
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structural analysis, solution equilibria and biological activity of rhodium(III) complexes with a quinquedentate polyaminopolycarboxylate†
Marija S. Jeremić a,
Hubert Wadepohlb,
Vesna V. Kojićc,
Dimitar S. Jakimovc,
Ratomir Jelićd,
Suzana Popovićd,
Zoran D. Matović*a and
Peter Comba*b
a,
Hubert Wadepohlb,
Vesna V. Kojićc,
Dimitar S. Jakimovc,
Ratomir Jelićd,
Suzana Popovićd,
Zoran D. Matović*a and
Peter Comba*b
aUniversity of Kragujevac, Faculty of Science, Department of Chemistry, R. Domanovića 12, 34000 Kragujevac, Serbia. E-mail: zmatovic@kg.ac.rs
bUniversität Heidelberg, Anorganisch-Chemisches Institut and Interdisciplinary Center for Scientific Computing (IWR), Im Neuenheimer Feld 270, D-69120, Heidelberg, Germany. E-mail: Peter.Comba@aci.uni-heidelberg.de
cOncology Institute of Vojvodina, Faculty of Medicine, University of Novi Sad, Dr Goldmana 4, 21204 Sremska Kamenica, Serbia
dUniversity of Kragujevac, Faculty of Medical Sciences, S. Markovića 69, 34000 Kragujevac, Serbia
First published on 17th January 2017
Abstract
Two rhodium(III) complexes [Rh(ed3a)(OH2)]·H2O (1) and Na[Rh(ed3a)Cl]·H2O (2) with ethylenediamine-N,N,N′-triacetate (ed3a) have been synthesized and characterized by elemental, spectroscopic and structural analyses. The crystal structure of (1) and (2) and the spectroscopic analysis of the two rhodium(III)–ed3a complexes are discussed in detail. The protonation constants of H3ed3a and the conditional stability constants of its RhIII complexes have been determined in aqueous solution by pH potentiometry and UV-Vis spectrophotometry. Molecular mechanics (MM) and density functional theory (DFT) have been used to model all possible geometric isomers, determine the global energy minimum and compare the computed with the experimentally observed structures. The cytotoxic activity of the new RhIII complexes was evaluated by an MTT assay against four human cancer lines (MCF-7, A549, HT-29 and HeLa) and a normal human cell line (MRC-5). A549, HT-29 and HeLa cells were sensitive to all compounds tested, while the breast carcinoma cell line MCF-7 was only sensitive to the reference compounds (doxorubicin and cisplatin). Western blot (WB) analysis of the effects of the tested compounds indicates that both complexes increase the expression of caspase 3 and consequently the involvement of this enzyme in apoptotic processes of the treated cells. WB also demonstrates proteolytic cleavage of poly-(ADP-ribose)polymerase (PARP) in HeLa cells after treatment with both tested substances. Flow cytometry confirmed apoptotic cell death and showed the induction of cell cycle termination as a possible promoter of apoptosis.
Introduction
For a multitude of reasons, transition metal complexes with edta-type aminopolycarboxylate ligands have attracted considerable attention and have been extensively investigated and reviewed.1,2 The best studied quinquedentate ligand system is ethylenediamine-N,N,N′-triacetate (ed3a) and its derivatives. For octahedral metal complexes with pentadentate coordinated symmetrical edta- (ethylenediamine-N,N,N′,N′-tetraacetate) or ed3a-type ligands three possible geometric isomers, cis-equatorial, trans-equatorial and cis-polar are possible (Fig. 1). Most of the reported [M(ed3a-type)X] complexes (M = CoIII, CrIII, CuII, NiII and X = monodentate ligand) have the cis-equatorial configuration.3–5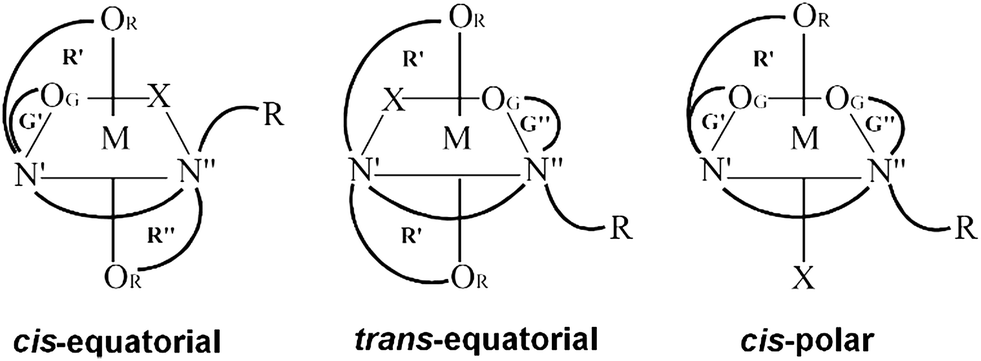 | ||
| Fig. 1 Geometrical isomerism of six-coordinate [M(ed3a-type)Xn] complexes: n = 1. | ||
The success of cisplatin as antitumor agent has stimulated enormous efforts to designing and preparing other clinically useful metal complexes.6–10 RhIII coordination compounds are isoelectronic with RuII and PtIV complexes, which provide a range of active antitumor agents.11,12 Generally, they are octahedral and inert but many RhIII complexes show considerable antitumor and antimicrobial activities.11,12 The first report of an antitumor active RhIII complex, RhCl3·3H2O,13 appeared before Rosenberg's discovery of cisplatin.6,7 Simple complexes such as mer-[RhCl3(NH3)3] or mer, cis-[RhCl3(DMSO)2(NH3)] are also known to be anticancer active.14,15 Early work up to 2002 on rhodium anticancer complexes in the oxidation states +1 to +3 was summarized in two review articles.16,17 A large number of complexes of rhodium has been explored for antitumor activity.18 Numerous half-sandwich complexes of Rh such as [(η5-C5Me5)Rh(LL)Cl], where (LL) is a bidentate polypyridyl ligand were prepared and studied in terms of their antiproliferative properties in human cancer cell lines.19–22 Rhodium(III) complexes appeared to be a good inhibitors of the kinase, inhibitors of other enzymes and inhibitors of protein–protein interactions.22 Data on the antimalignant activity of rhodium(III) chelated by EDTA-type ligands is very scarce.
Here, we report on the synthesis and coordination chemistry of RhIII complexes of H3ed3a and their medicinal/biological properties. The characterization is mainly based on the octahedral complexes [Rh(ed3a)(OH2)]·H2O (1) and Na[Rh(ed3a)Cl]·H2O (2). These contain fully deprotonated quinquedentate ligands, which may form four five-membered chelate rings. The IR, NMR and electronic spectra of these complexes are discussed in relation to their geometry, and the formation constants are used to estimate the in vivo stabilities of the rhodium(III) complexes. Also reported are the results of the two complexes' cytotoxicity in vitro towards diverse tumour cell lines and first results on the mechanisms of antiproliferative activity of the new RhIII complexes against human cervix adenocarcinoma cells. The effects on expression of proteins included in apoptotic signalling pathways (Bcl-2, Bax, caspase-3, and poly(ADP-ribose)polymerase, PARP) as well as cell cycle phase distribution of HeLa cells were monitored. A WB analysis was used to evaluate the expression levels of apoptosis-associated proteins.
Results and discussion
Coordination chemistry
Chelates of ed3a-type ligands can be prepared: (a) by the condensation method, starting from a neutralized α- or β-monohalogencarboxylic acid and the corresponding diamine, (b) by condensation of acrylic acid and a diamine for chelates with propionate arms or (c) by condensation of dihalogen derivatives of the diamine with various amino acids. The quinquedentate ed3a3− was prepared by the condensation method, from 1,2-diaminoethane and neutralized chloroacetic acid.5 The ligand was isolated as the calcium salt Ca3(ed3a)2·12H2O and transformed in the two complexes (1) and (2) as outlined in Scheme 1. The reaction of RhCl3·H2O and the quinquedentate ed3a3− produced a mixture of two complexes, and chromatography was used to separate the two complexes. The mixture was passed through a column of QAE A-25 Sephadex in the Cl− form. The yellow bands with different charges, i.e. neutral cis-equatorial-[Rh(ed3a)H2O]·H2O (1) and anionic cis-equatorial-Na[Rh(ed3a)Cl]·H2O (2), were separated. After desalting by passage through a Sephadex G-10 column, we were able to isolate crystals of (1) and (2) suitable for X-ray analysis, and the complexes were also characterized by elemental analysis, IR, UV-Vis and NMR spectroscopy.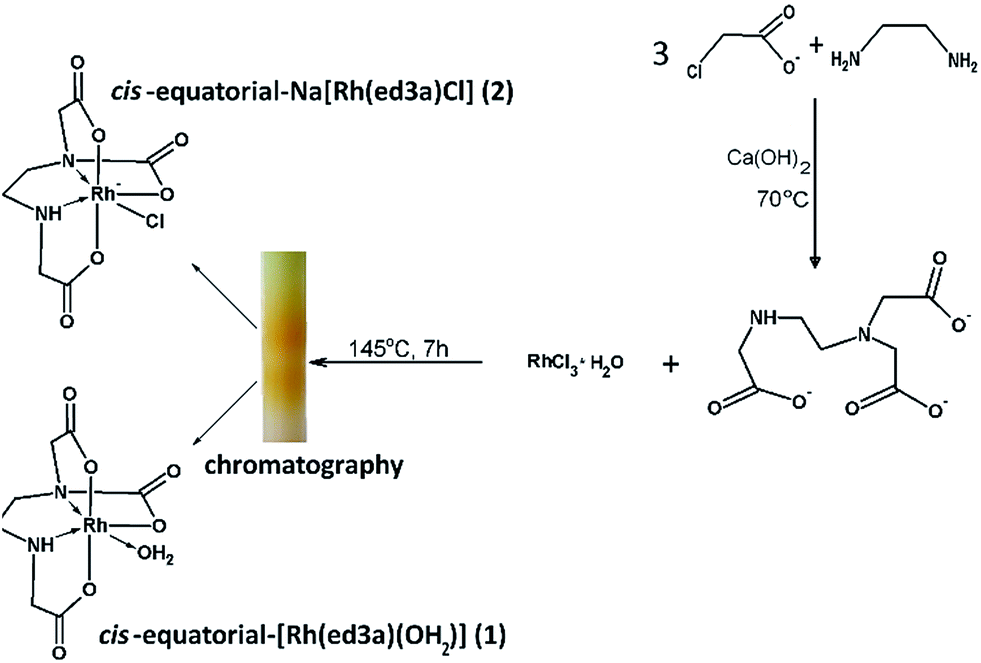 | ||
| Scheme 1 Synthesis of ed3a3− and the corresponding RhIII complexes (1) and (2). | ||
Description of the crystal structures
A structural diagram of the cis-equatorial-[Rh(ed3a)(OH2)]·H2O (1) and cis-equatorial-Na[Rh(ed3a)Cl]·H2O (2) with the adopted atom-numbering scheme is shown in Fig. 2, along with the packing in the crystals of (1) and (2). Selected bond lengths and valence angles are listed in Table 1.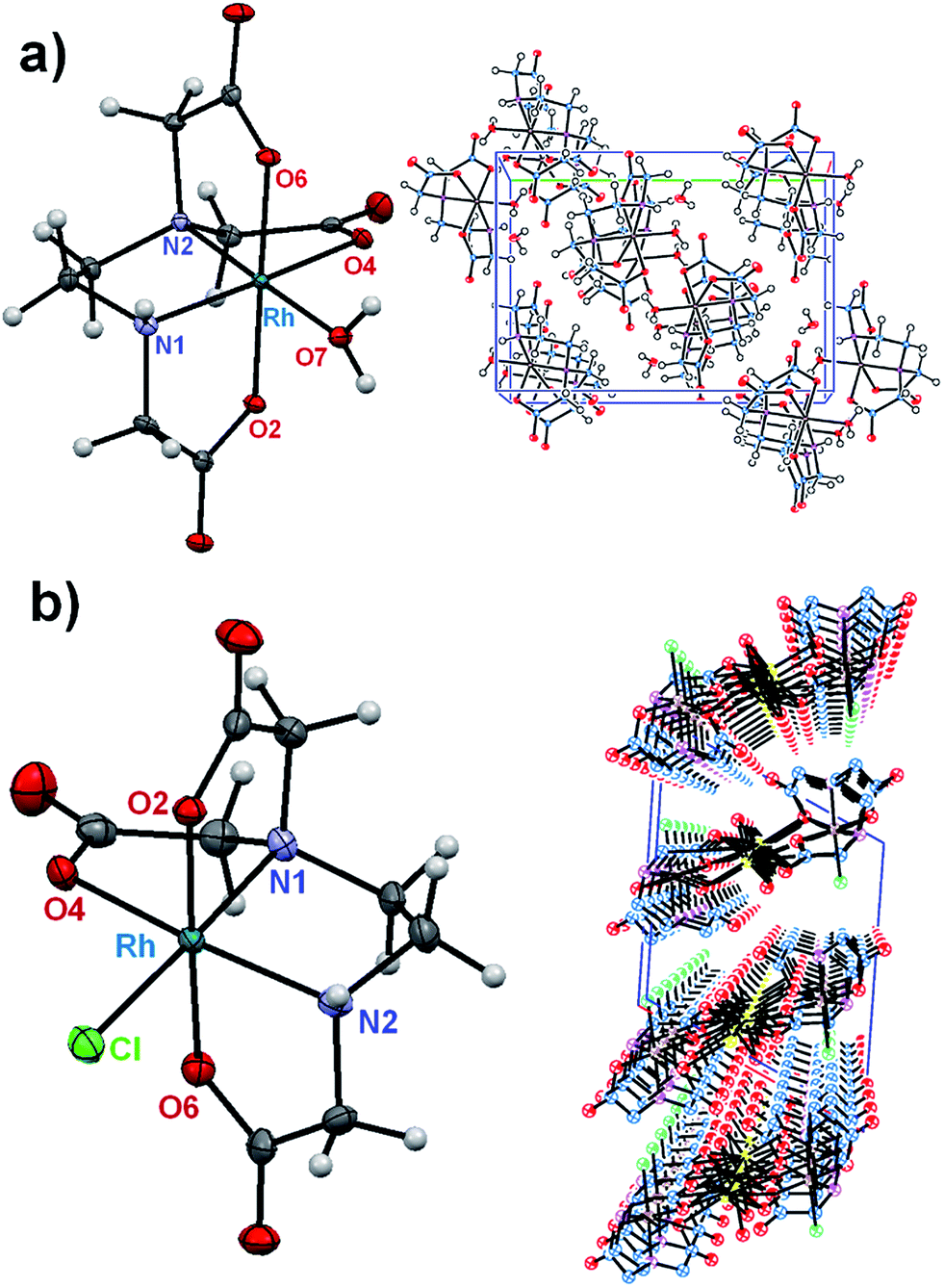 | ||
| Fig. 2 Ortep diagram of the [Rh(ed3a)(OH2)] molecular complex (a) and [Rh(ed3a)Cl]− complex anion (b) and crystal packing views along a axis (for [Rh(ed3a)(OH2)]) and b axis (for [Rh(ed3a)Cl]−) (50% probability ellipsoids). | ||
| (1) | (2) | ||
|---|---|---|---|
| M–L bond lengths (Å) | |||
| Rh–O(7) | 2.072(2) | Rh–Cl | 2.353(1) |
| Rh–O(2) | 2.018(2) | Rh–O(2) | 2.008(3) |
| Rh–O(4) | 2.050(2) | Rh–O(4) | 2.074(3) |
| Rh–O(6) | 2.010(2) | Rh–O(6) | 2.013(3) |
| Rh–N(1) | 2.027(2) | Rh–N(1) | 2.011(4) |
| Rh–N(2) | 1.996(2) | Rh–N(2) | 2.031(4) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Valence angles (°) | |||
| cis angles | |||
| O(2)–Rh–O(4) | 92.01(7) | O(2)–Rh–Cl | 91.11(10) |
| O(2)–Rh–O(7) | 89.03(7) | O(2)–Rh–O(4) | 90.05(12) |
| O(2)–Rh–N(1) | 83.06(7) | O(2)–Rh–N(1) | 85.55(14) |
| O(4)–Rh–O(7) | 98.62(7) | O(2)–Rh–N(2) | 95.39(13) |
| O(6)–Rh–O(4) | 86.64(7) | O(4)–Rh–Cl | 97.46(10) |
| O(6)–Rh–O(7) | 91.45(7) | O(6)–Rh–Cl | 89.99(10) |
| O(6)–Rh–N(1) | 98.21(8) | O(6)–Rh–O(4) | 91.05(12) |
| N(1)–Rh–O(7) | 91.14(8) | O(6)–Rh–N(2) | 83.30(14) |
| N(2)–Rh–O(2) | 93.66(7) | N(1)–Rh–O(4) | 82.97(14) |
| N(2)–Rh–O(4) | 83.09(7) | N(1)–Rh–O(6) | 93.34(14) |
| N(2)–Rh–O(6) | 85.90(7) | N(1)–Rh–N(2) | 86.90(16) |
| N(2)–Rh–N(1) | 87.42(8) | N(2)–Rh–Cl | 93.02(12) |
| trans angles | |||
| O(6)–Rh–O(2) | 178.62(7) | O(2)–Rh–O(6) | 178.33(13) |
| N(2)–Rh–O(7) | 176.77(8) | N(1)–Rh–Cl | 176.63(10) |
| N(1)–Rh–O(4) | 169.01(8) | N(2)–Rh–O(4) | 168.09(14) |
A coordination number of six is attained by RhIII in both complexes, via three deprotonated oxygen atoms of the carboxylate groups, two nitrogen atoms of the diamine, and the sixth coordination place occupies a water (1) or chloride ligand (2). In the crystals of (1) a hydrogen bonding network is formed, which involves the carboxylate and amino groups of the ed3a3− ligand as well as the coordinated and solvent water molecules. In (2) a polymeric structure is formed, with two of the carboxylate groups bridging RhIII and Na+. The water molecules are primarily involved in Na–O–Na bridging via their oxygen atoms but further hydrogen bridges are formed to the coordinated chloride and carboxylate ligands. Na+ is seven coordinate; intermolecular hydrogen bridges are also formed between amino (H donor) and carboxylate groups (H acceptor).
The positions of the carboxylate groups define the cis-equatorial geometry. Two acetate R rings (out-of-plane glycinate ring) occupy axial positions and the equatorial plane includes one five-membered acetate G ring (in-plane glycinate ring), the five-membered ethylenediamine E ring and a water molecule (1) or chloride ion (2). Complex (1) includes one longer equatorial Rh–O(7) bond (2.072(2) Å, to the water ligand), while complex (2) has one longer Rh–Cl bond (2.353(1) Å). The other metal–donor distances are within the expected range of 1.996(2) Å to 2.050(2) Å (1) and 2.008(3) Å to 2.074(3) Å (2).3–5,23 The cis angles are in the range of 83.0(7)° to 98.6(7)° for (1) and 83.0(1)° to 97.4(1)° for (2), and the trans angles vary between 169.0(8)° and 178.6(7)° for (1) and 168.1(1)° and 178.3(1)° for (2). The equatorial ethylenediamine (E) ring is in an envelope conformation. The puckering parameters q2 and φ2 (the ideal values for an envelope conformation are q2 > 0 Å, φ2 = 0°, and for a twisted conformation q2 > 0 Å, φ2 = 90° [π/2]),24 which relate to deviations of the ring atoms from the mean plane, are q2 = 0.443(1) Å and φ2 = 281.0(2)° for (1) and q2 = 0.455(2) Å and φ2 = 259.1(3)° for (2). The three five-membered acetate rings have a twisted conformation in (1). Their puckering parameters are q2 = 0.433(5) Å, φ2 = 153.1(7)° (RhO4C6C5N2); q2 = 0.301(0) Å, φ2 = 159.1(6)° (RhO2C1C2N1) and q2 = 0.087(4) Å, φ2 = 228.8(0)° (RhO6C8C7N2). In complex (2), one of the axial five-membered acetate rings is nearly planar and the second adopts an envelope conformation (R rings). The equatorial acetate ring (G) is in an envelope conformation. The puckering parameters are: q2 = 0.416(8) Å, φ2 = 147.6(2)° (RhO4C4C3N1); q2 = 0.290(8) Å, φ2 = 171.3(4)° (RhO6C8C7N2).
Spectroscopic analysis
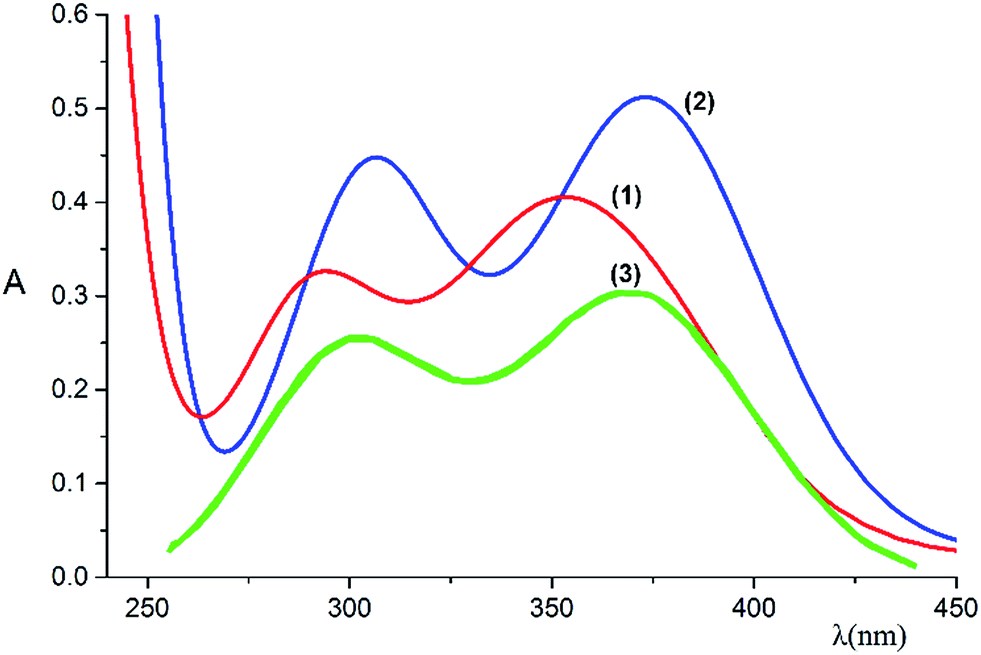 | ||
| Fig. 3 Electronic absorption spectra of RhIII complexes in aqueous solution: _____ [Rh(ed3a)(OH2)]·H2O (1), _____ [Rh(ed3a)Cl]−·H2O (2), _____ [Rh(Hedta)(OH2)] (3). | ||
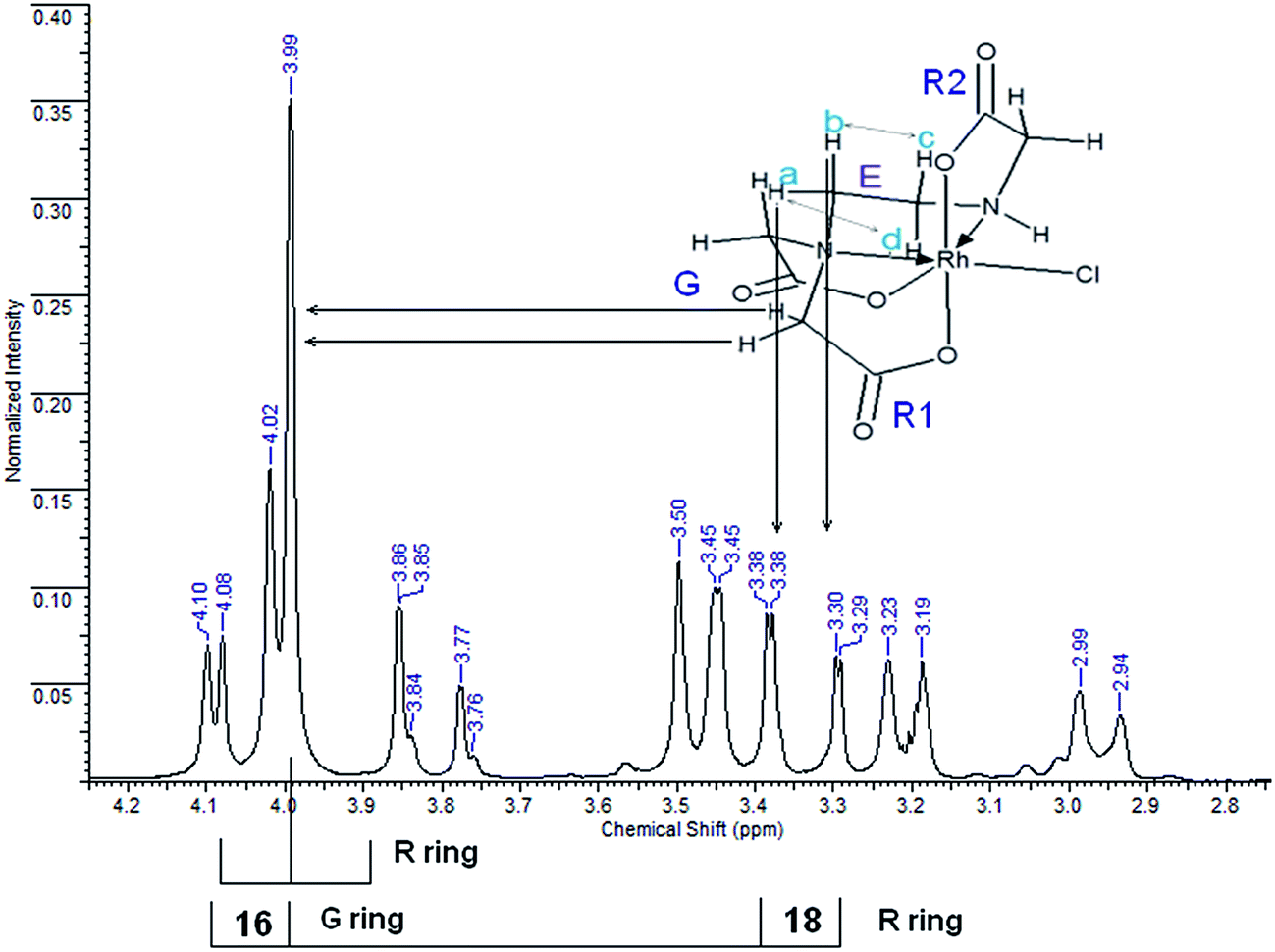 | ||
| Fig. 4 1H NMR spectrum of [Rh(ed3a)Cl]−. | ||
Some of the latter resonances (a complex ABCD pattern) are superimposed on the AB-type resonances of acetate. The tentative assignment of the low-field part of the AB pattern is complicated by the superposition with the singlet, obscuring one of the resonances. The symmetry of the molecule, assuming cis-equatorial configuration (see Fig. 1), with two acetate R rings (R1 and R2 above and below the Rh–ed3a nitrogen plane) and one G ring in the Rh–ed3a nitrogen plane, should result in three different acetate-type AB signals.
If the chemical shifts of two acetate protons on one of the rings are very similar, as might be expected for R1, the AB pattern collapses into a strong signal with very weak side peaks. Two acetate protons on R2 (Fig. 4) are symmetric with respect to the C–N bond in this ring and experience nearly identical shielding by the C–N bonds of the G and E rings. Therefore, the splitting of these two protons should be minimal. The collapsed AB pattern appearing as a single absorption at 3.99 ppm is attributed to this effect, and the weak side peaks can in fact be observed. The 1H NMR spectrum of (1) does not differ significantly from that in Fig. 4, except that the collapsed singlet at 4.05 ppm along with the weak side peaks at 4.02 ppm shows reversed shifts (Fig. S3, ESI†). Further, Fig. S4 (ESI†) and S5 (ESI†) show the corresponding 13C NMR spectra.
Solution studies
KHi) of ed3a were determined by pH potentiometry and are reported in Table 3 together with those of edta for comparison (standard deviations in parentheses). The protonation constants are defined by eqn (1):
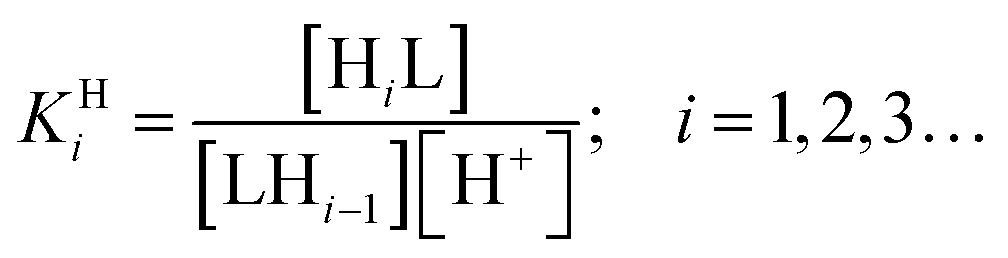 | (1) |
The first two protonation events occur at the nitrogen atoms and the logKH1 and logKH2 values were found to be 9.72 and 5.81. The logKH3 value (2.89) corresponds to the protonation of a carboxylate. The logKH4 and logKH5 values of two additional carboxylate groups are below 2 and could therefore not be determined potentiometrically. A comparison of the protonation constants of ed3a3− and edta4− obtained in similar media reveals that the logKH3 values are similar but the logKH1 and logKH3 are significantly different. The first and second protonation constants of ed3a3− are lower by 0.47 and 0.35 logK units than those of edta4−, which can be explained by one additional acetate group in edta4−. The acetate group with its positive inductive effect reduces the acidity of the proton at an amine nitrogen atom.
The distribution diagram of ed3a3− is given in Fig. S6 (ESI†). The fully deprotonated species, ed3a3− exists in solution at pH higher than 7. Protonated species Hed3a2−, H2ed3a− and H3ed3a are present in solution from pH 2 to 12.
Potentiometric titrations. The conditional stability constants of the RhIII complex with H3ed3a were determined potentiometrically at 25 °C in 0.1 M aqueous NaCl. The experimental data obtained are shown in Fig. S7 (ESI†).
To find the model that gives the best fit to the experimental data, various complexes and combinations thereof were included in Hyperquad2006 calculations.27 The model selected was that which gave the best statistical fit and which was chemically consistent with the titration data.28 The sample standard deviation, s, and the χ2-statistics were used as criteria for the selection of the complex models. The results obtained are listed in Table 4.
| logβp,q,r ± σ |
|||
|---|---|---|---|
| Potentiometric | Spectrophotometric | ||
| 25 °C | 25 °C | After heating | |
| a The water/chloride exchange in RhIII complex is likely to occur,18d,30 accordingly in the formulas, the water or Cl− monodentates are omitted. | |||
| [Rh(Hed3a)]+ | 12.16(4) | 12.39(8) | 12.54(9) |
| [Rh(ed3a)] | 5.18(3) | 5.26(6) | 7.26(3) |
| [Rh(ed3a)2]3− | 8.87(8) | 8.18(12) | — |
| [RhOH(ed3a)]− | −2.93(5) | −3.04(7) | — |
| Statistics | χ2 = 11.28, s = 1.50 | s = 0.032 | s = 0.024 |
Spectrophotometric titrations. Spectrophotometric data were obtained from RhIII–ed3a solutions, cooled to 25 °C, where both RhIII and ed3a concentrations were kept constant, while the pH was varied by addition of standard HCl or NaOH solutions, as appropriate. All corresponding UV-Vis spectra are presented in Fig. S8–S12, ESI.† The spectroscopic data were evaluated with the HypSpec2014 program,29 where the complexes found by potentiometry were included in HypSpec calculations, and the corresponding conditional stability constants were optimized. The resulting parameters are given in Table 4.
The distribution diagram of the RhIII–ed3a3− system (concentration ratio [ed3a]/[Rh] = 3:1) is shown in Fig. 5. The dominating complex at low pH values is [Rh(Hed3a)]+ with the maximum concentration of 45% at pH = 6.3.
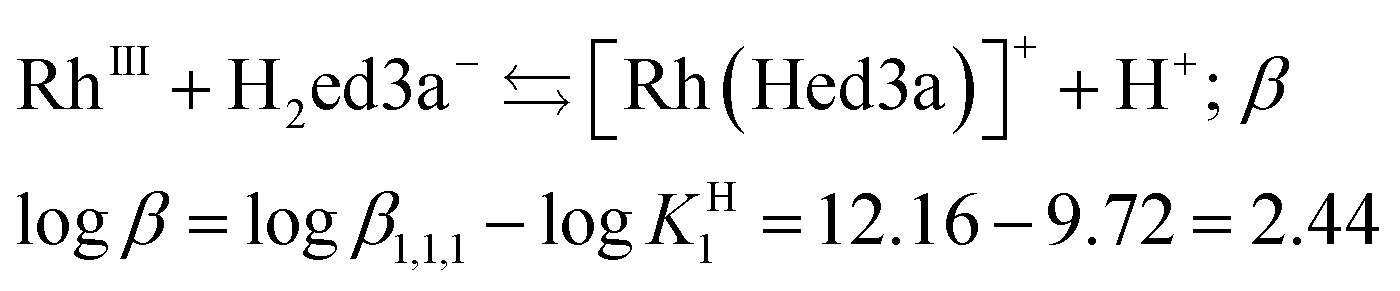 | (2) |
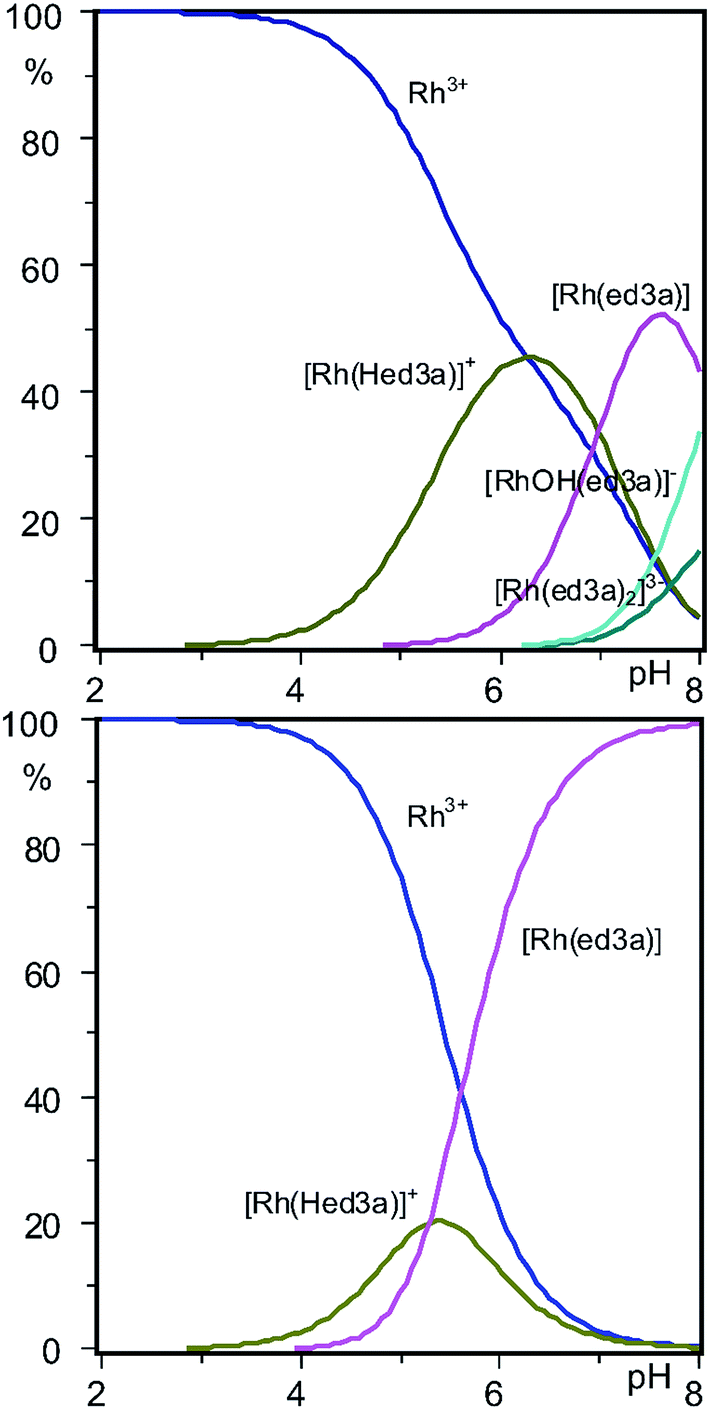 | ||
| Fig. 5 Concentration distribution diagrams of RhIII–ed3a3− complexes at concentrations of 2.0 mM for RhIII, 6.0 mM for ed3a3− and 100.0 mM NaCl, obtained by: (top) potentiometry and spectrophotometry at 25 °C; (bottom) spectrophotometry after heating in a closed vessel to 145 °C (see Experimental). | ||
The conditional stability constant (logβ = 2.44) implies that one nitrogen and carboxylate oxygen donors do not coordinate to rhodium in [Rh(Hed3a)]+ (Scheme 2). Here, two pathways are proposed for the formation of complexes in solution, i.e. 1 ⇄ 2 ⇄ 4 and 1 ⇄ 2 ⇄ 3. The first pathway emerges from the results of the potentiometric and spectrophotometric titration, while the second describes the results of the spectrophotometric titration after heating in a closed vessel to 145 °C.
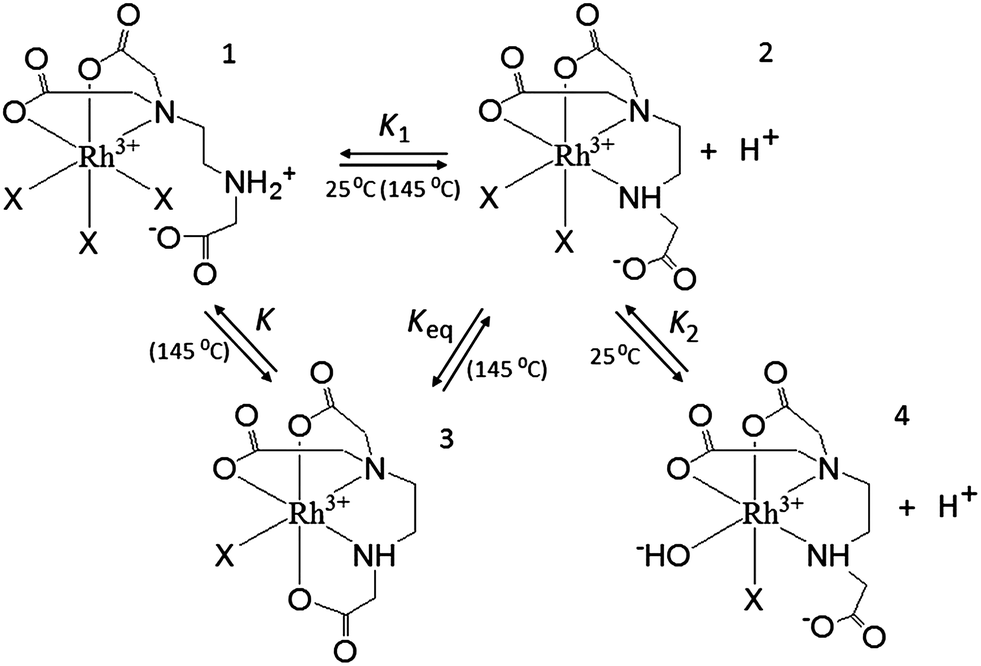 | ||
| Scheme 2 Dissociation of [Rh(Hed3a)X3]+. X = H2O or Cl−. | ||
With increasing pH [Rh(Hed3a)]+ releases a proton (the equilibrium 1 ⇄ 2 ⇄ 4), and forms [Rh(ed3a)] in which the ed3a3− is fully deprotonated, with a maximum of 52% concentration at pH = 7.6 (Fig. 5 and Scheme 2). The equilibrium constant, K1, for this reaction may be calculated from the overall conditional stability constants of [Rh(Hed3a)]+ and [Rh(ed3a)], logK1 = logβ1,1,1 − logβ1,0,1 = 12.16 − 5.18 = 6.98. This value is similar to the protonation constant logKH2. This means that the amine nitrogen loses a proton and coordinates to rhodium(III). Since the stability of the [Rh(ed3a)] is small (logβ1,0,1 = 5.18), the remaining free carboxylate group of ed3a3− is only weakly coordinated to rhodium(III). The equilibrium constant, logK, for the model 1 ⇄ 2 ⇄ 3 is 5.28 which is similar to logKH2 (see Table 3). After heating the solution of complex 2 (Scheme 2), the monodentate donor X is replaced by the COO− group, and the formation of a quinquedentate ligand complex with rhodium(III) and ed3a occurs. The equilibrium constant (equilibrium 2 ⇄ 3) logKaq = 2.08 can be compared with the formation constant of a hexacoordinated complex between rhodium(III) and EDTA.31
Upon increasing the pH (the model 1 ⇄ 2 ⇄ 4), [Rh(ed3a)(H2O)X] releases a proton and forms [RhOH(ed3a)X], which begins to form at pH = 6.2, and its concentration increases with further increase of pH. The complex [RhOH(ed3a)X], upon increasing of pH, binds another Hed3a2− ligand and forms [Rh(ed3a)2]3− (see Fig. 5 and S6, ESI†):
| [RhOH(ed3a)X] + Hed3a2− = [Rh(ed3a)2]3− + H2O + X | (3) |
[Rh(ed3a)2]3− starts to form at pH 6.5 and has a maximum concentration at pH = 9. Our current research deals with complexes with pd3a3− ligand (pd3a stands for propanediamine-N,N,N′-triacetate). The ligand pd3a3− belongs to ed3a-type chelates. The [Rh(pd3a)2]3− complex was isolated and its structure was confirmed by X-ray analysis. However, these results have not been published yet (Jeremić et al., unpublished results). Bearing that in mind, we believe that existence of [Rh(ed3a)2]3− is highly probable as well.
Computational chemistry
Table 6 contains selected geometric parameters for the optimized structures of (1) and (2), comparing experimental, DFT and force field data of Rh–N in-plane, Rh–O in-plane, Rh–O axial bonds, the averaged coordination cis- and trans-angles and the averaged Rh–O–C angles. It emerges that the observed bond lengths and angles are in excellent agreement with the experimental structures.
| X-ray:DFT:MM |
||
|---|---|---|
| (1) | (2) | |
| a Water molecule.b Average value.c RMSD values have been calculated on heavy atoms of overlaid X-ray and MOMEC optimized geometries. | ||
| Rh–N (Å) in-plane | 1.996:2.015:2.055 |
2.011:2.055:2.054 |
| 2.027:2.084:2.061 |
2.033:2.075:2.062 |
|
| Rh–O (Å) in-plane | 2.050:2.043:2.004 |
2.074:2.063:2.004 |
| 2.072:2.143:2.018a |
— | |
| Rh–O (Å) axial | 2.018:2.039:2.001 |
2.008:2.046:1.999 |
| 2.010:2.020:1.996 |
2.013:2.035:1.994 |
|
| Rh–Cl (Å) | — | 2.353:2.399:2.363 |
| Rh–O–C (°)b | 112.7:114.3:113.0 |
112.8:114.5:112.9 |
| cis-ang. (°)b | 90.0:90.0:90.1 |
90.0:90.0:90.1 |
| trans-ang. (°) | 178.6:178.0:176.3 |
178.3:176.2:176.1 |
| 176.8:177.4:174.6 |
176.6:176.9:173.2 |
|
| 169.0:168.1:167.0 |
168.2:166.8:166.8 |
|
| RMSD (Å)c | 0.0700 | 0.0910 |
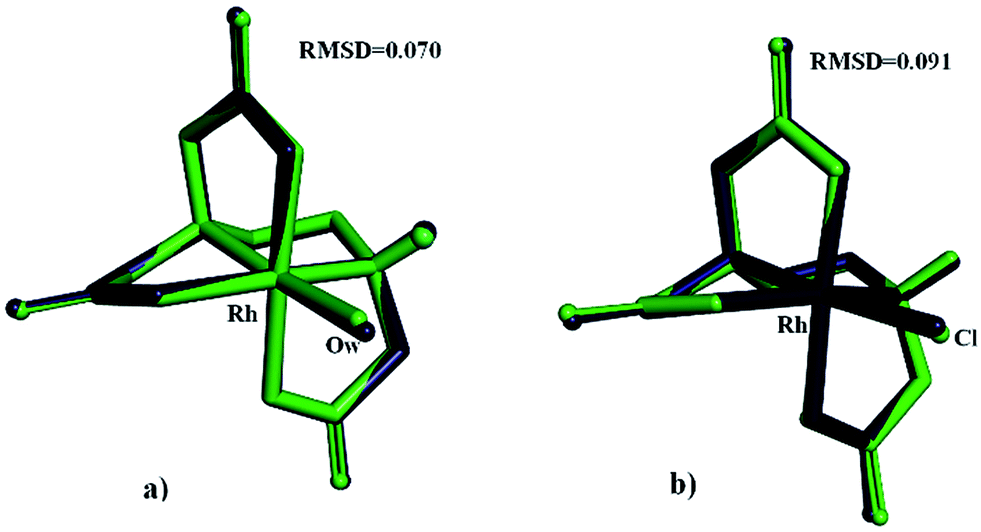 | ||
| Fig. 6 An overlay of X-ray and MOMEC optimized structures for [Rh(ed3a)(OH2)] (a) and [Rh(ed3a)Cl]− (b) complexes: blue = MOMEC optimized structures, green = X-ray structures. | ||
From MM as well as from DFT it emerges that, as expected from a large body of data of aminocarboxylate ligands coordinated to transition metal complexes (see Introduction), the observed cis-equatorial isomer is the most stable. MM suggests that the minima are more shallow than those predicted by DFT and that it might be possible to isolate other isomers. However, isomer conversion is slow for an inert metal center such as RhIII, and for the biological tests reported below, the isomer observed experimentally in the solid state is the only relevant species.
All three possible structures of the (1) and (2) are chiral (see Fig. 1) and therefore may adopt Λ or Δ configuration and therefore increase the number of possible isomers by a factor of 2. Also, the diaminoethane five-membered chelate ring may adopt λ or δ conformation, and this may also increase the number of isomers. However, changing the absolute configuration requires the inversion of N atoms and therefore a ligand exchange, which is not likely to occur around an inert rhodium(III) center. Conformational flexibility is not unlikely but is a fast process with energy barriers in the 20 kJ mol−1 range. It therefore appears that these isomeric possibilities are not of importance for the biological tests reported below, i.e. the only relevant structures are those observed by experiment.
Biological tests
Antiproliferative activity may results from cytostatic (effect on cell cycle) or cytotoxic effects and both can contribute to apoptosis. The Bcl-2 family of proteins are crucial in the regulation of apoptotic processes. Apoptosis depends on the balance between pro- and anti-apoptotic Bcl-2 proteins. The anti-apoptotic protein Bcl-2 plays a key role in apoptosis. Its suppressive activity in apoptotic process may contribute to drug resistance of tumor cells.35–37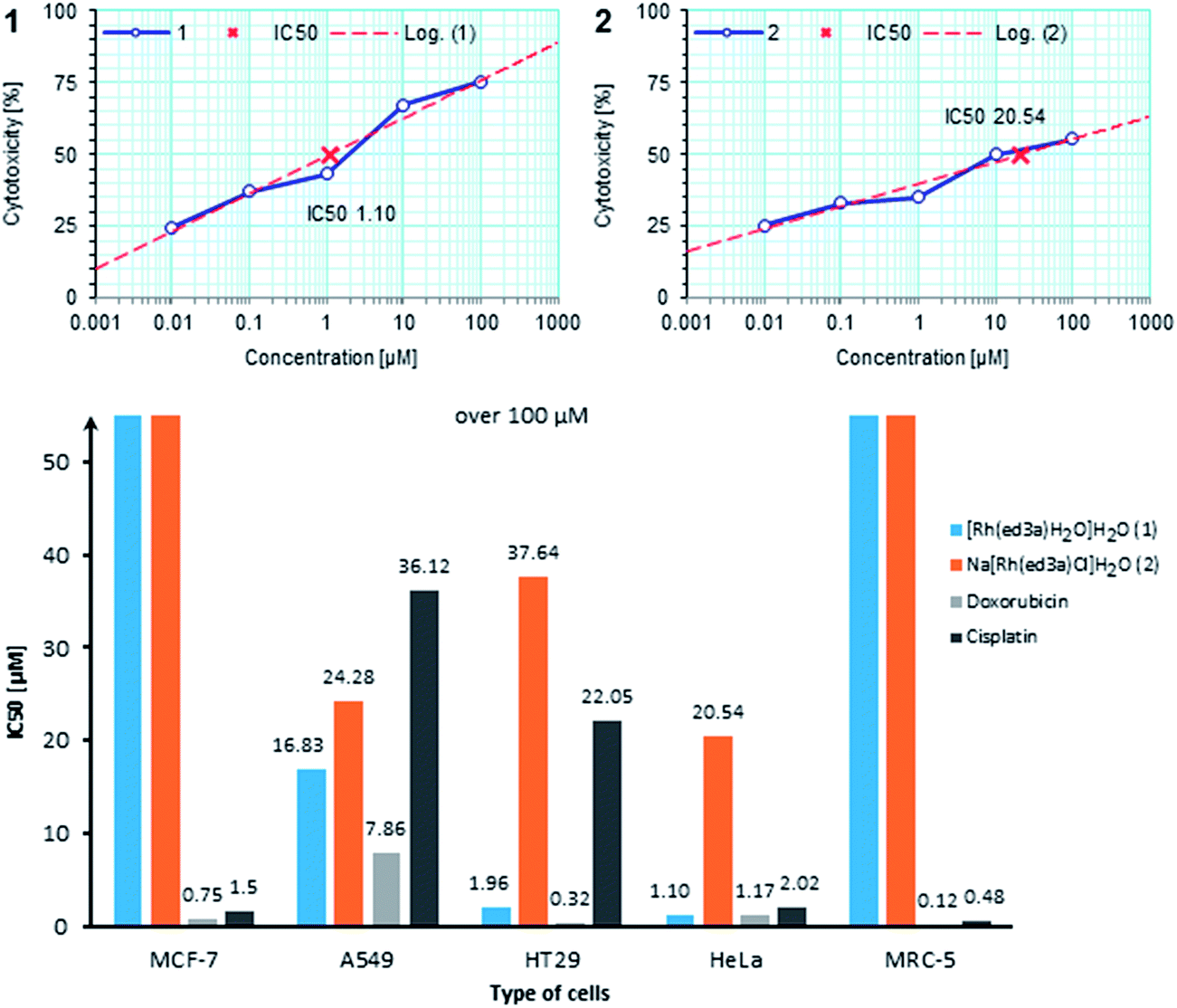 | ||
| Fig. 7 Linear and dose-dependent cytotoxic effect of complexes (1) and (2) against HeLa cells through the range of applied concentrations and graphical presentation of IC50 values (50% growth inhibitory concentration, μM) of selected RhIII complexes, doxorubicin and cisplatin for a set of malignant cells and MRC-5 cell line; each point is the mean of two independent experiments, each done in quadruplicate. | ||
| Compound | IC50, μM | ||||
|---|---|---|---|---|---|
| MRC-5 | MCF-7 | A549 | HT-29 | HeLa | |
| (1) | >100 | >100 | 16.83 | 1.96 | 1.10 |
| (2) | >100 | >100 | 24.28 | 37.64 | 20.54 |
| Na2Hed3a | >100 | >100 | 18.01 | >100 | 13.86 |
| Doxorubicin | 0.12 | 0.75 | 7.86 | 0.32 | 1.17 |
| Cisplatin | 0.45 | 1.5 | 36.12 | 22.05 | 2.02 |
Exposure of phosphatidylserine at the outer surface of cell membrane is one of the first events when a cell is committed to apoptosis. Due to the fact that Annexin V (a calcium-dependent phospholipid-binding protein) shows affinity to bind to the cell membrane, this molecule was used for detecting apoptotic cells.38 7-AAD, a fluorescent dye with high affinity for DNA, was employed for distinguishing between viable and late apoptotic or necrotic cells, since this large molecule cannot pass through an intact cell membrane. Therefore, flow cytometry of Annexin V-FITC/7-AAD stained cells is considered as golden standard for detecting apoptosis. Our results show that (1) and (2) induce apoptosis in HeLa cells (Fig. 8). The majority of cells were early apoptotic (39.38% and 28.71%, respectively), a small percentage of cells were in late apoptosis (2.1% and 1.88%, respectively), while minor a percentage of cells were necrotic (0.17; 0.19).
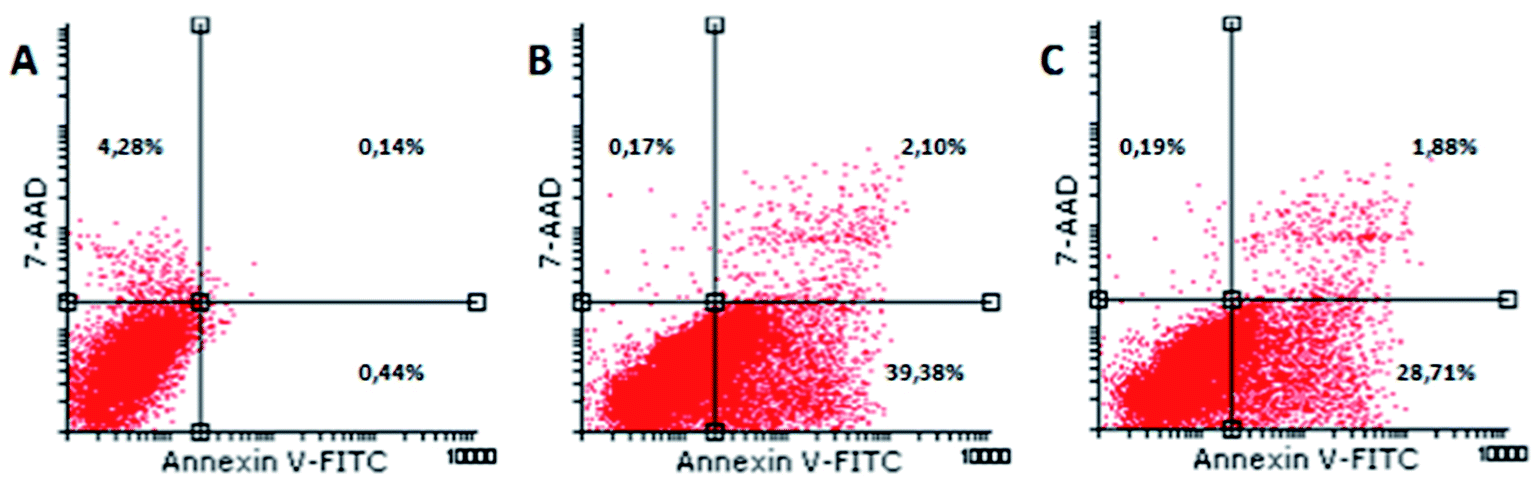 | ||
| Fig. 8 Flow cytometric analysis of Annexin V-FITC/7-AAD staining. Dot plots present percentages of viable (Annexin V−7-AAD−), early apoptotic (Annexin V+7-AAD−), late apoptotic (Annexin V+7-AAD+) and necrotic cells (Annexin V−7-AAD+) in untreated HeLa cells (A) and cells treated with IC50 concentration of (1) (B) and (2) (C). | ||
Although exposure of PS on the outer leaflet of the cell membrane is considered as hallmark of apoptosis, translocation of PS can occur in other types of cell death.39 Therefore, more than one method has to be utilized to verify apoptosis. Cell morphology assessment may be the most reliable method for discrimination of apoptosis.40 When the apoptotic program is started, cells shrink and become rounded, chromatin is condensing and fragmenting. Using AO/EB staining, we confirmed that cells treated with the tested substances show all morphological changes typical for apoptosis (Fig. 9). AO/EB staining enabled discrimination between viable, early apoptotic, late apoptotic and necrotic cells after 24 h treatment with the tested substances.
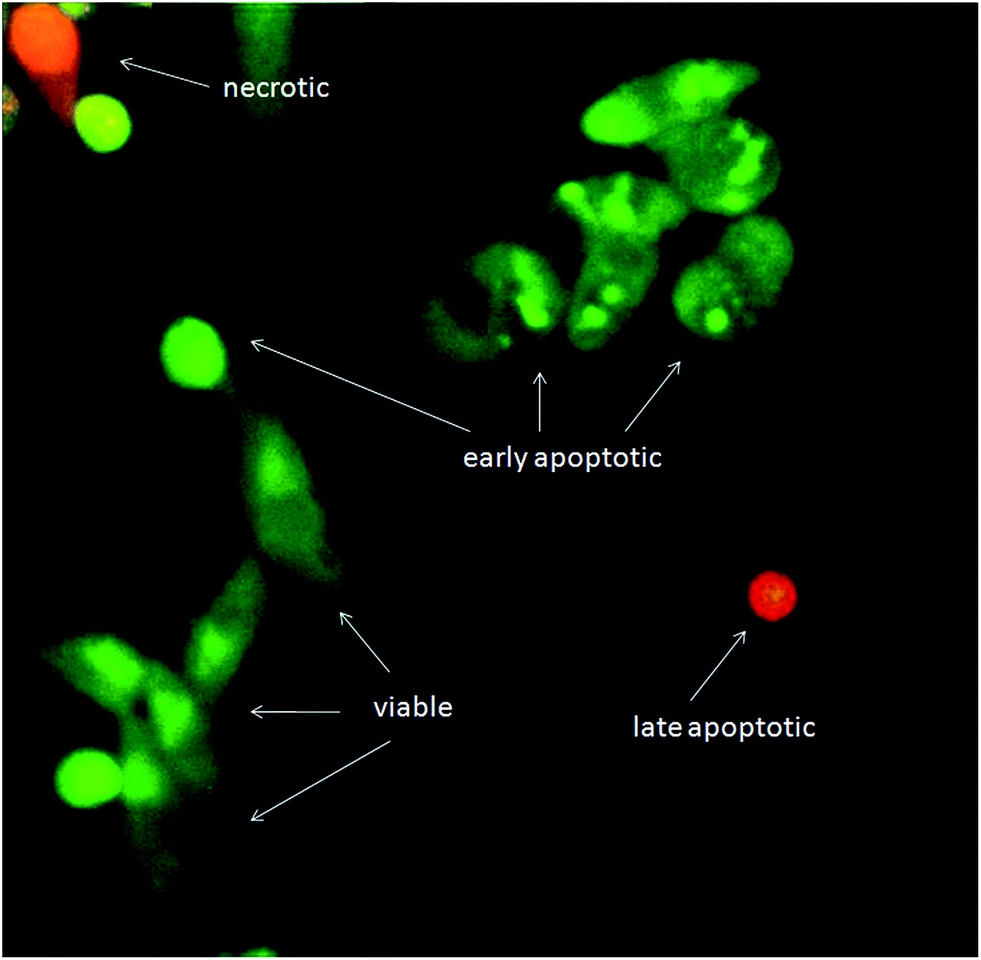 | ||
| Fig. 9 Changes in morphology of HeLa cells after treatment visualized by AO/EB staining. Both (1) and (2) induced alterations typical for apoptosis. Nuclei of viable cells are green with organized structure, whereas nuclei of early apoptotic cells are bright green and of late apoptotic cells bright orange to red with condensed chromatin. Necrotic cells have normal morphology and orange to red nuclei with organized structure. | ||
WB analysis of the effects of the tested compounds (Fig. 9 and 10) indicates that, in comparison with the control, they decrease the amount of the Bcl-2 protein, similar to doxorubicin; Bax protein expression is only increased with [Rh(ed3a)(OH2)]·H2O (1). Both RhIII complexes increase the expression of caspase 3 (Fig. 10 and 11), which indicates the involvement of caspase 3 in apoptotic processes of the investigated cell line. WB also demonstrate proteolytic cleavage of poly-(ADP-ribose)polymerase (PARP) in HeLa cells, after treatment with both (1) and (2) (Fig. 9 and 10). Actin was used as an internal control and shows uniform expression in all samples. The variations were within a range of ±5%, compared to the control.
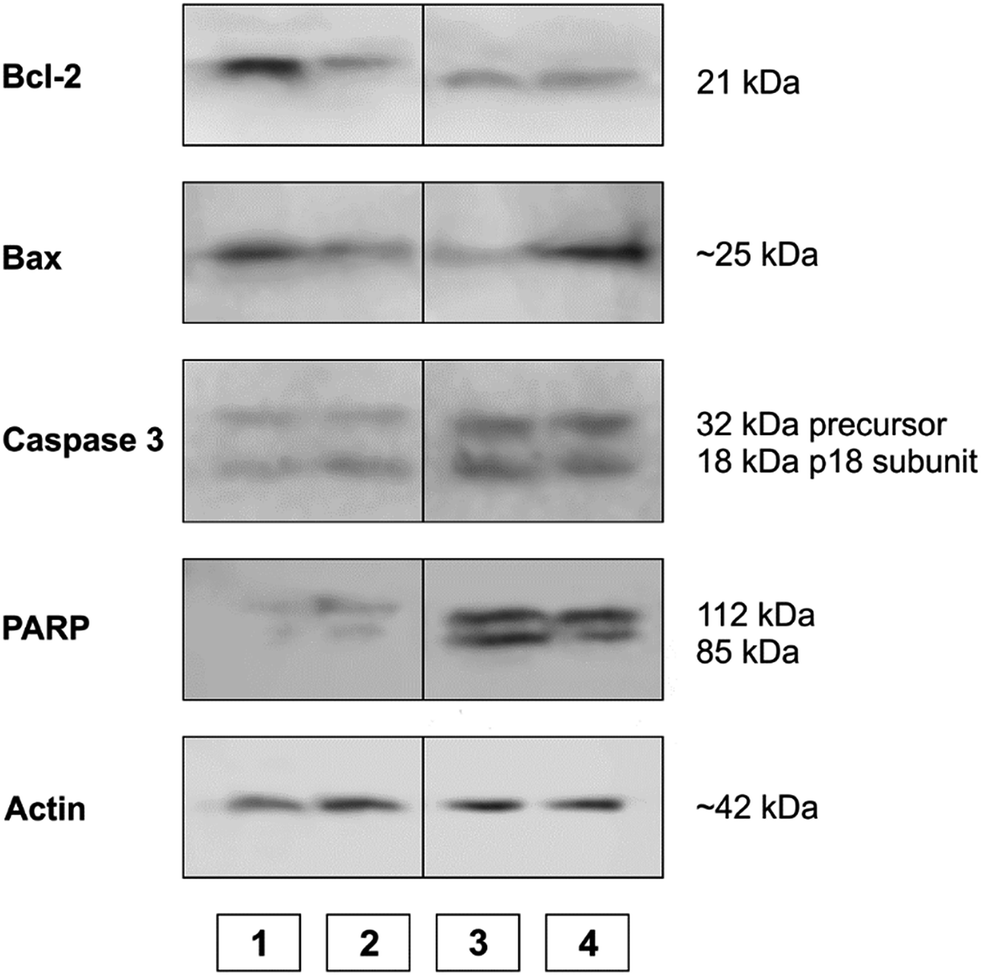 | ||
| Fig. 10 The expression of apoptotic proteins investigated by Western blot analysis. (1) Control sample; (2) doxorubicin; (3) Na[Rh(ed3a)Cl]·H2O (2); (4) [Rh(ed3a)(OH2)]·H2O (1). | ||
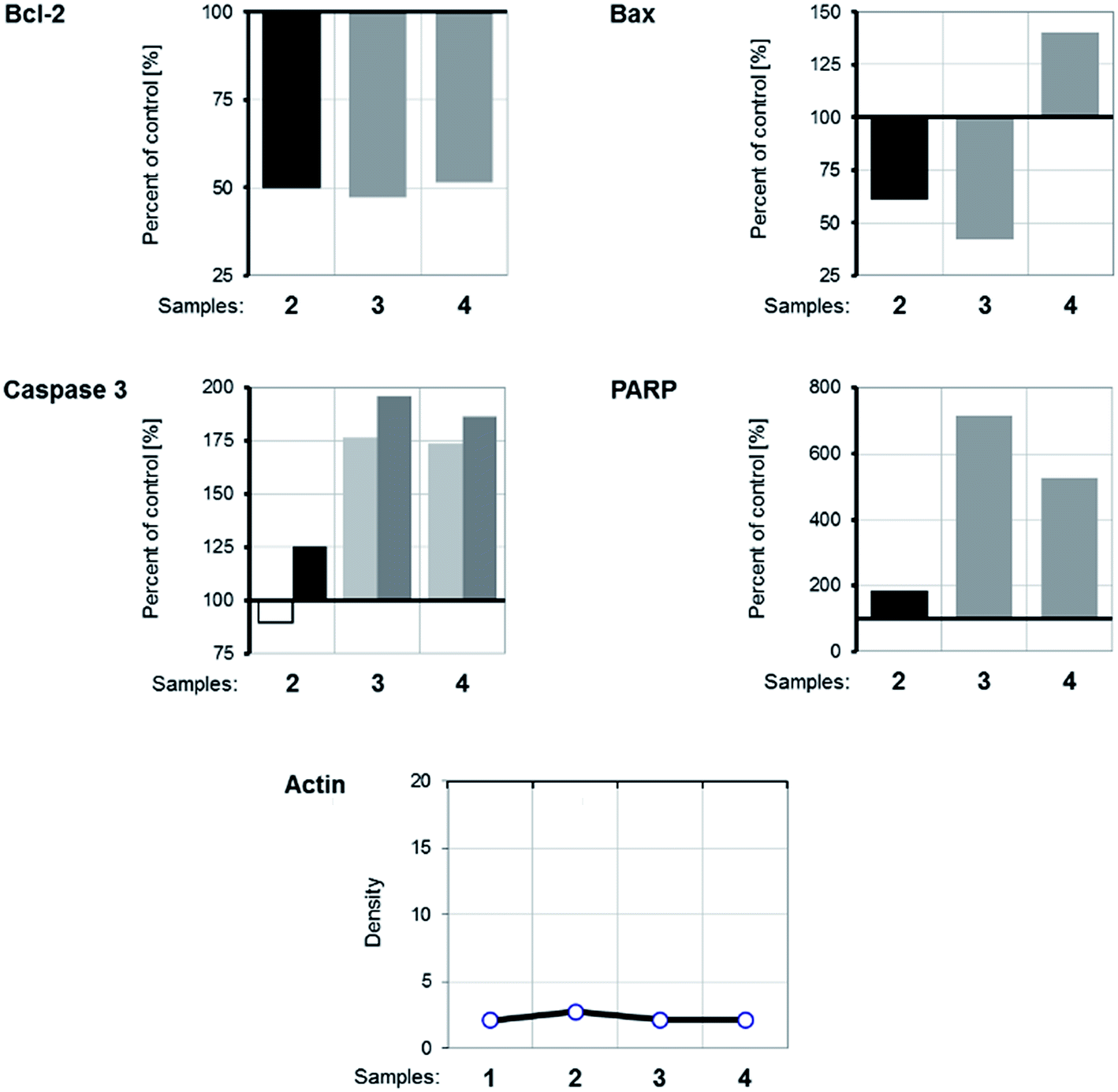 | ||
| Fig. 11 Graphical presentation of protein expression densitometry data obtained by WB analysis and processed with ImageJ [http://imagej.nih.gov]. Expression of proteins belonging to an apoptotic signaling pathway in the samples are compared to untreated samples and presented as percentage of control. The densitometry of actin expression, which serves as internal control, is presented as measured. (1) Control; (2) doxorubicin; (3) Na[Rh(ed3a)Cl]·H2O (2); (4) [Rh(ed3a)(OH2)]·H2O (1). | ||
The expression pattern of the investigated proteins of the apoptotic signaling pathway of the most affected tumor cell line HeLa reveals a conducted apoptosis. This is confirmed by the detection of PARP protein cleavage in samples treated with both RhIII complexes (1) and (2). The reduction of Bcl-2 protein expression after the treatment was observed in samples. Also, a much larger increase of Bax protein expression relative to the control was observed after treatment with [Rh(ed3a)(OH2)]·H2O (1) (sample 4). Bcl-2 and Bax proteins are important members of the Bcl protein family and are located at the beginning of the apoptotic signaling pathway.41,42
Factors that can influence their balance may instigate cells to survival or death. Antiapoptotic Bcl-2 proteins block the intrinsic apoptosis pathway. Their concentration is increased in human cancer cells and they are important targets for new therapies.41,42 Therefore, the observed decrease (down regulated) Bcl-2 expression as a result of the two new RhIII complexes (1) and (2), as well as the upregulation of Bax expression by complex (1) are of pharmacological importance.
Protein expression analysis also gives a picture of increased caspase 3 activity, which suggests that the caspase-dependent apoptosis underlies the observed cytotoxic effect.
Both cell cycle progression and apoptosis are crucial for maintaining tissue homeostasis. These sets of events are coupled and share certain regulatory molecules. Cellular damage and stress signals result in cell cycle termination that provide the cell a time to repair the damage. If the cell cannot recover, the apoptotic program is activated. Analysis of the cell cycle in HeLa cells treated with (1) and (2) (Fig. 12) showed that both substances induced G0/G1 cycle termination (from 74.97% in untreated cells to 81.70% in cells treated with complex (1) and 81.52% when treated with complex (2)). Concomitantly, the percentage of cells in the S phase decreased from 5.04% in control cells to 0.65% and 0.85%, respectively. These results indicate that blockade of DNA synthesis induced by the tested substances is a possible trigger of apoptosis.
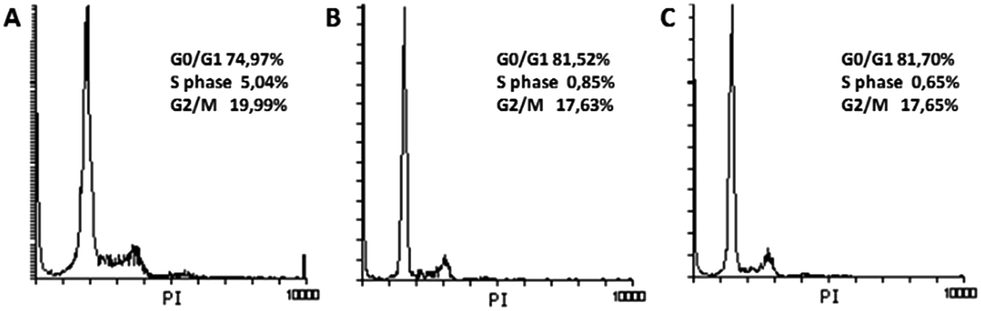 | ||
| Fig. 12 Cell cycle analysis. Histograms present cell cycle distribution in untreated HeLa cells (A) and cells treated for 48 h with (1) (B) and (2) (C). | ||
Conclusions
Two new RhIII–ed3a3− complexes, the neutral cis-equatorial-[Rh(ed3a)(OH2)]·H2O (1) and the anionic cis-equatorial-Na[Rh(ed3a)Cl]·H2O (2), were isolated and characterized using experimental and computational techniques. The cis-equatorial geometry was verified by X-ray crystallography for (1) and (2) and solution spectroscopy (UV-Vis, IR, NMR) indicates that, for (1) this is retained in solution and that it is also the geometry of (2), as expected from published strain energy analyses of similar systems.5 MM and DFT results are in agreement with the experimental X-ray and spectroscopic data and the observed isomer preference. The composition and conditional stability constants of RhIII complexes were determined by pH potentiometry and UV-Vis spectrophotometry. The speciation scheme of the RhIII–ed3a3− system consists of four species: [Rh(Hed3a)]+, [Rh(ed3a)], [Rh(ed3a)2]3− and [Rh(OH)(ed3a)]−. At physiological pH the dominant species in the system are [Rh(Hed3a)]+ and [Rh(ed3a)] complexes. Knowledge of the composition and stability of these complexes in the system can contribute to a better understanding of their physiological roles in different tissues and cell system. Biological tests demonstrated that the RhIII complexes (1) and (2) show an interesting cytotoxicity behaviour in comparison to the doxorubicin and cisplatin reference systems: in contrast to these, they are inactive against the MCF-7 human breast carcinoma and healthy MRC-5 cell lines but are very active against the human cervix adenocarcinoma HeLa cell line. Against HT-29 and A549 cells, the aqua complex (1) is significantly more active than the chloro complex (2). Flow cytometry and Western blot analysis revealed mechanism of antitumor activity of tested complexes: cytostatic as a result of DNA synthesis blockade and cytotoxic through induction of apoptosis.Experimental
Materials and methods
Reagent grade commercially available chemicals were used without further purification. The preparation of the calcium salt of ed3a3−, Ca3(ed3a)2·12H2O was reported previously.5 Monochloroacetic acid, ethylenediamine, calcium hydroxide, RhIII chloride hydrate, hydrochloric acid, sodium hydroxide and sodium chloride were purchased from Sigma-Aldrich. A rhodium(III) chloride stock solution was prepared by dissolving dried RhCl3·H2O, p.a. (Sigma-Aldrich), in doubly-distilled water. Elemental microanalyses for C, H, N were performed on a CHN-O-vario EL by the Microanalysis Laboratory at the chemical institutes at Heidelberg University. IR spectra were measured with a Perkin-Elmer 16 PC FTIR instrument as KBr pellets. NMR spectra were recorded at 200 MHz (1H) and 50 MHz (13C) on a Bruker Advance I 200 instrument with deuterated solvents as reference. Electronic absorption spectra were obtained from a Tidas II J&M spectrophotometer at concentrations of the RhIII complexes (aqueous solutions) of approx. 1.0 × 10−3 M. The potentiometric measurements were carried out using a Methrom 827 pH meter with a Titronic universal piston burette and combined glass electrode. Spectroscopic measurements were made with a double beam UV-Vis spectrophotometer model Cary 300 (Agilent Technologies, Santa Clara, USA) with 1.0 cm quartz cells. Melting points were determined using a Stuart digital melting point apparatus with accuracy ±1 °C.Synthetic procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 67.30, 64.59, 62.70, 56.53, 56.17 (CH2).O), 66.60, 63.72, 61.84, 57.37, 57.04 (CH2). The third band, remaining on top of the column after elution with 100 mM NaCl, was eluted with concentrated NaCl and was found to be a mixture of different hydroxo species with charges higher than −2.
O), 67.30, 64.59, 62.70, 56.53, 56.17 (CH2).O), 66.60, 63.72, 61.84, 57.37, 57.04 (CH2). The third band, remaining on top of the column after elution with 100 mM NaCl, was eluted with concentrated NaCl and was found to be a mixture of different hydroxo species with charges higher than −2.Crystal structure determination
Crystal data and details of the structure determinations are listed in Table 8. Full shells of intensity data were collected at low temperature with an Agilent Technologies Supernova-E CCD diffractometer (Mo-Kα radiation for complex (1) and Cu-Kα radiation for complex (2), microfocus tubes, multilayer mirror optics). Data were corrected for air and detector absorption, Lorentz and polarization effects;43 absorption by the crystal was treated numerically (Gaussian grid).43,44 The structures were solved by intrinsic phasing45 (complex (1)) or by the heavy-atom method combined with structure expansion by direct methods applied to difference structure factors46 (complex (2)) and refined by full-matrix least squares methods based on F2 against all unique reflections.47 All non-hydrogen atoms were given anisotropic displacement parameters. Hydrogen atoms were generally input at calculated positions and refined with a riding model.| (1) | (2) | |
|---|---|---|
| Formula | C8H15N2O8Rh | C8H13ClN2NaO7Rh |
| Mr | 370.13 | 410.55 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c |
| a/Å | 6.94313(8) | 13.4003(6) |
| b/Å | 15.71268(18) | 6.9326(3) |
| c/Å | 11.09315(11) | 14.9772(8) |
| β/° | 96.9886(10) | 115.328(6) |
| V/Å3 | 1201.22(2) | 1257.62(12) |
| Z | 4 | 4 |
| F000 | 744 | 816 |
| dc/Mg m−3 | 2.047 | 2.168 |
| X-radiation, λ/Å | Mo-Kα, 0.71073 | Cu-Kα, 1.5418 |
| μ/mm−1 | 1.462 | 13.639 |
| Max., min. transmission factors | 0.946, 0.792 | 0.815, 0.285 |
| Data collect. temp./K | 110(1) | 110(1) |
| θ range/° | 3.2 to 29.0 | 3.7 to 70.9 |
| Index ranges (indep. set) h, k, l | −9 … 9, −21 … 21, −15 … 15 | −16 … 16, −8 … 8, −17 … 15 |
| Reflections measured | 66435 |
24110 |
| Unique [Rint] | 3126 [0.0598] | 2410 [0.0938] |
| Observed [I ≥ 2σ(I)] | 2965 | 1861 |
| Data/restraints/parameters | 3126/0/87 | 2410/0/190 |
| GooF on F2 | 1.276 | 1.030 |
| R indices [F > 4σ(F)] R(F), wR(F2) | 0.0313, 0.0518 | 0.0332, 0.0739 |
| R indices (all data) R(F), wR(F2) | 0.0358, 0.0527 | 0.0510, 0.0807 |
| Difference density: max, min/e Å−3 | 0.520, −0.671 | 1.334, −0.647 |
The positions of some hydrogen atoms (those on N and O) were taken from difference Fourier syntheses and refined. CCDC 1412103 (for (1)), CCDC 1412104 (for (2)) contain the supplementary crystallographic data for this paper.†
Solution studies
All the equilibrium measurements were made at a constant ionic strength maintained by 0.1 M NaCl at 25 °C. To determine the protonation constants of ed3a3− and conditional stability constants of the complexes formed with ed3a3− were determined by potentiometric titrations. The metal-to-ligand ratios were 1:1.5 and 1:3. The concentration of the RhIII was 2 mM.
The potentiometric measurements were carried out using a Methrom 827 pH meter with a Titronic universal Piston burette and combined glass electrode. The temperature of the sample solutions (20 mL) was maintained at 25.0 °C by circulating thermostatically controlled water through the jacket of the titration vessel. The samples were stirred with a magnetic stirrer, and to avoid the effect of CO2. All of the measurements were performed under a nitrogen atmosphere.
Spectroscopic measurements were made on solutions in which the concentration of RhIII and ed3a3− were constant (CRh = 2.0 mM, Ced3a = 6.2 mM and Ced3a = 3.1 mM), while the pH was varied between 2.5 and 7.8. The pH of the test solutions was measured with a Methrom 827 pH meter. Stable values within 0.02 pH and 0.01 absorbance units, were attained for the first series of solutions after 1 h at 25 °C (these remained stable for 30 min), and for the second series after heating for 3 h at 145 °C (closed vessel, see above), and these remained stable for 3 h at the same temperature. Spectra of the test solutions were recorded in the 250–600 nm wavelength range. To calculate the equilibrium constants the HYPERQUAD2006 and HypSpec2014 programs were used.27,29 The concentration distribution diagrams were obtained using the program HYSS2006 under the experimental conditions described.48
Computational details
| Utotal = ∑(Eb + Eθ + EΦ + Enb + Eδ) | (4) |
Input coordinates were obtained from crystal structures data or produced with HyperChem 7.01.50 Parameters not reported before are given in Table 9. All other parameters are given in the literature.34
| Bond distance parameters | ||
|---|---|---|
| Bond type | Force constant (mdyn Å−1) | Strain-free bond distance (Å) |
| a dyn = 10−5 N. | ||
| Rh–Ndiamine | 1.75 | 2.05 |
| Rh–Cl | 1 | 2.35 |
| Rh–Ocarboxyl | 1.75 | 1.99 |
| Rh–Owater | 0.5 | 1.9 |
| Valence angle parameters | ||
|---|---|---|
| Valence angle type | Force constant (mdyn Å rad−2) | Strain-free valence angle (rad) |
| Ocarboxyl–Rh–Ocarboxyl | 0.026 | 1.571 |
| Cl–Rh–Ocarboxyl | 0.026 | 1.571 |
| Ocarboxyl–Rh–Ndiamine | 0.026 | 1.571 |
| Ocarboxyl–Rh–Owater | 0.026 | 1.571 |
| Cl–Rh–N(diamine) | 0.026 | 1.571 |
| Ndiamine–Rh–Ndiamine | 0.026 | 1.571 |
| Ndiamine–Rh–Owater | 0.026 | 1.571 |
| Rh–Owater–H | 0.100 | 1.915 |
| Rh–Ocarboxyl–Ccarboxyl | 0.026 | 1.970 |
| Torsion angle parameters | ||
|---|---|---|
| Bond torsion angle type | Force constant (mdyn Å) | Offset angle (rad) |
| Ocarboxyl–Rh | 0.0000 | 0.0000 |
| Ndiamine–Rh | 0.0000 | 0.0000 |
| Owater–Rh | 0.0000 | 0.0000 |
| Rh–Ocarboxyl–Ccarboxyl–Ocarboxyl | 0.0400 | 0.0570 |
Biological tests
MTT assay. Growth inhibition was evaluated by tetrazolium colorimetric MTT assay (SIGMA).51 Exponentially growing cells were harvested and plated into 96-well microtiter plates (Costar) at optimal seeding density of 10 × 103 cells per well. Tested substances and reference compounds doxorubicin and cisplatin, at tenfold the required final concentration, were added (10 μL per well) to all wells except to the control ones and microplates were incubated for 48 h. Three hours before the end of the incubation period, 10 μL of MTT solution (5 mg mL−1) was added to all wells. Acid-isopropanol (100 μL of 0.04 N HCl in isopropanol) was added to all wells and mixed thoroughly to dissolve the dark blue crystals. After a few minutes at room temperature, to ensure that all crystals were dissolved, the plates were read on a spectrophotometer plate reader (Multiscan MCC340, Labsystems) at 540/690 nm. Inhibition of growth was expressed as a percent of a control and cytotoxicity was calculated according to the formula: (1 − Atest/Acontrol) × 100. The substance potency was expressed as the IC50 (50% inhibitory concentration). Two independent experiments were set out with quadruplicate wells for each concentration of the compound. IC50 values were determined by Median effect analysis.52
Cytomics FC500 (Beckman Coulter, USA) and the percent of viable, apoptotic and necrotic cells was evaluated using Flowing Software (http://www.flowingsoftware.com/). The results were presented by dot plots.
Acknowledgements
The authors are grateful to the Serbian Ministry of Education, Science and Technological Development for the financial support (Project No. III41010). One of the authors (Marija Jeremić) is particularly grateful to the Erasmus Mundus Programme for the awarded scholarship.References
- (a) D. J. Radanović, Coord. Chem. Rev., 1984, 54, 159–261 CrossRef; (b) B. E. Douglas and D. J. Radanović, Coord. Chem. Rev., 1993, 128, 139–165 CrossRef CAS.
- (a) R. Meier, C. Platas-Iglesias, F. W. Heinemann, G. Linti, J. Schulte and S. K. Srivastava, Inorg. Chem., 2014, 53, 6684–6697 CrossRef CAS PubMed; (b) A. Brausam, J. Maigut, R. Meier, P. Á. Szilágyi, H. J. Buschmann, W. Massa, Z. Homonnay and R. van Eldik, Inorg. Chem., 2009, 48, 7864–7884 CrossRef CAS PubMed.
- C. Maricondi, S. Utsuno, D. J. Radanović, S. R. Trifunović, J. E. Abola and B. Douglas, Inorg. Chim. Acta, 1988, 142, 135–149 CrossRef CAS.
- S. Grubišić, M. Gruden-Pavlović, S. R. Niketić, S. Kaizaki and N. Sakagami-Yoshida, Inorg. Chem. Commun., 2003, 6, 1180–1184 CrossRef.
- (a) Z. D. Matović, A. Meetsma, V. D. Miletić and P. J. van Koningsbruggen, Inorg. Chim. Acta, 2007, 360, 2420–2431 CrossRef; (b) S. Belošević, M. Ćendić, A. Meetsma and Z. D. Matović, Polyhedron, 2013, 50, 473–480 CrossRef.
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- B. Rosenberg, L. Van Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- (a) R. B. Weiss and M. C. Christian, Drugs, 1993, 46, 360–377 CrossRef CAS PubMed; (b) G. Giaccone, Drugs, 2000, 59, 9–17 CrossRef CAS PubMed.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- Y. P. Ho, S. C. F. Au-Yeung and K. K. W. To, Med. Res. Rev., 2003, 23, 633–655 CrossRef CAS PubMed.
- L. Ronconi and P. J. Sadler, Coord. Chem. Rev., 2007, 251, 1633–1648 CrossRef CAS.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- A. Taylor and N. Carmichael, Cancer Studies, 1953, 2, 36–79 Search PubMed.
- M. J. Cleare and P. C. Hydes, Met. Ions Biol. Syst., 1980, 11, 1–62 CAS.
- G. Mestroni, E. Alessio, A. Sessanta o Santi, S. Geremia, A. Bergamo, G. Sava, A. Boccarelli, A. Schettino and M. Coluccia, Inorg. Chim. Acta, 1998, 273, 62–71 CrossRef CAS.
- I. Haiduc and C. Silvestru, “Cobalt, Rhodium and Iridium” in Organometallics in Cancer Chemotherapy, CRC Press, Boca Raton, Florida, 1990, pp. 169–207 Search PubMed.
- N. Katsaros and A. Anagnostopoulou, Crit. Rev. Oncol. Hematol., 2002, 42, 297–308 CrossRef CAS PubMed.
- (a) G. Gupta, E. Denoyelle-Di-Muro, J.-P. Mbakidi, S. Leroy-Lhez, V. Sol and B. Therrien, J. Organomet. Chem., 2015, 787, 44–50 CrossRef CAS; (b) H. Pruchnik, M. Latocha, A. Zielińska and F. P. Pruchnik, J. Organomet. Chem., 2016, 822, 74–79 CrossRef CAS; (c) M. Kalidasan, S. Forbes, Y. Mozharivskyj, M. Ahmadi, Z. Ahmadihosseini, R. M. Phillips and M. R. Kollipara, Inorg. Chim. Acta, 2014, 421, 349–358 CrossRef CAS; (d) O. Dömötör, S. Aicher, M. Schmidlehner, M. S. Novak, A. Roller, M. A. Jakupec, W. Kandioller, C. G. Hartinger, B. K. Keppler and É. A. Enyedy, J. Inorg. Biochem., 2014, 134, 57–65 CrossRef PubMed; (e) H.-R. Zhang, Y.-C. Liu, Z.-F. Chen, T. Meng, B.-Q. Zou, Y.-N. Liu and H. Liang, New J. Chem., 2016, 40, 6005–6014 RSC.
- S. Schäfer, I. Ott, R. Gust and W. S. Sheldrick, Eur. J. Inorg. Chem., 2007, 19, 3034–3046 CrossRef.
- M. A. Nazif, R. Rubbiani, H. Alborzinia, I. Kitanovic, S. Wölfl, I. Ott and W. S. Sheldrick, Dalton Trans., 2012, 41, 5587–5598 RSC.
- Y. Geldmacher, K. Splith, I. Kitanovic, H. Alborzinia, S. Can, R. Rubbiani, M. A. Nazif, P. Wefelmeier, A. Prokop, I. Ott, S. Wölfl, I. Neundorf and W. S. Sheldrick, J. Biol. Inorg. Chem., 2012, 17, 631–646 CrossRef CAS PubMed.
- Y. Geldmacher, M. Oleszak and W. S. Sheldrick, Inorg. Chim. Acta, 2012, 393, 84–102 CrossRef CAS.
- G. H. Y. Lin, J. D. Leggett and R. M. Wing, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 1023–1030 CrossRef CAS.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- U. Rychlewska, M. I. Djuran, B. Warżajtis, D. D. Radanović, M. M. Vasojević and D. J. Radanović, Polyhedron, 2003, 22, 3265–3276 CrossRef CAS.
- A. E. Martell and R. M. Smith, Critical Stability Constants, Plenum Press, New York, 1974, vol. 1–4 Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- P. Djurdjevic, R. Jelic, L. Joksovic, I. Lazarevic and M. Jelikic-Stankov, Acta Chim. Slov., 2010, 57, 386–397 CAS.
- P. Gans, A. Sabatini and A. Vacca, Ann. Chim., 1999, 89, 45–49 CAS.
- É. A. Enyedy, O. Dömötör, C. M. Hackl, A. Roller, M. S. Novak, M. A. Jakupec, B. K. Keppler and W. Kandioller, J. Coord. Chem., 2015, 68(9), 1583–1601 CrossRef.
- S. I. Bezzubov, V. D. Dolzhenko and Y. M. Kiselev, Zh. Anal. Khim., 2012, 67(2), 176–179 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearmark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- MOMEC97, a molecular mechanics program for coordination compounds adapted to HyperChem: http://www.uni-heidelberg.de/comba-group;%20molecular%20modeling.
- (a) P. V. Bernhardt and P. Comba, Inorg. Chem., 1992, 31, 2638–2644 CrossRef CAS; (b) P. Comba, T. W. Hambley and M. Ströhle, Helv. Chim. Acta, 1995, 78, 2042–2047 CrossRef CAS; (c) J. E. Bol, C. Buning, P. Comba, J. Reedijk and M. Ströhle, J. Comput. Chem., 1998, 19, 512–523 CrossRef CAS.
- (a) S. Kumar, Cell Death Differ., 2007, 14, 32–43 CrossRef CAS PubMed; (b) M. Vogler, D. Dinsdale, M. J. S. Dyer and G. M. Cohen, Cell Death Differ., 2009, 16, 360–367 CrossRef CAS PubMed.
- (a) C. Adrain and S. J. Martin, Sci. Signaling, 2009, 2, pe62 CrossRef PubMed; (b) S. E. Logue and S. J. Martin, Biochem. Soc. Trans., 2008, 36, 1–9 CrossRef CAS PubMed.
- R. Kim, M. Emi, K. Tanabe, Y. Uchida and K. Arihiro, Eur. J. Surg. Oncol., 2006, 32, 269–277 CrossRef CAS PubMed.
- G. Koopman, C. P. Reutelingsperger, G. A. Kuijten, R. M. Keehnen, S. T. Pals and M. H. van Oers, Blood, 1994, 84, 1415–1420 CAS.
- H. Lecoeur, M. C. Prévost and M. L. Gougeon, Cytometry, 2001, 44, 65–72 CrossRef CAS PubMed.
- D. Baskic, S. Popovic, P. Ristic and N. N. Arsenijevic, Cell Biol. Int., 2006, 30, 924–932 CrossRef CAS PubMed.
- L. Galluzzi, I. Vitale, J. M. Abrams, E. S. Alnemri, E. H. Baehrecke, M. V. Blagosklonny, T. M. Dawson, V. L. Dawson, W. S. El-Deiry, S. Fulda, E. Gottlieb, D. R. Green, M. O. Hengartner, O. Kepp, R. A. Knight, S. Kumar, S. A. Lipton, X. Lu, F. Madeo, W. Malorni, P. Mehlen, G. Nuñez, M. E. Peter, M. Piacentini, D. C. Rubinsztein, Y. Shi, H.-U. Simon, P. Vandenabeele, E. White, J. Yuan, B. Zhivotovsky, G. Melino and G. Kroemer, Cell Death Differ., 2012, 19, 107–120 CrossRef CAS PubMed.
- S. Fulda and K. M. Debatin, Oncogene, 2006, 25, 4798–4811 CrossRef CAS PubMed.
- CrysAlisPro, Agilent Technologies UK Ltd, Oxford, UK, 2011–2014 Search PubMed.
- W. R. Busing and H. A. Levy, Acta Crystallogr., 1957, 10, 180–182 CrossRef CAS.
- (a) G. M. Sheldrick, SHELXT, University of Göttingen and Bruker AXS GmbH, Karlsruhe, Germany, 2012–2014 Search PubMed; (b) M. Ruf and B. C. Noll, Application Note SC-XRD 503, Bruker AXS GmbH Karlsruhe, Germany, 2014 Search PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- (a) P. T Beurskens, G. Beurskens, R. de Gelder, J. M. M.Smits, S. Garcia-Granda and R. O. Gould, DIRDIF-2008, Radboud University Nijmegen, The Netherlands, 2008 Search PubMed; (b) P. T. Beurskens, in Crystallographic Computing 3, ed. G. M. Sheldrick, C. Krüger and R. Goddard, Clarendon Press, Oxford, UK, 1985, pp. 216–266 Search PubMed.
- (a) G. M. Sheldrick, SHELXL-20xx, University of Göttingen and Bruker AXS GmbH Karlsruhe, Germany, 2012–2014 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- L. Alderighi, P. Gans, A. Lenco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- HyperChem™, Hypercube, Inc., 1115 NW 4th Street, Gainesville, Florida 32601, USA.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- T. C. Chou and P. Talalay, Adv. Enzyme Regul., 1984, 22, 27–55 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further solution study details, IR, 1H NMR, 13C NMR data. CCDC 1412103 and 1412104 for the new compounds are included. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26199j |
| This journal is © The Royal Society of Chemistry 2017 |