
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCellular-membrane inspired surface modification of well aligned ZnO nanorods for chemosensing of epinephrine†
M. A. Mohsin,
B. D. Liu *,
X. L. Zhang,
W. J. Yang,
L. S. Liu and
X. Jiang*
*,
X. L. Zhang,
W. J. Yang,
L. S. Liu and
X. Jiang*
Shenyang National Laboratory for Materials Sciences (SYNL), Institute of Metal Research (IMR), Chinese Academy of Sciences (CAS), No. 72, Wenhua Road, Shenyang, 110016, China. E-mail: baodanliu@hotmail.com; xjiang@imr.ac.cn
First published on 12th January 2017
Abstract
Neurological disorders have always triggered the interest of scientists due to the effects they have on the quality of life and the complexity they provide in their perception. Herein, we report a cellular-membrane inspired surface modification to fabricate a chemosensor for selective epinephrine detection. For this, well aligned ZnO nanorod arrays were grown on a silicon substrate using a feasible hydrothermal method and further utilized for the immobilization of a lipid membrane incorporated with calix[6]arene (in a 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) after modification with 1-octadecanthiol. Such surfaces were employed for selective epinephrine detection using electrochemical impedance spectroscopy as a transduction method. The detection process was based on the Lock & Key mechanism. A distinctive increase in charge transfer resistance was observed for epinephrine as compared to dopamine, leading to the conclusion that the sensor is sensitive and selective to epinephrine. This selectivity for epinephrine can be attributed to the slight differences in the molecular sizes of dopamine and epinephrine. Such electrochemical sensors can provide a very useful platform for the manufacturing of devices for monitoring the physiological concentrations of epinephrine.
:1 ratio) after modification with 1-octadecanthiol. Such surfaces were employed for selective epinephrine detection using electrochemical impedance spectroscopy as a transduction method. The detection process was based on the Lock & Key mechanism. A distinctive increase in charge transfer resistance was observed for epinephrine as compared to dopamine, leading to the conclusion that the sensor is sensitive and selective to epinephrine. This selectivity for epinephrine can be attributed to the slight differences in the molecular sizes of dopamine and epinephrine. Such electrochemical sensors can provide a very useful platform for the manufacturing of devices for monitoring the physiological concentrations of epinephrine.
Introduction
There are approximately 100 neurotransmitters that have been discovered up until now which play vital role in the cerebral system.1 Among these, epinephrine (EP) controls the smooth muscle contraction and relaxation in skin, lungs and heart. It also plays an important role in cardiovascular fluid transportation.2 From the point of view of a sensor, epinephrine detection is mainly accomplished by means of its ease in oxidization.3–6 However, the fact that EP and dopamine (DA) have nearly the same oxidation potential poses a great challenge in EP detection.7 Moreover, the oxidation product of EP forms a polymeric compound which blocks the electrode surface.3 Hence its detection in real samples is very difficult and sometimes not without ambiguity. Moreover, in a blood sample its concentration is diminutive (i.e., in the few μM concentration range) as compared to other interfering compounds such as uric acid (UA) and ascorbic acid (AA) whose concentrations are 15 μM and 100 μM, respectively.8,9 It has been established that the vital cause in neurological disorders is an irregularity in the concentration of neurotransmitters such as dopamine and epinephrine.10,11 Moreover, the lifetime of these neurotransmitters in the extra cellular space is short because of metabolism and cellular uptake. Therefore, for the analysis of these neurotransmitters, a chemical sensor is generally required which can provide accurate information at the micro dimension and in the millisecond timescale over which the communication occurs. However, widespread use of such chemical sensors is limited by conceptual and technical challenges. Nonetheless it becomes very important to monitor the minute concentrations of these neurotransmitters in various sample complexes, such as in cerebrospinal fluid, blood, and urine. A real-time analysis by a chemical sensor may assist in early diagnostics and/or an understanding of the prognosis of a treatment for such neurological disorders.A bilayer lipid membrane (BLM) is considered to be a very important aspect of the well-known sensor design in which the receptor molecule is incorporated into the supported bilayer lipid membrane (sBLM) system.12,13 The sBLM has achieved extraordinary approval as a chemical sensor because of its intrinsic usage of cellular membrane like molecules to develop the sensing surfaces.14,15 Consequently the use of sBLM incorporated with a suitable receptor molecule offers a unique biosensor as perceived in Nature, thus allowing the detection of biologically natural compounds in their characteristic natural environment. The immobilization interaction between the BLM and the solid support is only weak and depends upon the concentration and size ratios of the head groups of the lipid molecule. Hence, such “Nature based sensor” systems have an inherent limitation of fragility and therefore their lack of utility for long-term applications has hindered their commercialization.8,16,17
In this paper we have utilized a needle like nano array of crystalline zinc oxide nanorods (NRs) to immobilize a lipid membrane containing calixarene as a receptor molecule. Herein, we propose to use the ZnO NRs array which can provide enhanced stability due to the fact that the interspaces between the NRs offer excellent holding spaces. Likewise these BLM are fluid like substances and can rearrange themselves at the rough surface of the ZnO NRs, such as the crevasses lying between the nanorods which provide holding spaces for these BLM, enhancing the mechanical stability of the sBLM. However, even a very careful preparation of the sBLM does not provide complete coverage of the surface because of the presence of pinholes (defects) in the structure of the sBLM.18 Also the presence of the calixarene molecules in the BLM provides an easy ion transport, which could lead to the oxidation of the aforementioned compounds under electrochemical detection.19,20 In order to overcome this limitation, the ZnO NRs are covered with a 1-octadecanthiol (ODT) layer (which acts as a linker between the sensing layer and the semiconductor metal support, i.e., in this case ZnO NRs). Therefore, any pinholes left in the sBLM containing the receptor molecules which can cause erroneous results can be removed due to the presence of the linker layer of ODT. This study is further continued to understand the relative surface areas provided by each of the substrate phased surfaces of the ZnO crystalline NRs for adsorption of the linker layer between the substrate surfaces and the sensing layer.
The semiconductor nature of ZnO, with its biocompatibility, wide band gap, high electron mobility and most importantly low cost, makes it an excellent candidate for improving the substrate design of a chemical sensor for real-time analysis.21–24 In addition, the formation of the sensor surface in a 3D conformation can render the chemical sensor an exceptional detectability. In addition, such a sensor system provides a label-free suitable chemical detection methodology in addition to its ease of usage, fast response time, and cost efficiency, and it can be regenerated for multiple uses or be used in a single use format such as disposable sensors.
Experimental
ZnO nanorods synthesis and characterization
The ZnO NRs were synthesized by using RF magnetron sputter to prepare a seed layer on a well-cleaned silicon substrate by using the ZnO as a target. Subsequently a hydrothermal method was employed to grow the ZnO nanorods, as reported in our previous work.25 The crystallinity and the morphology of the straight aligned ZnO NRs were confirmed by an X-ray powder diffractometer (XRD, Rigaku RINT 2000) operating at 40 kV and 40 mA by using Cu Kα radiation (λ = 1.54056 Å) and high-resolution transmission electron microscope (HR-TEM) images were obtained on JEOL, JEM-3000F. The scanning electron microscope micrographs of the ZnO NRs were obtained on the JEOL, JSM-6700F with an accelerating potential at 20 kV, and a magnification of 5 to 25k× at the axial and top view positions.1-Octadecanthiol layer formation on ZnO nanorods and characterization
The resulting NRs were further treated with 1-octadecanthiol (ODT) 1 mM for the formation of an ultra-thin film. The ODT layer formation was analyzed at different immersion times i.e., 3 s, 5 minutes, 30 minutes, 1 hour and 24 hours at room temperature. The optical contact angles were determined on a Contact Angle System OCA instrument by using the sessile drop method. The instrument was operated using SCA-20 software for PCA and OCA. A static water drop was formed at the surface of the bare ZnO NRs and similarly on the different ODT modified ZnO NRs samples. A 0.5 μL drop of water was formed at a slow rate (0.5 μL s−1) at the tip of the syringe. A circular shaped drop formed, which was allowed to come into contact with the surfaces of the modified and unmodified ZnO NRs. The contact angles were computed by tangent and circle fitting. The images of the surfaces were taken with an Olympus BX51 microscope at various magnifications. The images were obtained both under a UV lamp and in visible light, as shown in the ESI (Fig. S1†).The ODT modified surface topography was obtained on a Bruker Innova® atomic force microscope (AFM) in the tapping mode and the data acquisition parameters were set as applied amplitude 3.5 V, scan size 25 μm2 and scan rate 4 μm s−1.
We obtained X-ray photoelectron spectra on an ESCALAB 250 XPS (Thermo Scientific Instruments) equipped with a monochromatic Al Kα X-ray source and a non-monochromatic X-ray source (Al/Mg). The spectra were referenced to C(1s) at 284.7 eV with a separation between the Zn(2p3/2) and Zn(2p1/2) of 23 eV. Survey spectra were recorded with a pass energy of 1.0 eV, 500 μm spot, and 150 W electron beam power with an acquisition time of 1 min and 8 seconds.
Sensor layer immobilization, response and characterization
For the immobilization of the sensor layer, such as the SBPC containing the receptor molecule, the liposome fusion method was employed, which gives a self-assembled monolayer of the lipid membrane on the ZnO nanorod array which was previously coated with ODT.26 From the wettability analysis Fig. S2 (ESI†), an increase in the hydrophobicity of the ZnO nanorods is evident after modification with the ODT at different insertion times. Therefore, it is most likely that self-rearrangement of the LM on the ODT hydrophobic layer occurs, with the hydrophobic chain end pointing towards the ODT whereas the hydrophilic part points towards the aqueous medium. A schematic illustration is given in Fig. S3.† A homemade electrochemical cell equipped with a three electrode setup confined in a Faraday cage connected to an electrochemical station (PGSTAT 302N) operated with Nova 1.8 software, was used for the study of the chemical sensor formation and response. Ag|AgCl|KCl (3 M) was used as the reference electrode and 1 mm diameter platinum wire was used as the counter electrode. Measurements were carried out at room temperature (20 ± 1 °C).Results and discussion
To fabricate a chemical sensor with an outstanding interaction capability between the analyte and the sensing surface, crystalline and well-aligned ZnO nanorods with a large surface area are considered crucial. In this work, we synthesize the ZnO nanorods through an easy and accessible hydrothermal method on a Si substrate. As shown in Fig. 1A and B, the ZnO nanorods exhibit a bundle-like morphology from the bird's-eye-view SEM image. The magnified SEM image shown in the inset (Fig. 1A) clearly presents the hexagonal shape of wurtzite-type ZnO. From the cross-sectional SEM image, one can see that the initial nucleation of ZnO is in the form of a solid and continuous film, which gradually evolves into separated ZnO nanorods with a shrinking diameter (Fig. 1B). All the ZnO nanorods are vertically standing on the Si surface and demonstrate good alignment.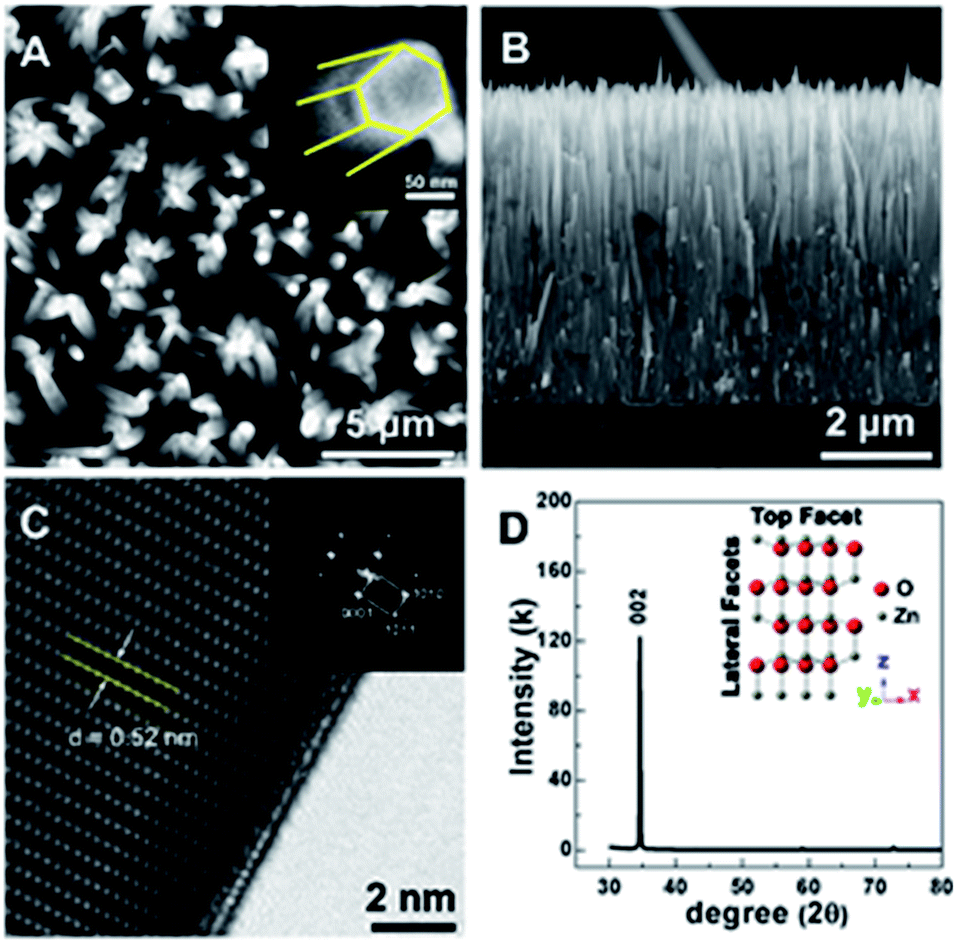 | ||
| Fig. 1 Morphology and crystal structure of the ZnO nano rods. (A and B) are the top and lateral view of the ZnO nanorods and (C and D) are the HRTEM and XRD pattern with inset showing the crystal structure, respectively. | ||
Microstructural analysis using high resolution transmission electron microscope (HRTEM) and Fast Fourier transform (FFT) further confirms the single crystalline characteristic of the as-prepared ZnO nanorod array (Fig. 1C). The distance of 0.52 nm between adjacent lattice planes matches well with the value of the [0002] plane in wurtzite-type ZnO. In addition, the preferential orientation of the ZnO nanorods can also be verified from the strong diffraction peak of the [0002] plane in the X-ray diffraction (XRD) pattern shown in Fig. 1D. As a result, a suitable substrate is obtained for the immobilization of the sensor surface.
A distinctive change is difficult to observe after the modification of the ZnO NRs with the ODT in the TEM images, as shown in Fig. S4;† however, it gives good insight into the faceted structural nature of the ZnO NRs. In the inset of Fig. 1D, a proposed unit cell of the ZnO crystal structure is illustrated. The schematic of the unit cell is derived from the XRD data and it is evident that the lateral facets of the crystal cell are composed of both Zn and O atoms, whereas the top facet is made up of either Zn or O atoms. The polar nature of the ZnO surfaces has been reported in the literature.27–34 The relative difference in surface activity of the polar surfaces [0001] (Zn-term surface) and [000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] (O-term surface) as compared to nonpolar
] (O-term surface) as compared to nonpolar 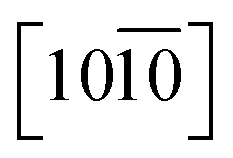 and
and 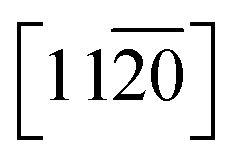 surfaces containing both the O and Zn terminated end atoms is well known. Moreover, ODT is a polar compound with a relatively higher solubility in polar organic solvents compared to nonpolar. Therefore, the rate of formation of a self-assembled monolayer for ODT is faster in non-polar solvents such as n-hexane or n-butane as compared to tetrahydrofuran, dimethylformamide, acetonitrile, cyclooctane, toluene and ethanol.35–38 Similarly S in the ODT has stronger tendency to form stable covalent bonds with the Zn as compared to the O atom.39–43 Therefore, the relative ODT chemisorption ability on the ZnO nanorods' top and lateral facets is different. Furthermore, it can also be claimed from the data that the facet surface with only a Zn terminated end group has a higher tendency to form a densely packed ODT layer as compared to the side facets of the ZnO NRs which contain both oxygen and zinc terminated end groups. This allows us to safely assume that the ZnO NRs are polar in nature and can react sufficiently strongly with the polar ODT to form stable monolayers. In order to further illustrate the chemisorption of the ODT on the facets of ZnO NRs, detailed X-ray photoelectron spectroscopy (XPS) studies were carried out. We performed a series of controlled experiments to reveal the concentration of the various elements present at the surface of the ZnO NRs array, as shown by the depth profile in Fig. 2A, which shows a decrease in the carbon content at the surface as the etching time increases and vice versa for Zn. This confirms the orientation and the extent of coverage by the ODT monolayer. The hydrophobic part (i.e., the methyl group) is pointing outward at the surface of the ZnO NRs. A lower constant concentration of the sulfur proves that the sulfur is lying adjacent to the surface of the ZnO NRs. A further probe of the ZnO NR surface was necessary to illustrate the relative chemisorption ability of the ODT at various facets of the ZnO NRs. The angles of the incident X-ray beam to probe the surface of the ZnO NRs were carefully chosen to be −7°, 0°, 6.9°, 10.5° and 14.9° with respect to the actual X-ray beam angle originating from X-ray beam gun. A careful selection of the angles was made more on the positive side, namely 0° to 14.9°, whereas only one negative angle (−7°) was chosen because our ZnO nanorod array is straightly aligned i.e., grown normal to the surface of the substrate. Therefore, in order to have a clear illustration of the surface analysis by the XPS measurement, more specifically the surfaces lying in between any two adjacent nanorods, more positive angles were considered more suitable, which allow the incident X-ray to illuminate deep in between the straightly aligned nanorod array. Whereas only one negative angle was considered sufficient to give the surface composition. More negative angles could also have been taken; however, they would result in surface analysis only of the top parts of the nanorods array. The experimental schematics are shown in Fig. S5 (ESI†). This adaptation in experimentation aided in probing the same 500 μm area of the ZnO NRs covered with ODT. A relative intensity profile for the elements under consideration is shown in Fig. 2B and also shown in Fig. S6(A).† A decrease in relative intensity occurs for all the elements at the −7° angle (i.e., the substrate is tilted towards the angle of incidence of the X-ray beam i.e., 45°) and as the tilt angle for the sample holder containing the sample is increased away from the incident X-ray beam, the relative intensity for all the elements increases. An increase in the relative intensity implies a greater concentration of the ODT monolayer at the specific orientation of the sample. An unmitigated explanation for such an observation can be given as that at lower angles, such as at −7°, the X-ray beam may not reach the spaces lying in between the NRs, because the tilt angle of the sample holder is moved away from the surface normal. Therefore, such intensities can be assigned to be emanating from the top of the ODT coated ZnO NRs and some part of the lateral side. Moreover, it is suggested that with the increase in sample holder tilt angle, the X-ray beam can penetrate more deeply in between the NRs and, hence, shows the increase in the relative intensities. However, in the case of carbon this relative intensity decreases with the increase in angle, which implies that the lateral facets of the ZnO NRs have less carbon content as compared to the top part of the ZnO NRs. As a result, we can safely assume the fact that the top end of the ZnO NRs are zinc terminated end groups, allowing more compact ODT layer formation as compared to the lateral facets of the ZnO NRs which contain both zinc and oxygen terminated end groups. Similarly Fig. S6(B)† shows the XPS spectra for the 24 hour treated ODT/ZnO NRs array, which indicates the high purity of the ODT layer formation. The values of the binding energy agree well with the reported literature.39 This modification of the ZnO NRs renders it suitable for chemical sensor layer immobilization (Table 1). The formation of a self-assembled monolayer of thiol on noble metal surfaces has been the subject of research in many previous studies.35,41,44 In this work the formation of a stable thiol monolayer at the surface of the ZnO NRs is considered crucial for the immobilization of LM containing calixarene as a receptor molecule. Moreover, it is anticipated that ODT can hinder the electroactive compounds reaching the ZnO NRs surface. So it can assist in preventing the oxidation of the epinephrine and/or dopamine under electrochemical investigations. However, it needs to be mentioned that a complete coverage of the metallic surface depends upon various factors such as the concentration, solvent system used, temperature conditions and of course the chemical nature and morphology of the substrate. Since ODT is a long chain organic compound, when bonded to a metal surface it orients outward at a specific angle to the metallic surface depending on the substrate. For example in the case of a gold surface the chain tilt angle is ∼30° and for similar types of elements such as Ag, Cu and Pt the angle ranges from 10 to 15°.45 However, the situation is not the same for ZnO, as mentioned earlier. The chain tilt angle for the ZnO surface is 0°, as reported by Zhang et al.44 They calculated the chain tilt angle by using electrochemical impedance spectroscopy (EIS). However, a dissimilarity in the chain tilt angle was observed by Hedberg et al. They used a sum frequency generation (SFG) technique to find out the chain tilt angles at the oxidized and reduced Zn surfaces, suggesting that for the oxidized Zn sample the angles range from 120° to 174°, depending on the insertion time.41 It is also suggested from previous studies that the surface coverage by the ODT on the ZnO surface depends crucially on the insertion time. As in the case of Au, a longer insertion time gives a more ordered and complete coverage of the metallic surface. However, it may limit the closely compact ordered ODT layer formation. Therefore, a comparative study as shown by the wetting profile (Fig. S2†) of the ZnO NR surface proves the formation of the hydrophobic layer at the ZnO NRs surfaces. Furthermore, it is also suggested from a wettability property study of the ZnO NRs array that the water molecules may reside in between the NRs where they come into contact with the surface, hence giving a very small contact angle value of 9.1°. And after the ODT layer formation on the ZnO NRs, a clear change in the wettability property is observed; hence the surface becomes hydrophobic. Since ODT is a polar compound, the wettability profile also proves that the methyl chain ends are pointing outward and the thiol group is attached at the surface of the ZnO NRs. Thus the water molecules are repelled by the methyl chain end groups, resulting in the hydrophobic characteristics of the modified surface.
surfaces containing both the O and Zn terminated end atoms is well known. Moreover, ODT is a polar compound with a relatively higher solubility in polar organic solvents compared to nonpolar. Therefore, the rate of formation of a self-assembled monolayer for ODT is faster in non-polar solvents such as n-hexane or n-butane as compared to tetrahydrofuran, dimethylformamide, acetonitrile, cyclooctane, toluene and ethanol.35–38 Similarly S in the ODT has stronger tendency to form stable covalent bonds with the Zn as compared to the O atom.39–43 Therefore, the relative ODT chemisorption ability on the ZnO nanorods' top and lateral facets is different. Furthermore, it can also be claimed from the data that the facet surface with only a Zn terminated end group has a higher tendency to form a densely packed ODT layer as compared to the side facets of the ZnO NRs which contain both oxygen and zinc terminated end groups. This allows us to safely assume that the ZnO NRs are polar in nature and can react sufficiently strongly with the polar ODT to form stable monolayers. In order to further illustrate the chemisorption of the ODT on the facets of ZnO NRs, detailed X-ray photoelectron spectroscopy (XPS) studies were carried out. We performed a series of controlled experiments to reveal the concentration of the various elements present at the surface of the ZnO NRs array, as shown by the depth profile in Fig. 2A, which shows a decrease in the carbon content at the surface as the etching time increases and vice versa for Zn. This confirms the orientation and the extent of coverage by the ODT monolayer. The hydrophobic part (i.e., the methyl group) is pointing outward at the surface of the ZnO NRs. A lower constant concentration of the sulfur proves that the sulfur is lying adjacent to the surface of the ZnO NRs. A further probe of the ZnO NR surface was necessary to illustrate the relative chemisorption ability of the ODT at various facets of the ZnO NRs. The angles of the incident X-ray beam to probe the surface of the ZnO NRs were carefully chosen to be −7°, 0°, 6.9°, 10.5° and 14.9° with respect to the actual X-ray beam angle originating from X-ray beam gun. A careful selection of the angles was made more on the positive side, namely 0° to 14.9°, whereas only one negative angle (−7°) was chosen because our ZnO nanorod array is straightly aligned i.e., grown normal to the surface of the substrate. Therefore, in order to have a clear illustration of the surface analysis by the XPS measurement, more specifically the surfaces lying in between any two adjacent nanorods, more positive angles were considered more suitable, which allow the incident X-ray to illuminate deep in between the straightly aligned nanorod array. Whereas only one negative angle was considered sufficient to give the surface composition. More negative angles could also have been taken; however, they would result in surface analysis only of the top parts of the nanorods array. The experimental schematics are shown in Fig. S5 (ESI†). This adaptation in experimentation aided in probing the same 500 μm area of the ZnO NRs covered with ODT. A relative intensity profile for the elements under consideration is shown in Fig. 2B and also shown in Fig. S6(A).† A decrease in relative intensity occurs for all the elements at the −7° angle (i.e., the substrate is tilted towards the angle of incidence of the X-ray beam i.e., 45°) and as the tilt angle for the sample holder containing the sample is increased away from the incident X-ray beam, the relative intensity for all the elements increases. An increase in the relative intensity implies a greater concentration of the ODT monolayer at the specific orientation of the sample. An unmitigated explanation for such an observation can be given as that at lower angles, such as at −7°, the X-ray beam may not reach the spaces lying in between the NRs, because the tilt angle of the sample holder is moved away from the surface normal. Therefore, such intensities can be assigned to be emanating from the top of the ODT coated ZnO NRs and some part of the lateral side. Moreover, it is suggested that with the increase in sample holder tilt angle, the X-ray beam can penetrate more deeply in between the NRs and, hence, shows the increase in the relative intensities. However, in the case of carbon this relative intensity decreases with the increase in angle, which implies that the lateral facets of the ZnO NRs have less carbon content as compared to the top part of the ZnO NRs. As a result, we can safely assume the fact that the top end of the ZnO NRs are zinc terminated end groups, allowing more compact ODT layer formation as compared to the lateral facets of the ZnO NRs which contain both zinc and oxygen terminated end groups. Similarly Fig. S6(B)† shows the XPS spectra for the 24 hour treated ODT/ZnO NRs array, which indicates the high purity of the ODT layer formation. The values of the binding energy agree well with the reported literature.39 This modification of the ZnO NRs renders it suitable for chemical sensor layer immobilization (Table 1). The formation of a self-assembled monolayer of thiol on noble metal surfaces has been the subject of research in many previous studies.35,41,44 In this work the formation of a stable thiol monolayer at the surface of the ZnO NRs is considered crucial for the immobilization of LM containing calixarene as a receptor molecule. Moreover, it is anticipated that ODT can hinder the electroactive compounds reaching the ZnO NRs surface. So it can assist in preventing the oxidation of the epinephrine and/or dopamine under electrochemical investigations. However, it needs to be mentioned that a complete coverage of the metallic surface depends upon various factors such as the concentration, solvent system used, temperature conditions and of course the chemical nature and morphology of the substrate. Since ODT is a long chain organic compound, when bonded to a metal surface it orients outward at a specific angle to the metallic surface depending on the substrate. For example in the case of a gold surface the chain tilt angle is ∼30° and for similar types of elements such as Ag, Cu and Pt the angle ranges from 10 to 15°.45 However, the situation is not the same for ZnO, as mentioned earlier. The chain tilt angle for the ZnO surface is 0°, as reported by Zhang et al.44 They calculated the chain tilt angle by using electrochemical impedance spectroscopy (EIS). However, a dissimilarity in the chain tilt angle was observed by Hedberg et al. They used a sum frequency generation (SFG) technique to find out the chain tilt angles at the oxidized and reduced Zn surfaces, suggesting that for the oxidized Zn sample the angles range from 120° to 174°, depending on the insertion time.41 It is also suggested from previous studies that the surface coverage by the ODT on the ZnO surface depends crucially on the insertion time. As in the case of Au, a longer insertion time gives a more ordered and complete coverage of the metallic surface. However, it may limit the closely compact ordered ODT layer formation. Therefore, a comparative study as shown by the wetting profile (Fig. S2†) of the ZnO NR surface proves the formation of the hydrophobic layer at the ZnO NRs surfaces. Furthermore, it is also suggested from a wettability property study of the ZnO NRs array that the water molecules may reside in between the NRs where they come into contact with the surface, hence giving a very small contact angle value of 9.1°. And after the ODT layer formation on the ZnO NRs, a clear change in the wettability property is observed; hence the surface becomes hydrophobic. Since ODT is a polar compound, the wettability profile also proves that the methyl chain ends are pointing outward and the thiol group is attached at the surface of the ZnO NRs. Thus the water molecules are repelled by the methyl chain end groups, resulting in the hydrophobic characteristics of the modified surface.
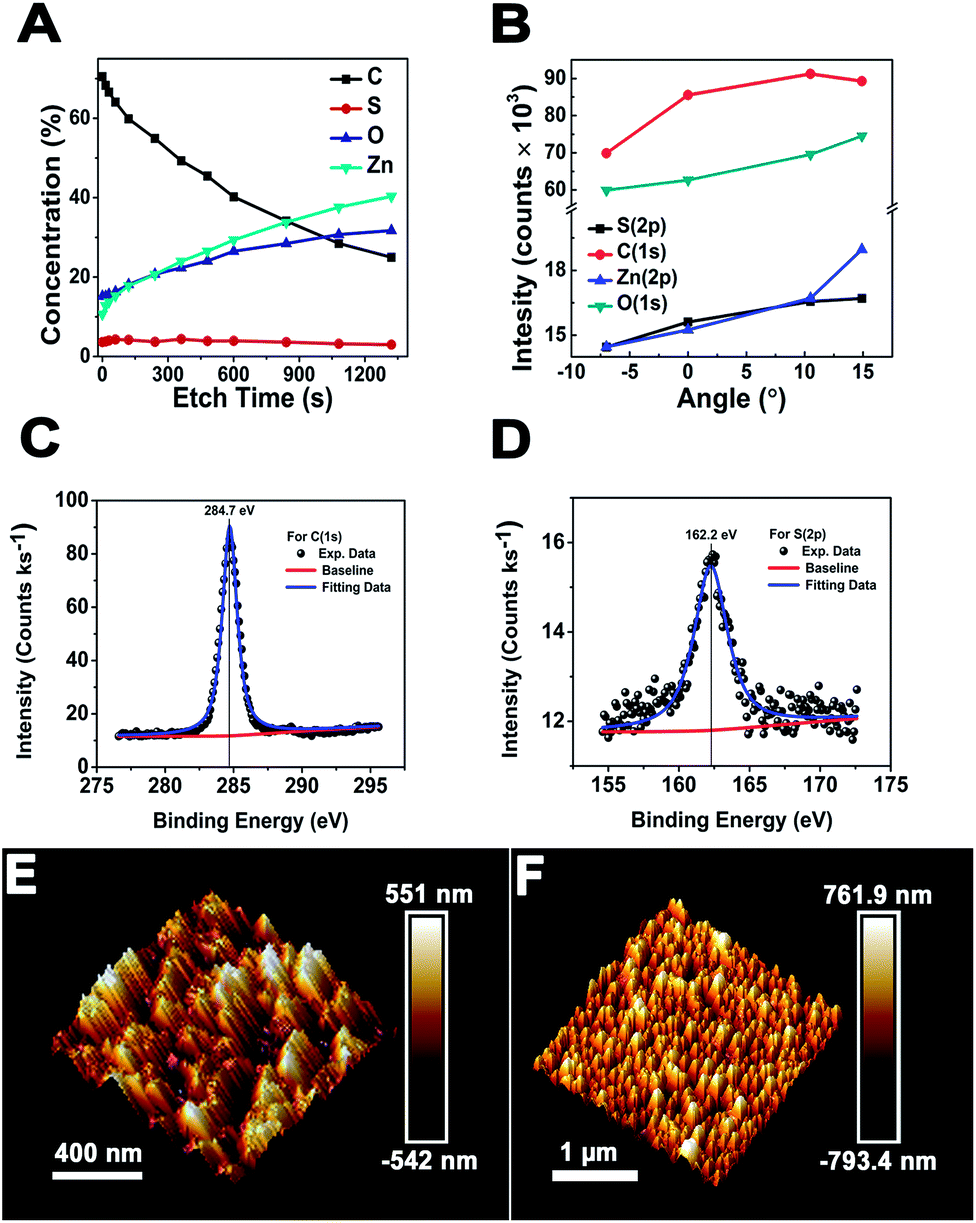 | ||
| Fig. 2 (A–D) are the XPS analysis in which (A) shows the depth profile, (B) is the surface analysis at different angle of incidence of the X-ray beam and (C and D) are the high resolution XPS signal for C(1s) and S(2p) at 0° angle for 24 h ODT modified ZnO nanorods respectively. (E and F) AFM images of the bare and modified for 24 h ODT ZnO nanorods, respectively. | ||
| S(2p) | C(1s) | O(1s) | Zn(2p3/2) | Zn(2p1/2) |
|---|---|---|---|---|
| 162.6 | 284.7 | 530.6 | 1021.6 | 1044.6 |
In order to confirm the morphology of the modified surface, atomic force microscopy (AFM) in tapping mode is carried out. In previous studies AFM is used for the illustration of formation of ODT on the metallic surface46,47 in which a contact mode is usually applied to scratch the ODT layer formed at the smooth surface. However, we have employed the tapping mode in this case because the contact mode could damage the straight aligned ZnO NRs. Moreover, higher values of the applied scratching force could result in breaking the NRs from the surface. A rough estimate of the thickness of the ODT layer was made by using the surface depth analysis, which was done with the help of the NovaScope Analysis 1.5 software (©Bruker Corporation). The surface image was cleaned, flattened and then the depth analysis was done. The peak to peak distance before and after the modification reveals a decrease in distance of ∼41.3 nm (as shown in Table S1†) and similarly the morphology of the surface retains the characteristics of the straight aligned ZnO NRs array shown in the Fig. 2F. A thin layer of ODT formation on the ZnO NRs is confirmed by the morphological image obtained by the AFM in tapping mode before and after the modification with ODT to observe the clear differences in the surface morphology (Fig. 2E and F). A surface roughness analysis (Rq values) was done to illustrate the change in surface roughness before and after the modification, given as 164 nm and 214 nm, respectively. This is because the ODT is a long chain organic molecule with its chain lengths extended outwards on the surface of the ZnO nanorods. Therefore an increase in the surface roughness after the modification also suggests uniform ODT layer formation. The formation of a thin layer is considered necessary so as to retain the characteristic crystallinity of the ZnO NRs.
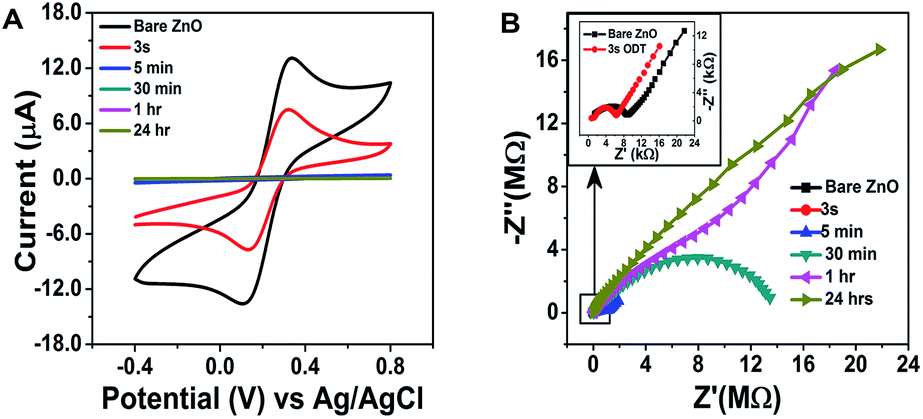
In order to check the surface modification suitability for sensor layer preparation and the sensor response, electrochemical impedance spectroscopy (EIS) and cyclic voltammetry were recorded, as shown in Fig. 3A and B. The surface modification of the ZnO NRs with the ODT was studied using the redox probe [Fe(CN)6]3/4− (10 mM). For bare ZnO NRs this probe gives well-resolved reduction and oxidation peaks with a peak separation of 90 mV (Fig. 3A). The disappearance of the redox peaks at different immersion times of the ZnO NRs in ODT solution reveals the relative time dependence for the formation of the stable ODT layer on the ZnO NRs. It also proves the restricted access of the redox probe to the ZnO NRs surface due to the formation of a hydrophobic layer. In order to assess quantitatively the electrical properties of the ODT modification on ZnO NRs, the EIS experiments were measured in the same background electrolyte and redox probe. The EIS response was analyzed by means of an equivalent circuit, as shown in the inset to Fig. S7† which shows that the data can be fitted well with the proposed equivalent circuit (Fig. S7†).48 The equivalent circuit consists of the cell resistance in series with the resistance to charge transfer and the space-charge capacitance. The Nyquist plot for the bare ZnO NRs shows a semi-circle at higher frequency values and linearity at lower frequencies, as shown in the inset of Fig. 3B. The diameter of the semicircle is proportional to the charge transfer resistance RCT whereas the linear part represents the mass diffusion of the redox probe which is represented by the Warburg impedance. The RM value after the modification increases from 0.855 kΩ to 1.450 kΩ after only a very small immersion time of 3 s (as shown in Table S2†). The RCT is a relatively high 7.22 kΩ for the bare ZnO in comparison to the gold electrode (which is in the orders of few hundred Ω), suggesting a characteristic semiconductor property.49 RSC and CSC are the resistance and capacitance related to the semiconductor characteristics of the ZnO. A 1.03 kΩ RSC value for the bare ZnO shows a low interference caused by the semiconductor nature of the ZnO, which further approves the choice of ZnO NRs for immobilization of the LM containing the calixarene. An important feature of the Nyquist plot which needs attention is that only at small immersion times were a semi-circle and linear part shaped spectrograph obtained and at 30 minute immersion time the linear part of the Nyquist plot disappears and only a semicircle is seen and at still larger immersion times incomplete semicircular plots are observed. These findings confirm the formation of a monolayer of ODT with sufficiently complete surface coverage of the ZnO NRs at 30 min and 1 hour ODT immersion time. Therefore, for further sensor layer preparation ZnO/30 min ODT and ZnO/1 h ODT were used.
 | ||
| Fig. 3 Electrochemical investigations of the modification of ZnO nanorods. (A and B) show the cyclic voltammograms and electrochemical impedance spectra at different modification times. | ||
In order to examine the integrity of the sensor layer formation on the ODT modified ZnO NRs similar experimental conditions were utilized, as mentioned earlier. From the Nyquist plot (Fig. 4A–C) it is evident that the charge transfer resistance decreases so that there is an increase in the ease with which the redox probe can pass through the sensing layer composed of CX:SBPC (1:100) as compared to the ZnO nanorods modified by 1 h ODT. This decrease in charge transfer resistance observed from the Nyquist plot also suggests that the calixarene is successively incorporated into the LM, thus providing ion transport for the negatively charged redox probe (Fe(CN)64/3−, 10 mM, (1:1)), due to its cavity like structure. Furthermore, an increase in the charge (as shown in Fig. S8 (ESI†)) is also observed after the formation of the sensing layer on the ODT coated ZnO nanorods. This is because SBPC contains a choline group which implies a positive charge on the phospholipid.50 Therefore, an increase in charge is in accordance with the Nyquist plot measurements. We calculated the surface density Γ of the sensor layer by using the equation:51
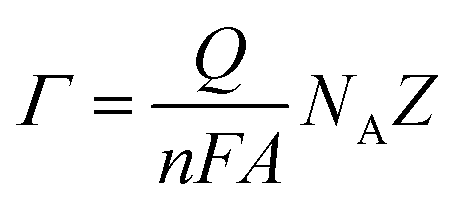 | (1) |
487 C), NA is Avogadro's number, Z is charge on the redox probe, and A is the electroactive surface area (which is 4.91 mm2). The calculations resulted in 7 × 1012 molecules per mm. This value is almost about three times higher than the value reported in the literature51 which can be attributed to the larger surface area provided by the ZnO NRs. Therefore, such a sensor layer is suitable for efficient chemical detection. The incorporated calixarene in the LM provides excellent cavities for binding with the target molecules.
 | ||
| Fig. 4 (A–C) Nyquist plots and (D) cyclic voltammograms of bare ZnO NRs and those modified by ODT and sBLM incorporated with calixarene in CX:SBPC (1:100). Exp. conditions: 0.05 M phosphate buffer pH 7.2 for bare ZnO and for the other modified surfaces McIlvaine buffer pH 5 is used, (1:1) redox probe 10 mM, 120 s Ar gas, Ag/AgCl in 3 M KCl ref. electrode. | ||
Furthermore, electrochemical impedance spectroscopy plots are shown in Fig. 5A and B for the addition of epinephrine and dopamine for the sensor surfaces prepared as ZnO/1 h ODT/CX:SBPC (1:100), respectively. It can be seen that the sensor shows a much more prominent response for the EP addition as compared to the DA, because calixarenes are macromolecules with a cavity-like shape. Therefore, a Lock & Key mechanism is proposed for the interaction of epinephrine and dopamine with the sensing surface, because sensor surfaces which contained only SBPC were non-responsive to the dopamine addition. However, only a small amount of calixarene in the sensor surface shows a distinctive change in the impedance spectra (Fig. 5A). The impedance spectra for the epinephrine addition show a much higher change in RCT at higher frequencies and at lower frequencies show a smaller diffusion resistance of the redox probe which indicates that the cavities provided by the calixarene molecules incorporated in the LM are suitable for the epinephrine molecule, and charge transfer due to the redox probe is hindered significantly. Whereas in the case of dopamine not much significant charge transfer resistance is observed. Rather, with each addition of dopamine an increase in diffusion resistance is evident (Fig. 6A). This suggests that the cavity size of the calix[6]arene is much bigger and can accommodate more dopamine molecules and as a result the diffusion through the sensor surface becomes large with the successive addition of dopamine. This observation can be attributed to the fact that dopamine and epinephrine have slightly different molecular sizes. Likewise, we assume that the SBPC LM incorporated with calix[6]arene is similar to an ion-channel sensor,19 since the cavity provided by the calix[6]arene holds a site for the ion transport. Such sensor surfaces offer an electrostatic interaction (either attractive or repulsive) to the analytes, after recognition of the host by the guest, forming an insulation complex, which hinders the redox marker from reaching the sensor surface. In this case, the sensor surface interacts with the protonated NH4+ moiety of the target molecules (either EP or DA) with the hydroxyl functional groups of the calix[6]arene pointing outwards towards the analytes. Therefore, as shown in the Nyquist plots (Fig. 5A and B), the hydroxyl groups present in the EP when attached to the sensor surface offer more charge transfer resistance (RCT) to the redox probe with each successive addition, whereas there almost none for the DA. Similarly, the secondary amine in the EP molecule lies close to the electron-withdrawing hydroxyl group, thus making it more electropositive compared to the DA molecule (as shown in the molecular structure in Fig. S9†). Therefore, the interaction between the more electropositive amine group of the EP and the hydroxyl groups of the calix[6]arene is more favoured as compared to the DA. Likewise the diameter of the calix[6]arene molecule is 5 to 25 nm, as reported in the literature,52 and normally the analyte interacts with the π stacking of the aromatic walls of calix[6]arene. Also Nikolelis and co-worker reported that a sensor based on solid supported lipid membranes with incorporated calix[4]resorcinarene is highly selective toward catecholamines.53–55 In our case we used calix[6]arene which has a bigger cavity size than calix[4]arene. Therefore, the CX:SBPC (1:100)/1 h ODT/ZnO nanorod array shows a more selective and sensitive sensor response for the epinephrine as compared to the dopamine. Nevertheless, the sensor shows excellent detectability of the aforementioned biologically natural compounds. It is believed that the convenient methodology developed here can be further applied to the cellular-membrane inspired surface modification of various semiconductor nanostructures for chemosensing of biologically important compounds. The highly sensitive sensor platform can be adjusted for device fabrication and can perform sensitive and selective detection of epinephrine at micro level domains in nano level concentrations.
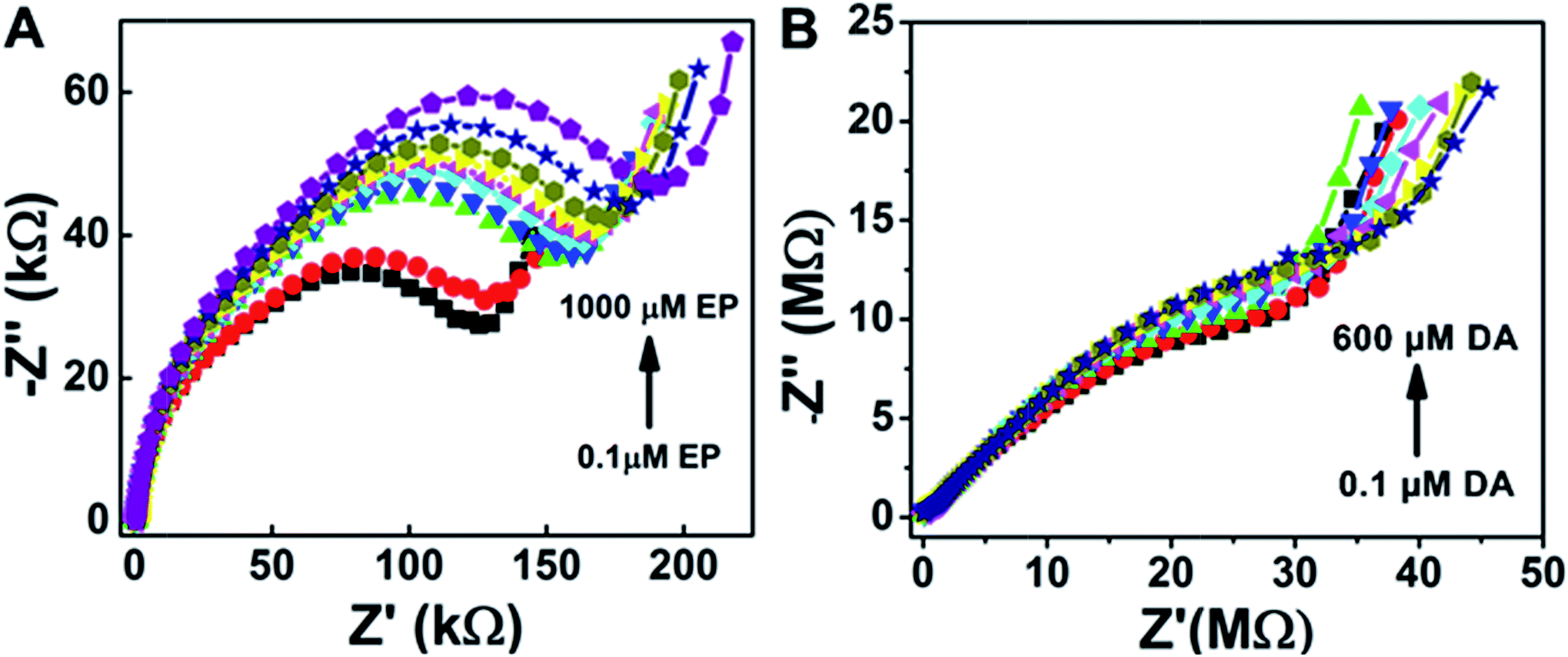 | ||
| Fig. 5 Nyquist plots for the sensor response for ZnO/1 h. ODT/CX:SBPC (1:100) under epinephrine addition (A) and dopamine addition (B) in McIlvaine buffer pH 5, Fe(CN)63−/4− (1:1) 10 mM, at 0.235 V bias voltage, amplitude 5 mV and frequency from 100 kHz to 0.04 Hz. | ||
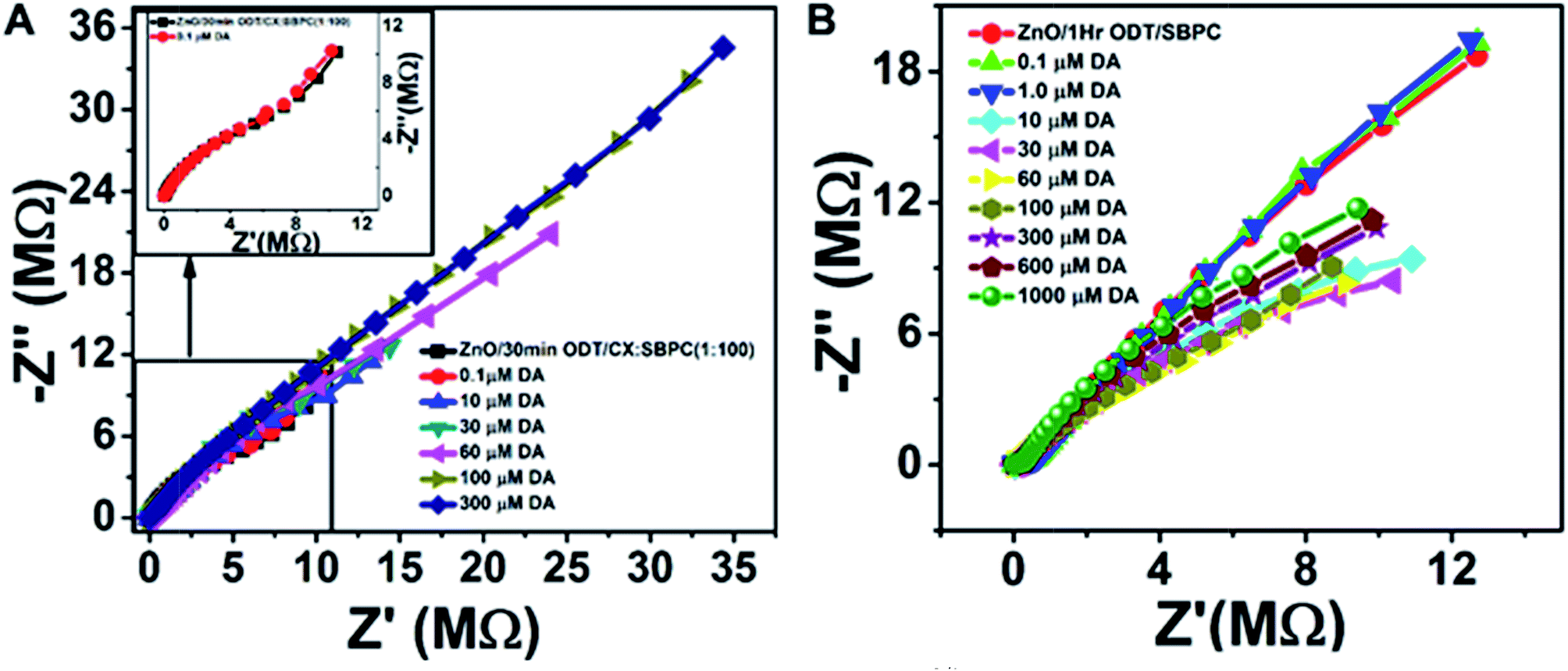 | ||
| Fig. 6 Sensor response with respect to the dopamine (DA) addition in McIlvaine buffer pH 5, Fe(CN)63−/4− (1:1) 10 mM, at 0.235 V bias voltage, amplitude 5 mV and frequency from 100 kHz to 0.04 Hz. (A) is the sensor surface made of ZnO/30 min ODT/CX:SBPC (1:100) and (B) ZnO/1 h ODT/only SBPC. | ||
Conclusions
We have synthesized a ZnO NRs array using magnetron sputter associated with the hydrothermal method to get a high purity crystalline NRs array and utilized this array to immobilize LM incorporated with calixarene. The ZnO NRs were modified with an ODT layer to form a stable and ordered monolayer. These ODT coated ZnO NRs were investigated with XPS, AFM and electrochemical methods. The XPS results suggest that the hexagonal crystalline NRs have relative chemisorption of the ODT. Therefore, the calixarene incorporated LM form a stable sensor layer on the ODT coated ZnO NRs. The sensor surface and sensor response were evaluated using EIS and CV analysis. We have successfully employed sLM incorporated with calixarene based sensors on the ZnO NRs array for the detection of epinephrine. These findings open the possibility of using ZnO NRs arrays for the selective and sensitive detection of epinephrine. It also provides a very useful platform for device fabrication which can be used for biomedical applications and improve neurological disease prognoses.Acknowledgements
B. D. Liu would like to thank the Knowledge Innovation Program of Institute of Metal Research (IMR), Chinese Academy of Science (CAS) (grants No. Y2NCA111A1 and No. Y3NCA111A1) and the Youth Innovation Promotion Association, Chinese Academy of Sciences (Grant No. Y4NC711171) for the support of this work. The author (M. A. Mohsin) acknowledges the financial support from the CAS President's International Fellowship for Postdoctoral Researchers, Grant No. 2015PM022.References
- T. Pradhan, H. S. Jung, J. H. Jang, T. W. Kim, C. Kang and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4684–4713 RSC
.
- W. H. Hartung, Chem. Rev., 1931, 9, 389–465 CrossRef CAS
- T. Łuczak, Electrochim. Acta, 2009, 54, 5863–5870 CrossRef
- T. C. Canevari, M. Nakamura, F. H. Cincotto, F. M. de Melo and H. E. Toma, Electrochim. Acta, 2016, 209, 464–470 CrossRef CAS
- B. O. Agboola and K. L. Ozoemena, Electroanalysis, 2008, 20, 1696–1707 CrossRef CAS
- H.-W. Chu, R. Thangamuthu and S.-M. Chen, Electrochim. Acta, 2008, 53, 2862–2869 CrossRef CAS
- H.-H. Li, H.-H. Wang, W.-T. Li, X.-X. Fang, X.-C. Guo, W.-H. Zhou, X. Cao, D.-X. Kou, Z.-J. Zhou and S.-X. Wu, Sens. Actuators, B, 2016, 222, 1127–1133 CrossRef CAS
- D. P. Nikolelis, T. Hianik and G.-P. Nikoleli, Electroanalysis, 2010, 22, 2747–2763 CrossRef CAS
- L. Lin, J. Chen, H. Yao, Y. Chen, Y. Zheng and X. Lin, Bioelectrochemistry, 2008, 73, 11–17 CrossRef CAS PubMed
- E. Krook-Magnuson, J. N. Gelinas, I. Soltesz and G. Buzsáki, JAMA Neurol., 2015, 72, 823–829 CrossRef PubMed
- J. Ng, A. Papandreou, S. J. Heales and M. A. Kurian, Nat. Rev. Neurol., 2015, 11, 567–584 CrossRef CAS PubMed
- V. Patil, K. S. Mayya, S. D. Pradhan and M. Sastry, J. Am. Chem. Soc., 1997, 119, 9281–9282 CrossRef CAS
- C. Satriano and M. E. Fragala, RSC Adv., 2012, 2, 3607–3610 RSC
- F. Ostermaier, L. Scharfenberg, K. Schneider, S. Hennig, K. Ostermann, J. Posseckardt, G. Rödel and M. Mertig, Phys. Status Solidi A, 2015, 212, 1389–1394 CrossRef CAS
- V. De Leo, L. Catucci, A. Falqui, R. Marotta, M. Striccoli, A. Agostiano, R. Comparelli and F. Milano, Langmuir, 2014, 30, 1599–1608 CrossRef CAS PubMed
- S. Das, A. Carnicer-Lombarte, J. W. Fawcett and U. Bora, Prog. Neurobiol., 2016, 142, 1–22 CrossRef CAS PubMed
- P. Lavalle, F. Boulmedais, P. Schaaf and L. Jierry, Langmuir, 2016, 32, 7265–7276 CrossRef CAS PubMed
- Z. Tang, Y. Wang, P. Podsiadlo and N. A. Kotov, Adv. Mater., 2006, 18, 3203–3224 CrossRef CAS
- K. Kurzątkowska, S. Sayin, M. Yilmaz, H. Radecka and J. Radecki, Sens. Actuators, B, 2015, 218, 111–121 CrossRef
- J. P. Buttress, D. P. Day, J. M. Courtney, E. J. Lawrence, D. L. Hughes, R. J. Blagg, A. Crossley, S. E. Matthews, C. Redshaw, P. C. Bulman Page and G. G. Wildgoose, Langmuir, 2016, 32, 7806–7813 CrossRef CAS PubMed
- Y. K. Tseng, C. J. Huang, H. M. Cheng, I. N. Lin, K. S. Liu and I. C. Chen, Adv. Funct.
Mater., 2003, 13, 811–814 CrossRef CAS
- E. Molaakbari, A. Mostafavi, H. Beitollahi and R. Alizadeh, Analyst, 2014, 139, 4356–4364 RSC
- H. Y. Yue, S. Huang, J. Chang, C. Heo, F. Yao, S. Adhikari, F. Gunes, L. C. Liu, T. H. Lee, E. S. Oh, B. Li, J. J. Zhang, T. Q. Huy, N. V. Luan and Y. H. Lee, ACS Nano, 2014, 8, 1639–1646 CrossRef CAS PubMed
- A. Tarlani, M. Fallah, B. Lotfi, A. Khazraei, S. Golsanamlou, J. Muzart and M. Mirza-Aghayan, Biosens. Bioelectron., 2015, 67, 601–607 CrossRef CAS PubMed
- X. Zhang, B. Liu, W. Yang, W. Jia, J. Li, C. Jiang and X. Jiang, Nanoscale, 2016, 8, 17573–17580 RSC
- V. M. Mirsky, M. Mass, C. Krause and O. S. Wolfbeis, Anal. Chem., 1998, 70, 3674–3678 CrossRef CAS PubMed
- M. A. Lahmer, Mater. Chem. Phys., 2016, 182, 200–207 CrossRef CAS
- M. Kunat, U. Burghaus and C. Woll, Phys. Chem. Chem. Phys., 2003, 5, 4962–4967 RSC
- A. Calzolari and A. Catellani, J. Phys. Chem. C, 2009, 113, 2896–2902 CAS
- J. D. Prades, A. Cirera and J. R. Morante, Sens. Actuators, B, 2009, 142, 179–184 CrossRef CAS
- C. Wöll, Prog. Surf. Sci., 2007, 82, 55–120 CrossRef
- Y. Tong, Y. Liu, C. Shao, Y. Liu, C. Xu, J. Zhang, Y. Lu, D. Shen and X. Fan, J. Phys. Chem. B, 2006, 110, 14714–14718 CrossRef CAS PubMed
- B. Meyer and D. Marx, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 035403 CrossRef
- N. H. Moreira, A. Dominguez, T. Frauenheim and A. L. da Rosa, Phys. Chem. Chem. Phys., 2012, 14, 15445–15451 RSC
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321–335 CrossRef CAS
- N. Kemnade, Y. Chen, M. I. Muglali and A. Erbe, Phys. Chem. Chem. Phys., 2014, 16, 17081–17090 RSC
- H. Ron, H. Cohen, S. Matlis, M. Rappaport and I. Rubinstein, J. Phys. Chem. B, 1998, 102, 9861–9869 CrossRef CAS
- C. E. Inman, S. M. Reed and J. E. Hutchison, Langmuir, 2004, 20, 9144–9150 CrossRef CAS PubMed
- P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y. T. Tao, A. N. Parikh and R. G. Nuzzo, J. Am. Chem. Soc., 1991, 113, 7152–7167 CrossRef CAS
- J. Rickert, W. Gopel, W. Beck, G. Jung and P. Heiduschka, Biosens. Bioelectron., 1996, 11, 757–768 CrossRef CAS PubMed
- J. Hedberg, C. Leygraft, K. Cimatu and S. Baldelli, J. Phys. Chem. C, 2007, 111, 17587–17596 CAS
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem.
Rev., 2005, 105, 1103–1169 CrossRef CAS PubMed
- S.-J. Ding, B.-W. Chang, C.-C. Wu, M.-F. Lai and H.-C. Chang, Anal. Chim. Acta, 2005, 554, 43–51 CrossRef CAS
- H. Zhang and S. Baldelli, J. Phys. Chem. B, 2006, 110, 24062–24069 CrossRef CAS PubMed
- J. C. Love, D. B. Wolfe, R. Haasch, M. L. Chabinyc, K. E. Paul, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 2003, 125, 2597–2609 CrossRef CAS PubMed
- J. X. Chen, R. E. Ruther, Y. Z. Tan, L. M. Bishop and R. J. Hamers, Langmuir, 2012, 28, 10437–10445 CrossRef CAS PubMed
- G. Kada, F. Kienberger and P. Hinterdorfer, Nano Today, 2008, 3, 12–19 CrossRef CAS
- T. Schubert, G. Steinhoff, H. G. von Ribbeck, M. Stutzmannn, M. Eickhoff and M. Tanaka, Eur. Phys. J. E, 2009, 30, 233–238 CrossRef CAS PubMed
- M. A. Mohsin, F. G. Banica, T. Oshima and T. Hianik, Electroanalysis, 2011, 23, 1229–1235 CrossRef CAS
- M. A. Mohsin, T. Hianik, F.-G. Banica, T. Oshima and P. D. Nikolelis, in Sensing in Electroanalysis, ed. K. Vytras, K. Kalcher and I. Svancara, University Press Centre, Pardubice, 2010, vol. 5, pp. 185–194 Search PubMed
- G. Evtugyn, U. Cherkina, A. Porfireva, J. Danzberger, A. Ebner and T. Hianik, Electroanalysis, 2013, 25, 1855–1863 CrossRef CAS
- S. Houmadi, D. Coquière, L. Legrand, M. C. Fauré, M. Goldmann, O. Reinaud and S. Rémita, Langmuir, 2007, 23, 4849–4855 CrossRef CAS PubMed
- D. P. Nikolelis, C. G. Siontorou, G. Theoharis and I. Bitter, Electroanalysis, 2005, 17, 887–894 CrossRef CAS
- D. P. Nikolelis, D. A. Drivelos, M. G. Simantiraki and S. Koinis, Anal. Chem., 2004, 76, 2174–2180 CrossRef CAS PubMed
- D. P. Nikolelis and G. Theoharis, Bioelectrochemistry, 2003, 59, 107–112 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: The procedure detail for the sensor layer preparation, figures of the TEM, XPS, contact angle measurements and optical microscope images and tables for the analysis result. See DOI: 10.1039/c6ra26250c |
| This journal is © The Royal Society of Chemistry 2017 |