (0 min, 75:25; 15 min, 60:40; 15.5 min, 75:25; 20 min, 75:25) at a flow rate of 0.5 mL min−1. The standard sets had good linearity within their own quantitative range [0.078–2.5 μM for berberrubine and demethyleneberberine, two major phase I metabolites of berberine30]. The accuracy, precision, recovery, and stability tests all met the requirements of the quantitative determination in biological samples.
Validated liquid chromatography tandem mass spectrometry (LC-MS/MS)30
Briefly, a Shimadzu Prominence UFLCXR series HPLC (Shimadzu, Japan) and a Thermo Scientific TSQ Quantum Ultra mass spectrometer (Thermo Scientific, Waltham, MA, USA) equipped with an electrospray ionization (ESI) source were used. To avoid the ion suppression caused by PEG400,31 palmatine, a homologous compound of berberine, was used as internal standard. The samples were precipitated with three volumes of acetonitrile. After centrifugation (24000 × g, 6 min, 4 °C), the supernatant was mixed with an equal volume of water, and 10 μL samples were injected into the LC-MS/MS system. The samples were eluted through a Hypersil Gold (C18) analytical column (5 μm, 100 × 2.1 mm) with a gradient of the aqueous phase (0.08% v/v formic acid and 2 mM ammonium acetate) and the acetonitrile phase (0 min, 85:15; 7 min, 32:68; 7.01 min, 85:15; 10 min, 85:15) at a flow rate of 0.3 mL min−1. The ESI source was set to positive ion mode. Data acquisition was performed in the multiple reaction monitoring mode of the selective mass transition for each compound. The transitions from the precursor ions to the protonated fragment product ions were monitored as follows: m/z 336.2 to m/z 322.3 for berberine; m/z 352.1 to m/z 336.1 for palmatine; m/z 322.1 to m/z 307.2 for demethyleneberberine; and m/z 324.1 to m/z 308.2 for berberrubine. The linear dynamic ranges in the biological samples were 0.98 to 500 ng mL−1 for berberine and 12.5 to 250 ng mL−1 for berberrubine and demethyleneberberine. The accuracy, precision, recovery, and stability tests all met the requirements of the quantitative determination in biological samples.
Mouse gut sacs studies30
Solubility of berberine in PEG400 solution
We analyzed the solubility of berberine in high-concentration PEG400 as previously described.32 Briefly, 10 mg berberine was added to 1 mL water or 50% PEG400. The berberine concentrations were in accordance with those used in the pharmacokinetic studies. The suspensions were subjected to sonication for 1 h and then kept at room temperature (about 22 °C) for 5 h. The samples were centrifuged at 24000 × g for 6 min. The supernatants were collected and diluted, and the berberine concentration in each sample was measured using the LC-MS/MS method. All experiments were performed in triplicate.
Effects of PEG400 on the in vitro metabolism of berberine30
In brief, reaction conditions, including the protein concentration and the incubation time, were optimized to ensure the linearity of the reactions.30 The reactions were conducted in 100 mM PBS buffer (pH 7.4) containing 1 mM NADPH, 0.5 mg of microsomal protein, various concentration of berberine (3.625, 6.25, 12.5, 25, 50, 100, 200, and 400 μM), and various concentration of PEG400 (0%, 0.3%, 1%, and 3%). The concentrations of PEG400 were chosen according to its in vivo exposure levels after receiving oral PEG400.33 After pre-incubation for 5 min, the reaction was initiated with addition of the NADPH solution. Samples were then incubated at 37 °C for 15 min, and the reactions were terminated by the addition of an equal volume of ice-cold methanol. After centrifugation (16000 rpm, 5 min, 4 °C), the samples were analyzed using the HPLC method. All reactions were performed in triplicate.
Effects of PEG400 on the in situ liver uptake of berberine34
Briefly, under light ether anesthesia with diethyl ether, berberine (100 μM) dissolved in saline or various concentrations of PEG400 (0.5%, 1.5%, and 5%, 0.04 mL kg−1 body weight) was injected rapidly into the portal vein of mice. The concentrations of PEG400 were chosen according to its in vivo exposure levels after receiving oral PEG400.33 The liver was excised 18 s later and weighed. The livers were homogenized, and the concentrations of berberine were detected using the LC-MS/MS method.Pharmacokinetics of berberine in mice
000 × g, 6 min, 4 °C), the plasma samples were collected, and the concentration of berberine was detected using the LC-MS/MS method.Assessment of intestinal membrane damage
The release of protein and LDH from the intestinal epithelial cells was measured as a marker of intestinal membrane damage as reported previously.38 Mice were divided into two groups (n = 4 mice per group: two males and two females). The mice were orally administered water or 50% PEG400 aqueous solution. The intestines were collected 4 h later and washed with blank the Krebs–Ringer buffer. LDH activity of the perfusate was determined using an LDH activity assay kit, and protein concentrations were determined using a QuantiPro BCA assay kit with bovine serum albumin as the standard.Statistical analysis
Results were expressed as the mean ± SD, and statistical significance was determined with one-way or two-way analysis of variance (ANOVA) for multiple comparisons with the minimum level of significance (P < 0.05).Results
Effects of PEG400 on the absorption and efflux of berberine across mouse gut sacs
As shown in Fig. 2A, the absorption of berberine at 60 min were 3.8 ± 3.1, 1.2 ± 0.6, 2.7 ± 1.3, 25.3 ± 8.7, and 0.19 ± 0.17 ng mL−1 cm sac in control, various concentrations of PEG400 (5%, 15%, or 50%) treated, and 50% PEG400 pretreated groups, respectively. The results showed that 5% PEG400 decreased while 50% PEG400 increased the absorption of berberine across mouse gut sacs (both p < 0.01). However, 15% PEG400 did not have any significant effects on berberine absorption (p > 0.05). Interestingly, pretreatment with 50% PEG400 decreased the absorption of berberine (p < 0.01).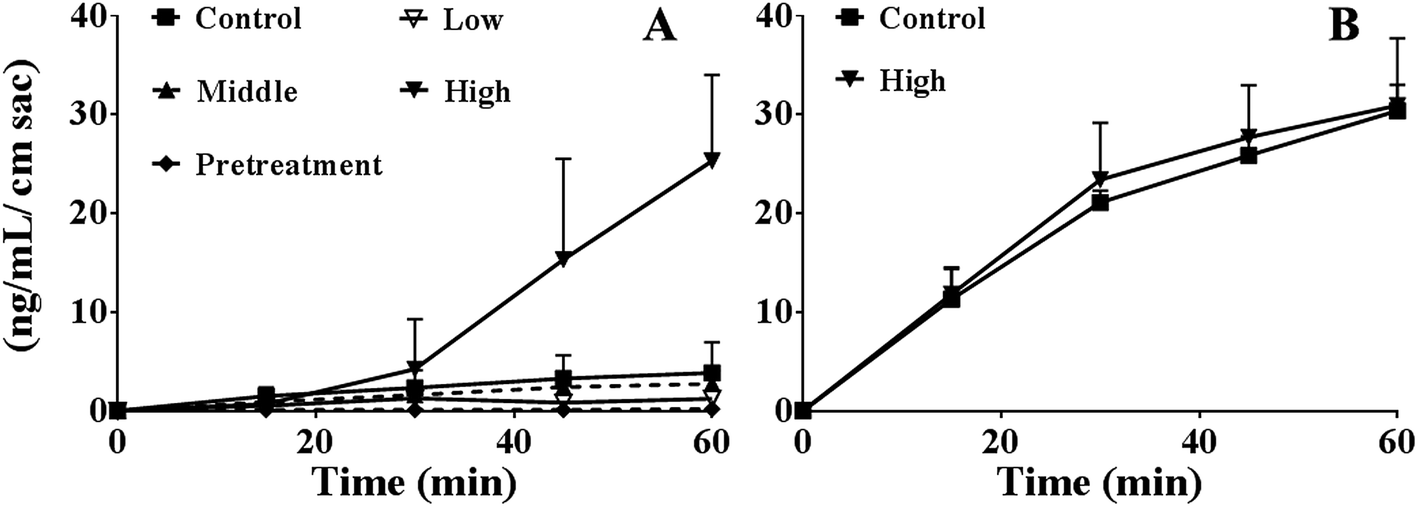 | ||
| Fig. 2 Effects of PEG400 on the permeability (A) and efflux (B) of berberine (50 μM) across mouse gut sacs (mean ± SD, n = 3–8). (A) Permeability of berberine across mouse gut sacs in the presence of various concentrations (low, middle, and high: 5%, 15%, and 50%, respectively) or after pretreatment with 50% PEG400 solution. (B) Efflux of berberine (50 μM) across mouse gut sacs in the presence of 50% PEG400 solution. | ||
As shown in Fig. 2B, the efflux of berberine at 60 min were 30.4 ± 2.6 and 30.9 ± 6.8 ng mL−1 cm sac in the control and 50% PEG400-treated groups, respectively. The results showed that 50% PEG400 did not inhibit the efflux of berberine across the gut sacs (p > 0.05).
Effects of high-concentration PEG400 on the absorption of FD4 across mouse gut sacs
As shown in Fig. 3A, the absorption of FD4 at 60 min were 0.38 ± 0.19, 0.24 ± 0.06, 0.18 ± 0.07, and 5.0 ± 2.0 ng mL−1 cm sac in control, 5%, 15%, or 50% PEG400 treated groups, respectively. The results showed that 50% PEG400 increased the absorption of FD4 across mouse gut sacs (p < 0.01).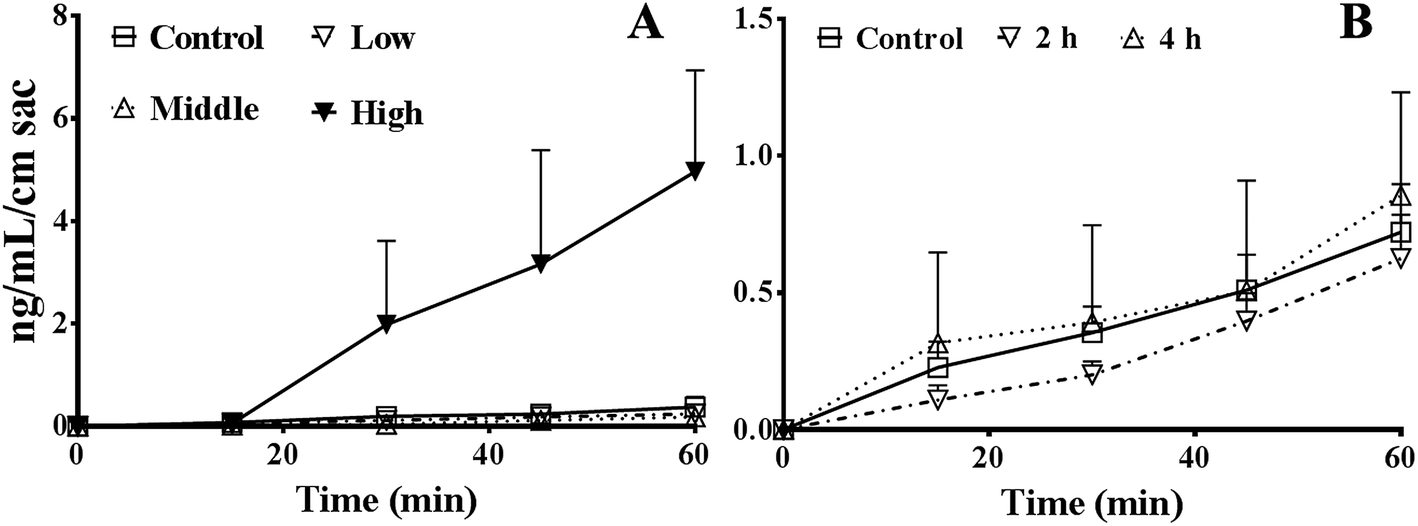 | ||
| Fig. 3 Effects of PEG400 on the permeability of FD4 (3 mg mL−1) across mouse gut sacs (mean ± SD, n = 3–4). (A) Permeability of FD4 across mouse gut sacs in the presence of various concentrations (low, middle, and high: 5%, 15%, and 50%, respectively) of PEG400. (B) Permeability of FD4 across mouse gut sacs after pretreatment with 50% PEG400 solution for 2 or 4 h. | ||
As shown in Fig. 3B, the absorption of FD4 at 60 min were 0.72 ± 0.18, 0.63 ± 0.16, and 0.85 ± 0.38 ng mL−1 cm sac in control mice and mice pretreated with 50% PEG400 for 2 or 4 h, respectively. The results showed that pretreatment with 50% PEG400 for 2 h decreased the absorption of FD4 (Fig. 3B, p < 0.05).
Effects of PEG400 on intestinal damage
As shown in Fig. 4, the total intestinal protein levels were not increased at 4 h after oral administration in the presence of 50% PEG400 (p > 0.05). The activities of LDH in the intestinal perfusates of both the control and 50% PEG400-treated groups were barely detectable.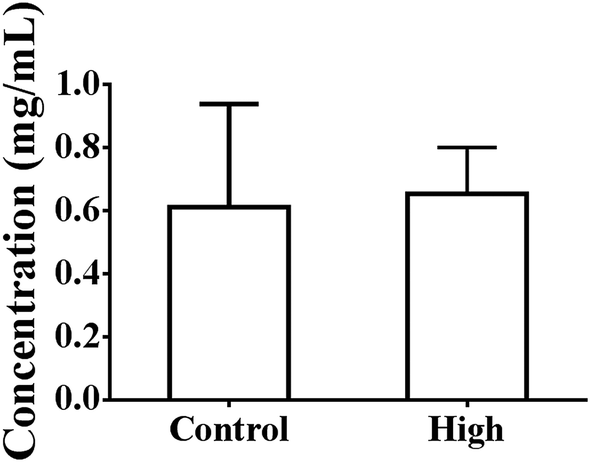 | ||
| Fig. 4 Total intestinal protein levels at 4 h after oral administration of PEG400 in mice (mean ± SD, n = 4). Low and high: 5% and 50% PEG400, respectively. | ||
Effects of PEG400 on the in situ hepatic uptake of berberine
As shown in Fig. 5, PEG400 did not influence the in situ hepatic uptake of berberine (p > 0.05).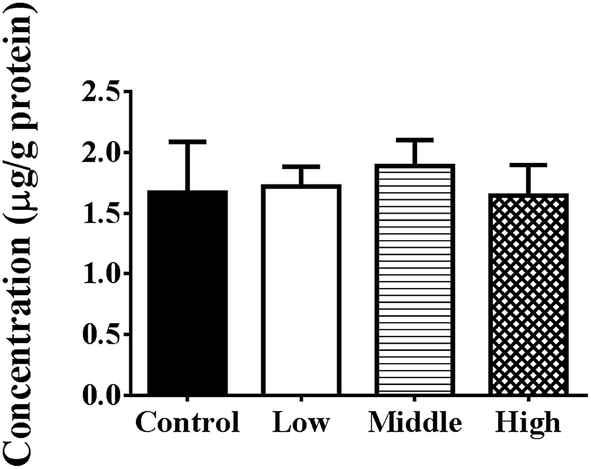 | ||
| Fig. 5 Effects of various concentrations of PEG400 on the in situ uptake of berberine in the liver (mean ± SD, n = 4). Berberine (100 μM) was dissolved in saline or various concentrations of PEG400 (0.5%, 1.5%, and 5%; 0.04 mL kg−1 body weight) and was injected rapidly into the portal vein of mice under light ether anesthesia with diethyl ether. The liver was excised 18 s later and weighed. The livers were then homogenized, and the concentration of berberine was detected using LC-MS/MS. | ||
Effects of PEG400 on the phase I metabolism of berberine
As shown in Fig. 6A and B, PEG400 significantly inhibited the formation of the major in vitro phase I metabolites (berberrubine and demethyleneberberine) of berberine in mouse hepatic microsomes in a concentration-dependent manner (both p < 0.01). However, the calculated metabolizing indexes, i.e., the ratios of the formed phase I metabolites (berberrubine plus demethyleneberberine) to the amount of berberine plus the formed metabolites in the plasma, showed that the in vivo metabolism of berberine was not influenced by PEG400 (p > 0.05, Fig. 6C).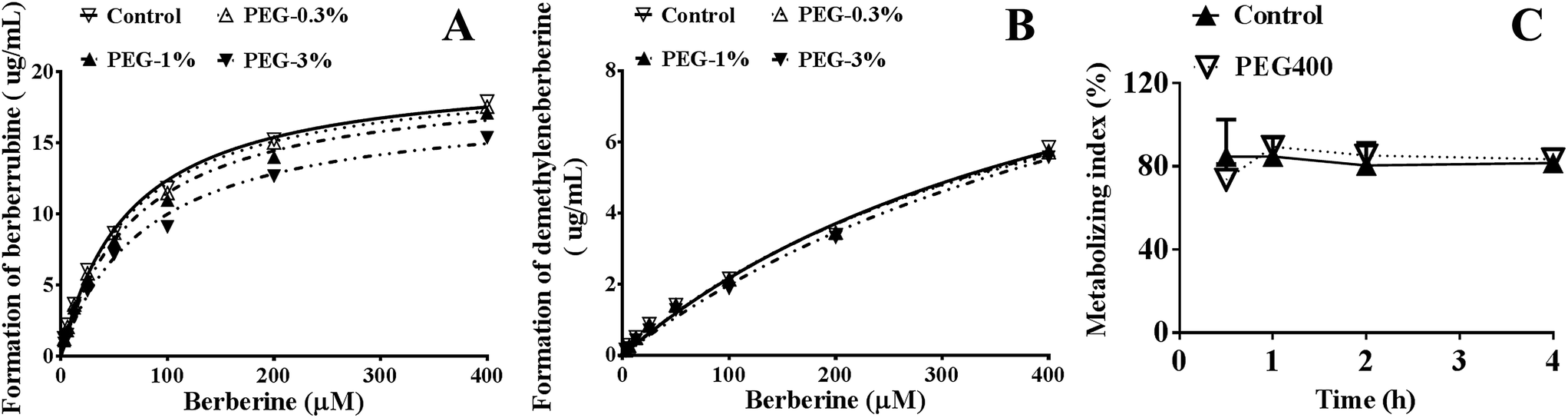 | ||
| Fig. 6 Effects of PEG400 on the in vitro and in vivo metabolism of berberine (mean ± SD, n = 3 for (A) and (B), n = 6 for (C)). (A and B) Effects of various concentrations of PEG400 (0.3%, 1%, and 3%) on the in vitro metabolism of berberine in mouse hepatic microsomes. (C) Effects of high-concentration (50%) PEG400 on the in vivo metabolic indexes of berberine. Data are shown as the ratios of the formatted phase I metabolites (berberrubine plus demethyleneberberine) to the amount of berberine plus the formed metabolites in plasma. | ||
Effects of PEG400 on the solubility of berberine
As shown in Fig. 7, berberine was completely dissolved in 50% PEG400 (10.5 mg mL−1), while its solubility in water was much lower (p < 0.01, 1.7 mg mL−1).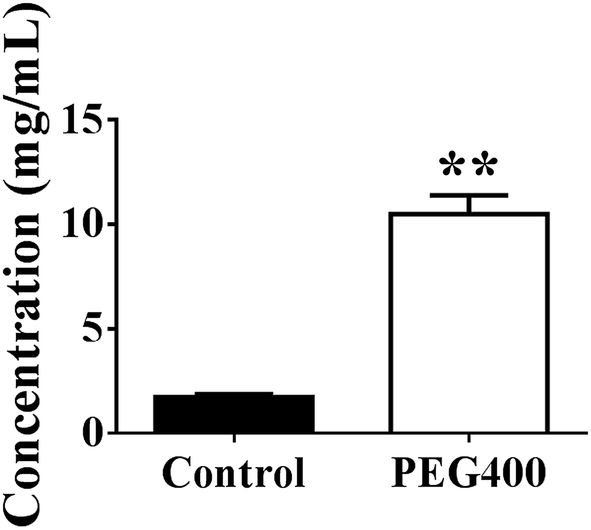 | ||
| Fig. 7 Solubility of berberine dissolved in water or 50% PEG400 (mean ± SD, n = 3). Ten milligrams of berberine was added to 1 mL water or 50% PEG400. The suspensions were then subjected to ultrasound for 1 h followed by sitting at room temperature (about 22 °C) for 5 h. The samples were centrifuged at 24000 × g for 6 min, and the supernatants were collected and measured using LC-MS/MS. ** p < 0.01 versus the control. | ||
Effects of PEG400 on the systemic exposure of berberine and related metabolites
As shown in Fig. 8A, the systemic exposure level (AUC0–4 h) of berberine in mice was largely increased by 50% PEG400 (p < 0.01, from 30.6 to 188.0 ng h mL−1). As shown in Fig. 8B, the total amount of berberine plus its related metabolites (berberrubine and demethyleneberberine) was also increased. The AUC0–4 h increased from 171.0 to 1158.1 ng h mL−1 (p < 0.01).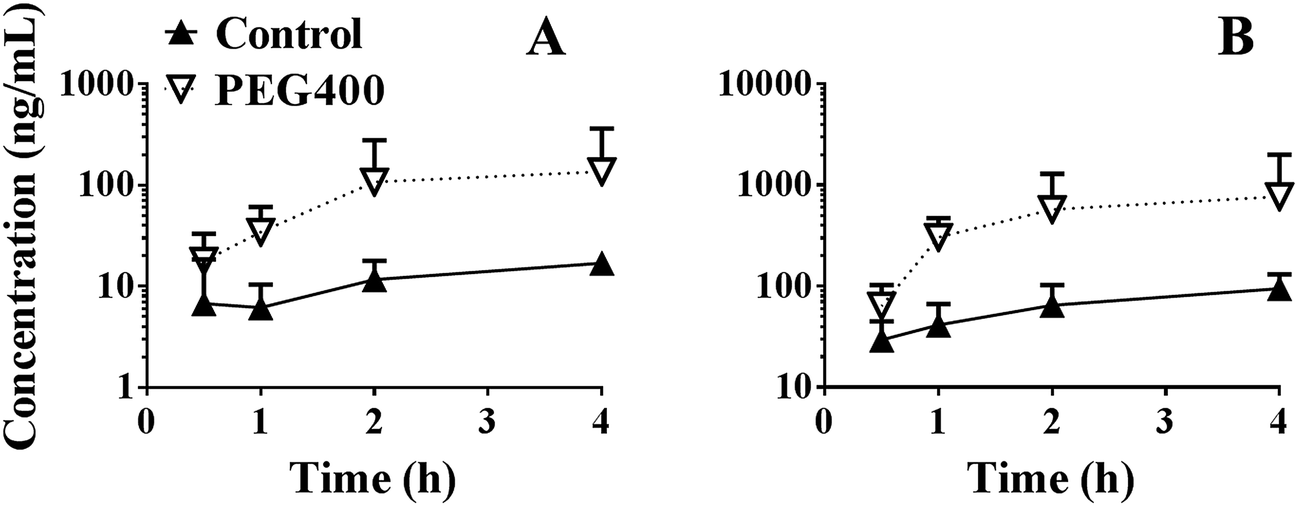 | ||
| Fig. 8 Effects of PEG400 on the systemic exposure of berberine (A) and total amount of berberine plus its phase I metabolites (B) (mean ± SD, n = 6). Mice were orally administered 200 mg kg−1 berberine dissolved in water or 50% PEG400. Blood was collected into heparinized tubes at the designated time points (0.5, 1, 2, and 4 h). The plasma was collected and analyzed using LC-MS/MS. | ||
Effects of cycloheximide pretreatment on the promotive effects of PEG400
As shown in Fig. 9, cycloheximide pretreatment significantly decreased the effects of PEG400 on the systemic exposure of berberine after oral administration in mice. The AUC0–3 h decreased from 388.0 to 106.2 ng h mL−1 (p < 0.01).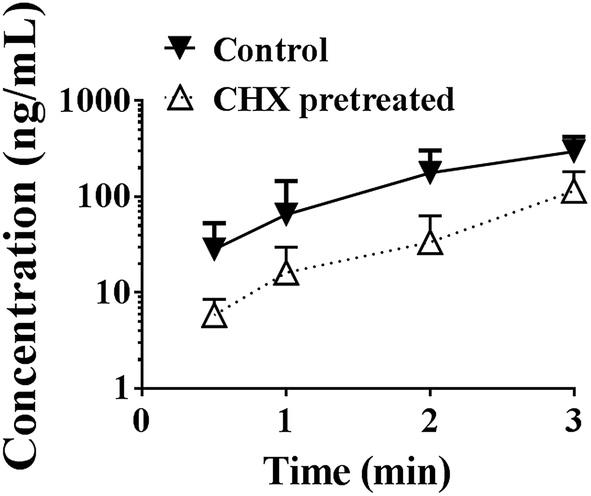 | ||
| Fig. 9 Effects of chylomicron flow blocking on the effects of PEG400 on systemic exposure of oral berberine in mice (mean ± SD, n = 6). Mice received intraperitoneal injection of saline or cycloheximide (10 mg kg−1). Two hours later, the mice were orally administered 200 mg kg−1 berberine dissolved in 50% PEG400 at the dose volume of 0.2 mL/10 g body weight. Plasma samples were prepared and analyzed. CHX, cycloheximide. | ||
Discussion
Given the extremely low bioavailability of berberine (0.36%) which is mainly caused by its extensive metabolism,24 slight inhibition on metabolizing capacity towards berberine may lead to a significant increase in its systemic exposure. PEG400 was reported to inhibit drug metabolizing enzymes, particularly phase I enzymes.6,7 Therefore, it was likely that the effect of PEG400 on the enhanced exposure of berberine would be associated with its effects on drug metabolizing enzymes. However, our results revealed that although PEG400 inhibited the in vitro metabolism of berberine in a concentration-dependent manner, it did not influence the in vivo metabolism of berberine. Although the detail mechanism remains to be elucidated, the inconsistent outcomes between in vitro and in vivo studies might be associated with the intrinsic drawbacks of the in vitro test systems.39 For example, liver microsomes, the most widely used in vitro system for drug metabolism and inhibition studies, do not represent the true in vivo situation because they contain only the endoplasmic reticulum-localized enzymes, which means that metabolism via other enzymes will not be detected with this system.40PEG400 has been reported to inhibit the function of P-gp,4 which pumps out berberine during its intestinal absorption.25 We also found that PEG400 (0.1–5%) decreased the efflux of berberine from HepG2 and Caco-2 cells in a concentration-dependent manner (data not shown). However, our in vitro absorption results indicated that increased berberine absorption across gut sacs was not due to decreased berberine efflux. Given that PEG400 is a potent inhibitor of both OATP1A2 (ref. 5) and P-gp,4 which mediate the uptake41 or excretion of berberine,26 respectively, we hypothesize that these results may be explained by the concerted inhibition of both OATP1A2 and P-gp.
In general, drugs move across epithelial barriers by the transcellular and the paracellular pathway. Hydrophilic drugs usually take the second route, which is regulated by the tight junctions (TJs). Strategies which open TJs by using chemical permeation enhancers in a reversible manner for paracellular drug delivery has got arisen interest.42 FD4 is primarily transported across the intestinal membrane via a paracellular pathway, allowing for easy estimation of intestinal epithelium barrier function.30,38 Our results confirmed with the intestinal transport of FD4 suggested that the effects of PEG400 on the enhanced absorption of berberine resulted from the reversible opening of paracellular TJs. These results were consistent with a report by Nakanishi et al. (1983), who found that the decreased rectal permeability induced by 50% PEG400 returned to the normal state within 2 h.43 TJs are composed of proteins including transmembrane proteins (like occludin and claudins) and cytoplasmic plaque proteins (like ZO-1, ZO-2, ZO-3, cingulin, and 7H6).44 Chemical permeation enhancers may act directly and specifically on the extracellular loops of TJ proteins and the TJ-associated membrane microdomains.44 However, the mechanism of PEG400 in terms of opening the paracellular TJs remained to be elucidated.
After being absorbed and transiting across the enterocytes, drugs enter into the blood capillaries or lymph capillaries.45 The former is the more common pathway because the fluid flow of the portal blood is 500-fold higher than that of the intestinal lymph.45 However, the latter pathway is more permeable, and drugs transported via the intestinal lymphatic system can bypass the liver and thus avoid hepatic first-pass metabolism.45 Indeed, lymphatic drug transport has been reported to be a contributor to the oral bioavailability of various lipophilic drugs and xenobiotics.45 Cycloheximide pretreatment is a widely used method to block chylomicron flow and does not affect other absorption pathways.37,46–48 Our results confirmed by cycloheximide pretreatment indicated that the lymphatic system played an important role in the enhanced absorption of berberine by PEG400. We assumed that increased absorption of berberine by PEG400 may increase berberine exposure to the lymphatic pathway, thereby contributing to the systemic exposure of orally available berberine. Given that chylomicrons consist of a core of triglyceride (85–92%) and cholesterol, and a monolayer of phospholipid (6–12%), cholesterol (1–3%) and protein (1–2%),37 lipid solubility therefore has a major role in lymphatic drug transport. For a weak acid or weak base like berberine, its lipid solubility may be affected by environmental conditions. For example, changes of pH values along the gastrointestinal tract may lead to different degree of ionization and lipid solubility of the compound. Its lipid solubility may also be affected by co-existed materials. Reportedly, polyethylene glycol 400 increased the partition coefficient of oxybenzone in a concentration dependent manner.49 We assumed that PEG400 improved the lymphatic transport of berberine via increasing its lipid solubility. However, it remained to be verified.
Oral PEG400 is generally considered a highly safe organic co-solvent.50 For example, repeated oral administration of PEG400 (5, 50, or 100%) at a volume of 5 mL per kg per day for 15 days induces mucosal changes in the stomach, but not the intestine of rats.51 In the study, no significant changes in the general condition, body weight, food consumption, hematology, or gross findings at necropsy were observed during the treatment period, with the exception of loose stools in rats treated with 50 or 100 v/v% PEG400.51 We had verified that oral 50% PEG400 at 20 mL kg−1 did not cause acute toxicity in mice (data not shown). In this study, the results of LDH and protein assays suggested that PEG400 did not cause intestinal membrane damage. Although these data were not enough to warrant the safety of high concentration of PEG400 especially in clinical applications, they did encourage more studies in this field.
Briefly speaking, our results provided new insights into the pharmaceutical effects of PEG400 from following aspects. First, our results confirmed that PEG400 is not an inert solubilizing agent as was believed,50 on the contrary, it showed profound effects on the intestinal absorption and systemic exposure of oral drugs like berberine. Second, previous studies have suggested that PEG400 shows concentration-dependent effects on the absorption of oral drugs,12,52 however, our results showed that PEG400 had biphasic effects on the absorption of oral berberine. That's to say, lower concentration of PEG400 decreased but higher concentration increased the absorption of berberine. Third, given that berberine is an alkaloid, the result was also contradictory to the study which showed that PEG400 decreases the absorption of weak bases.13 Fourth, our results discovered new mechanisms for the enhancing effects of PEG400 on oral drugs: promoting the paracellular absorption as well as lymphatic transportation of berberine.
Conclusion
In conclusion, our data showed that PEG400 could act as more than a co-solvent. Briefly, high concentration of PEG400 increased the systemic exposure of berberine by increasing its solubility, prompting its intestinal paracellular absorption and lymphatic transport. Given that PEG400 was widely considered to be inert, our results provide new insights into the effects and mechanisms of PEG400 on the systemic exposure of oral drugs. Especially, the results indicated that high concentration of PEG400 could act like chemical permeation enhancers, which reversibly promote the paracellular drug delivery. Although its potency and mechanism should be further explored and its safety should be carefully evaluated, our results did encourage further studies on the application of PEG400 in drug delivery.Conflicts of interest
The authors state no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 81202986 and No. 81273658).References
- G. L. Amidon, H. Lennernas, V. P. Shah and J. R. Crison, Pharm. Res., 1995, 12, 413–420 CrossRef CAS.
- H. D. Williams, N. L. Trevaskis, S. A. Charman, R. M. Shanker, W. N. Charman, C. W. Pouton and C. J. Porter, Pharmacol. Rev., 2013, 65, 315–499 CrossRef PubMed.
- D. A. Ashiru, R. Patel and A. W. Basit, Pharm. Res., 2008, 25, 2327–2333 CrossRef CAS PubMed.
- B. M. Johnson, W. N. Charman and C. J. Porter, AAPS PharmSciTech, 2002, 4, E40 Search PubMed.
- A. Engel, S. Oswald, W. Siegmund and M. Keiser, Mol. Pharm., 2012, 9, 2577–2581 CrossRef CAS PubMed.
- D. R. Mudra and R. T. Borchardt, J. Pharm. Sci., 2010, 99, 1016–1027 CrossRef CAS PubMed.
- M. M. Cotreau-Bibbo, L. L. von Moltke and D. J. Greenblatt, J. Pharm. Sci., 1996, 85, 1180–1185 CrossRef CAS PubMed.
- A. W. Basit, J. M. Newton, M. D. Short, W. A. Waddington, P. J. Ell and L. F. Lacey, Pharm. Res., 2001, 18, 1146–1150 CrossRef CAS.
- J. D. Schulze, W. A. Waddington, P. J. Eli, G. E. Parsons, M. D. Coffin and A. W. Basit, Pharm. Res., 2003, 20, 1984–1988 CrossRef CAS.
- L. E. Riad and R. J. Sawchuk, Pharm. Res., 1991, 8, 491–497 CrossRef CAS.
- A. Beig, J. M. Miller and A. Dahan, Eur. J. Pharm. Biopharm., 2012, 81, 386–391 CrossRef CAS PubMed.
- G. E. Flaten, K. Luthman, T. Vasskog and M. Brandl, Eur. J. Pharm. Sci., 2008, 34, 173–180 CrossRef CAS PubMed.
- S. Bendels, O. Tsinman, B. Wagner, D. Lipp, I. Parrilla, M. Kansy and A. Avdeef, Pharm. Res., 2006, 23, 2525–2535 CrossRef CAS PubMed.
- G. Fan, M. Y. Zhang, X. D. Zhou, X. R. Lai, Q. H. Yue, C. Tang, W. Z. Luo and Y. Zhang, Anal. Chim. Acta, 2012, 747, 76–83 CrossRef CAS PubMed.
- P. R. Vuddanda, S. Chakraborty and S. Singh, Expert Opin. Invest. Drugs, 2010, 19, 1297–1307 CrossRef CAS PubMed.
- H. L. Zhao, Y. Sui, C. F. Qiao, K. Y. Yip, R. K. Leung, S. K. Tsui, H. M. Lee, H. K. Wong, X. Zhu, J. J. Siu, L. He, J. Guan, L. Z. Liu, H. X. Xu, P. C. Tong and J. C. Chan, Diabetes, 2012, 61, 933–943 CrossRef CAS PubMed.
- Y. Deng, J. Xu, X. Zhang, J. Yang, D. Zhang, J. Huang, P. Lv, W. Shen and Y. Yang, Autophagy, 2014, 10, 1776–1786 CrossRef CAS PubMed.
- Q. Liu, X. Xu, M. Zhao, Z. Wei, X. Li, X. Zhang, Z. Liu, Y. Gong and C. Shao, Mol. Cancer Ther., 2015, 14, 355–363 CrossRef CAS PubMed.
- C. Spatuzza, L. Postiglione, B. Covelli, M. Ricciardone, C. Benvenuti, P. Mondola and A. Belfiore, Front. Pharmacol., 2014, 5, 230 Search PubMed.
- S. Menees, R. Saad and W. D. Chey, Nat. Rev. Gastroenterol. Hepatol., 2012, 9, 661–674 CrossRef CAS PubMed.
- S. Song, M. Qiu, Y. Chu, D. Chen, X. Wang, A. Su and Z. Wu, Antimicrob. Agents Chemother., 2014, 58, 5068–5078 CrossRef PubMed.
- G. Derosa and P. Maffioli, Curr. Top. Med. Chem., 2014, 14, 200–206 CrossRef CAS PubMed.
- S. Spinozzi, C. Colliva, C. Camborata, M. Roberti, C. Ianni, F. Neri, C. Calvarese, A. Lisotti, G. Mazzella and A. Roda, J. Nat. Prod., 2014, 77, 766–772 CrossRef CAS PubMed.
- Y. T. Liu, H. P. Hao, H. G. Xie, L. Lai, Q. Wang, C. X. Liu and G. J. Wang, Drug Metab. Dispos., 2010, 38, 1779–1784 CrossRef CAS PubMed.
- X. Zhang, F. Qiu, J. Jiang, C. Gao and Y. Tan, Xenobiotica, 2011, 41, 290–296 CrossRef CAS PubMed.
- P. L. Tsai and T. H. Tsai, Drug Metab. Dispos., 2004, 32, 405–412 CrossRef CAS PubMed.
- C. S. Liu, Y. R. Zheng, Y. F. Zhang and X. Y. Long, Fitoterapia, 2016, 109, 274–282 CrossRef CAS PubMed.
- S. Y. Gui, L. Wu, D. Y. Peng, Q. Y. Liu, B. P. Yin and J. Z. Shen, Pharmazie, 2008, 63, 516–519 CAS.
- S. Pund, G. Borade and G. Rasve, Phytomedicine, 2014, 21, 307–314 CrossRef CAS PubMed.
- B. L. Ma, M. K. Yao, J. Zhong, Y. M. Ma, C. L. Gao, J. S. Wu, F. R. Qiu, C. H. Wang and X. H. Wang, Drug Metab. Dispos., 2012, 40, 381–388 CrossRef CAS PubMed.
- R. Weaver and R. J. Riley, Rapid Commun. Mass Spectrom., 2006, 20, 2559–2564 CrossRef CAS PubMed.
- V. Anuta, G. M. Nitulescu, C. E. Dinu-Pirvu and O. T. Olaru, Molecules, 2014, 19, 16381–16401 CrossRef PubMed.
- V. Vijaya Bhaskar, A. Middha, S. Tiwari and S. Shivakumar, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 926, 68–76 CrossRef CAS PubMed.
- A. Zhang, C. Wang, Q. Liu, Q. Meng, J. Peng, H. Sun, X. Ma, X. Huo and K. Liu, Drug Metab. Dispos., 2013, 41, 994–1003 CrossRef CAS PubMed.
- W. Xie, D. Gu, J. Li, K. Cui and Y. Zhang, PLoS One, 2011, 6, e24520 CAS.
- T. Lao-ong, W. Chatuphonprasert, N. Nemoto and K. Jarukamjorn, Pharm. Biol., 2012, 50, 1007–1012 CrossRef CAS PubMed.
- A. Dahan and A. Hoffman, Eur. J. Pharm. Sci., 2005, 24, 381–388 CrossRef CAS PubMed.
- K. A. Hamid, H. Katsumi, T. Sakane and A. Yamamoto, Int. J. Pharm., 2009, 379, 100–108 CrossRef CAS PubMed.
- E. F. Brandon, C. D. Raap, I. Meijerman, J. H. Beijnen and J. H. Schellens, Toxicol. Appl. Pharmacol., 2003, 189, 233–246 CrossRef CAS PubMed.
- N. Plant, Drug Discovery Today, 2004, 9, 328–336 CrossRef CAS PubMed.
- C. Chen, Z. T. Wu, L. L. Ma, X. Ni, Y. F. Lin, L. Wang, K. P. Chen, C. G. Huang and G. Pan, Xenobiotica, 2015, 45, 1138–1146 CrossRef CAS PubMed.
- L. Gonzalez-Mariscal, Y. Posadas-Torrentera, J. Miranda, P. Uc, J. M. Ortega-Olvera and S. Hernandez, Curr. Pharm. Des., 2016, 22, 5313–5346 CrossRef CAS PubMed.
- K. Nakanishi, M. Masada and T. Nadai, Chem. Pharm. Bull., 1983, 31, 4161–4166 CrossRef CAS PubMed.
- M. A. Deli, Biochim. Biophys. Acta, 2009, 1788, 892–910 CrossRef CAS PubMed.
- J. A. Yanez, S. W. Wang, I. W. Knemeyer, M. A. Wirth and K. B. Alton, Adv. Drug Delivery Rev., 2011, 63, 923–942 CrossRef CAS PubMed.
- S. Attili-Qadri, N. Karra, A. Nemirovski, O. Schwob, Y. Talmon, T. Nassar and S. Benita, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17498–17503 CrossRef CAS PubMed.
- Z. Zhang, F. Gao, H. Bu, J. Xiao and Y. Li, Nanomedicine, 2012, 8, 740–747 CAS.
- F. Gao, Z. Zhang, H. Bu, Y. Huang, Z. Gao, J. Shen, C. Zhao and Y. Li, J. Controlled Release, 2011, 149, 168–174 CrossRef CAS PubMed.
- C. J. Mbah, Pharmazie, 2007, 62, 38–40 CAS.
- R. G. Strickley, Pharm. Res., 2004, 21, 201–230 CrossRef CAS.
- Y. Ueda, M. Tsuboi, Y. Ota, M. Makita, T. Aoshima, M. Nakajima and I. Narama, J. Toxicol. Sci., 2011, 36, 811–815 CrossRef CAS PubMed.
- A. Dahan and J. M. Miller, AAPS J., 2012, 14, 244–251 CrossRef CAS PubMed.
Footnote |
| † The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |