DOI:
10.1039/C6RA26150G
(Paper)
RSC Adv., 2017,
7, 3136-3144
The controllable construction and properties characterization of organic–inorganic hybrid materials based on benzoxazine-bridged polysilsesquioxanes
Received
2nd November 2016
, Accepted 29th November 2016
First published on 13th January 2017
Abstract
Using a hydrolysis condensation of triethoxysilane to prepare a synthetic benzoxazine-bridged bis(triethoxysilane) compound (BS), a series of benzoxazine-bridged polysilsesquioxanes precursors were prepared with different degrees of hydrolysis condensation. Ring-opening thermal homopolymerization of the benzoxazine rings in these precursors was employed to obtain several organic–inorganic hybrid materials. The chemical structures of these synthetic compounds were characterized by Fourier transform infrared (FTIR) spectroscopy, nuclear magnetic resonance (1H, 13C, 29Si NMR) spectroscopy and elemental analysis (EA). For the morphologies of these hybrid materials, different features of the lamellar structures were explored by transmission electron microscopy (TEM). From the results of energy dispersive X-ray spectroscopy (EDS), these lamellar structures were found to be comprised of carbon-rich (C-rich) and silicon-rich (Si-rich) layers. From a view-point of the structure–property relationships, the optical (photoluminescence (PL)), thermal (glass transition temperature (Tg) and thermogravimetric analysis (TGA)) properties were well characterized and discussed. Based on these results, we proposed that this chemically controllable method has potential as an effective approach used to prepare organic–inorganic hybrid materials with controllable morphology.
1. Introduction
In comparison with disordered nanostructured composites, ordered composites have been created with fantastic physical, chemical and biological properties, which display high potential in biomedicine, catalysis, sensor, energy conversion, and so on.1–5 In order to obtain ordered nanostructured composites, increasing research has been focused on the method of controllable self-organization.6–8 Recently, organic–inorganic hybrid materials constructed using this approach have gained great attention.9–12 Composite nanomaterials with well-defined structures have been extensively explored to realize a combination of the respective properties of each component or achieve cooperatively enhanced performance. Structural control of organic bridged silsesquioxanes has been previously reported concerning self-assembly behaviour.13–16 As a class of versatile functional organic–inorganic hybrid materials, organic bridged polysilsesquioxanes with tunable properties can be prepared via sol–gel processing of the organic bridged silsesquioxanes and have been widely studied.17–20 The organic group acts as a bridge for the silsesquioxane forming groups, is covalently attached to the trifunctional silicon groups through Si–C bonds, and can be varied in length, rigidity, geometry of substitution, and functionality. Due to the presence of the organic bridging group as an integral part of the material, some modulate bulk properties, such as porosity morphology,21 thermal stability,22 dielectric constant,13 optical clarity,23 chemical resistance,24 hydrophobicity,25 and refractive index,26 can be realized by this variability. Based on this enlightenment, by taking advantage of the self-organization ability of the organic bridged polysilsesquioxanes, we speculated whether it was feasible to obtain organic–inorganic hybrid materials with controllable morphology.
We studied the methods of preparing organic bridged polysilsesquioxanes materials with long range order on the nanometer length scale; one promising approach is the use of monomers possessing organic bridging groups with intrinsic intermolecular affinity (e.g. hydrogen bonding, π–π interactions and even weak van der Waals forces) to produce materials with uniform nano-sized pores in the final product. Upon carefully controlled polymerization, the repeating units in the polymer are aligned to form an ordered nanostructure, which can be called the self-assembly without templates or “designed”.27–29 For this behavior of self-assembly, the combined effect of both organic bridging groups and the polysilsesquioxane moieties may result in the formation of a nano-scale morphology. In our previous studies,30,31 an investigation on benzoxazine was carried out. From a view-point of the structure–property relationships, the introduction of a heterocyclic benzoxazine moiety that can form strong intermolecular hydrogen bonds may be helpful for the self-assembly of bridged polysilsesquioxanes.31,32 Corriu et al.33 pointed out that polymerization of the bridging groups favored the arrangement of the organic fragments in the bridged polysilsesquioxanes. Hence, it is of interest to incorporate a benzoxazine group into the bridging unit of the bridged silsesquioxane compound for the preparation of organic–inorganic hybrid materials. We expect that thermally activated ring-opening addition polymerization can promote the formation of organic polymer domains; moreover, the appearance of a nano-size phase separation between organic domains and inorganic domains may be generated. Accordingly, we also speculated that some ordered nanostructures could be present in the inorganic–organic hybrid materials.
In this study, benzoxazine-bridged bis(triethoxysilane) (BS) was synthesized and its chemical structure was investigated by Fourier transform infrared (FTIR) spectroscopy, nuclear magnetic resonance (1H, 13C and 29Si NMR) spectroscopy and elemental analysis (EA). Using the hydrolysis condensation of triethoxysilane to prepare BS, a series of benzoxazine-bridged polysilsesquioxanes precursors with different degrees of hydrolysis condensation were prepared and monitored using FTIR spectroscopy and EA. Differential scanning calorimetry (DSC) and FTIR spectroscopy were applied to investigate the ring-opening thermal homopolymerization process and the products of the precursors, respectively. The effects of the different degrees of hydrolysis condensation in BS on the morphologies and properties of the hybrid materials have been researched. As expected, the different features of the lamellar structures were explored using TEM of the hybrid materials. The significant results can be explained by a phase separation due to a synergistic effect of the polymerization-induced enrichment of the benzoxazine organic fragments and the self-organization enrichment of the inorganic polysilsesquioxanes. Furthermore, changes in the PL properties and thermal stabilities of the hybrid materials indirectly demonstrate the existence of the lamellar structure. Based on these results, we propose that this research will provide an effective approach to prepare hybrid materials with controllable morphology.
2. Experimental
2.1 Materials
Bisphenol A (98%), (3-aminopropyl)triethoxysilane (95%), paraformaldehyde (96%), calcium hydride (CaH2) (AR), tetramethylammonium hydroxide (TMAH) (AR) and aniline (99%) were purchased from Aladdin Chemical Reagent Co., Ltd. Chloroform and 4-dioxane (99.9%) were obtained from Tianjin Chemical Reagent Co., Ltd and dried with molecular sieves prior to use.
2.2 Hybrid materials preparation
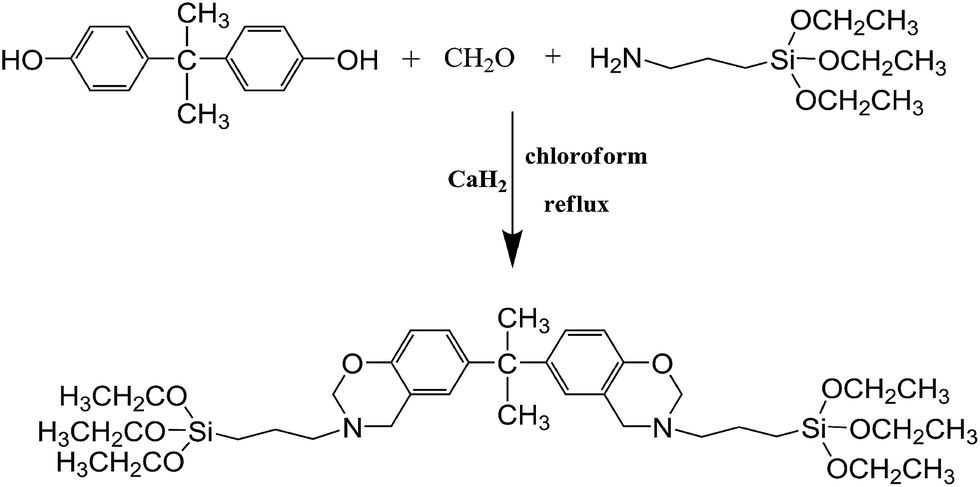
2.2.1 Synthesis of benzoxazine-bridged bis(triethoxysilane) monomer (BS). The synthetic pathways used to prepare BS are shown in Scheme 1. In a 500 mL three neck round bottom flask equipped with a heating mantle, magnetic stirrer, reflux condenser, water knockout vessel, nitrogen inlet/outlet and thermometer, CaH2 (12.6 g, 0.3 mol) and 200 mL of chloroform were added. The mixture was stirred for 20 min at room temperature. Then, paraformaldehyde (12.12 g, 0.4 mol) and (3-aminopropyl)triethoxysilane (44.3 g, 0.2 mol) were added and rapidly stirred at 60 °C. An hour later, bisphenol-A (22.8 g, 0.1 mol) was introduced into the abovementioned reaction system. The reaction mixture was stirred at 75 °C for 6 h under a N2 atmosphere, cooled and filtered. The volatile components of the resulting yellow transparent liquid were removed using a rotating evaporation instrument, and a bright yellow viscous liquid without further purification was obtained in 87% yield.
 |
| | Scheme 1 The synthetic route used to prepare benzoxazine-bridged bis(3-aminopropyltriethoxysilane) (BS). | |
2.2.2 Preparation of the hydrolysis products of BS (HBSn). BS (36 g, 0.4 mol) was dissolved in 1,4-dioxane and mixed with 10 wt% TMAH solution (the concentration of TMAH was 1.3 wt% in the reaction system). The molar ratio of water/BS in the reaction mixture was 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The reaction was carried out at room temperature with different hydrolysis times of 2 h, 4 h and 8 h. At the end of each reaction, the volatile components were removed immediately by the rotating evaporation instrument. The corresponding benzoxazine-bridged polysilsesquioxanes precursors were abbreviated as HBS2, HBS4 and HBS8. The colors of these products were as follows: HBS2 was translucent and yellow, HBS4 was opaque and brown-yellow and HBS8 was also opaque and brown.
:2. The reaction was carried out at room temperature with different hydrolysis times of 2 h, 4 h and 8 h. At the end of each reaction, the volatile components were removed immediately by the rotating evaporation instrument. The corresponding benzoxazine-bridged polysilsesquioxanes precursors were abbreviated as HBS2, HBS4 and HBS8. The colors of these products were as follows: HBS2 was translucent and yellow, HBS4 was opaque and brown-yellow and HBS8 was also opaque and brown.
2.2.3 Thermal homopolymerization of the benzoxazine precursor. The benzoxazine-bridged polysilsesquioxanes precursors (HBS2, HBS4 and HBS8) were immediately poured into stainless molds and then thermally cured at 100 °C for 1 h, 150 °C for 1 h, 180 °C for 2 h, 220 °C for 2 h, and 250 °C for 2 h. The three resulting polybenzoxazine/polysilsesquioxanes hybrid materials were abbreviated as PHBS2, PHBS4 and PHBS8. With the same thermal treatment as described above, the contrast material labeled as PBS (polybenzoxazine bridged bis(triethoxysilane)) was obtained from the curing of BS without hydrolysis.
2.3 Instrumental analysis
Proton (1H), carbon (13C) and silicon (29Si) nuclear magnetic resonance (NMR) spectra were measured on a Bruker AM-400S (400 MHz) spectrometer using tetramethylsilane (TMS) as an internal standard and deuterated chloroform (at 7.26 ppm) as solvent. Fourier transform infrared spectra (FTIR) were obtained on a Nicolet Avatar 320 FT-IR spectrophotometer. Samples were finely ground with KBr powder and pressed into disks. The quantitative analysis of C, H, N, O and Si were carried out on an F002 Heraeus CHN-O rapid elemental analyzer employing acetanilide as a standard. The dynamic homopolymerization reactions of these benzoxazine compounds were monitored on a Diamond/Pyris DSC (differential scanning calorimeter) under a dry nitrogen atmosphere. All the samples (about 10 mg) were heated from 50 °C to 300 °C at a heating rate of 10 °C min, during the study of the dynamic homopolymerization reactions. The morphologies and chemical components of the hybrid materials were characterized using a JEOL JEM-2010 TEM connected to energy dispersive X-ray spectrometer (EDS) at an acceleration voltage of 3 kV for elemental analysis. The electron beam energy was 50 keV in all cases. The samples were trimmed using an ultra-thin microtome machine, and the sectioned samples (ca. 50 nm in thickness) were placed on 200 mesh copper grids for observations. The thermal stabilities of the thermal homopolymerization samples were investigated using a Perkin-Elmer TGA-6 thermogravimetric analyzer at a heating rate of 10 °C min−1 from 50 °C to 800 °C under nitrogen atmosphere at a flow rate of 90 mL min−1 in all cases. The glass translation temperature (Tg) was determined using DSC under a dry nitrogen atmosphere. All the samples (about 10 mg) followed this thermal treatment process. First, they were quickly heated up to 330 °C for 5 min; second, they were cooled down to ambient temperature at a cooling rate of 20 °C min. After that, they were heated from ambient temperature to 330 °C at a heating rate of 20 °C min−1 again for the detection of the Tg. Photoluminescence spectra were measured on a Hitachi 4500 fluorescence spectrophotometer. The size of samples for the fluorescence test was 10 mm × 40 mm × 1 mm (width × length × thickness).
3. Results and discussion
3.1 Preparation and characterization
3.1.1 Characterization of BS. As shown in Scheme 1, BS was prepared over two steps. In the first step, formaldehyde was reacted with (3-aminopropyl)triethoxysilane to form a triazine compound with H2O as the by-product. In order to avoid the hydrolysis reaction between Si–O–C and H2O, chloroform as a dispersant played the role of preventing H2O from accessing the Si–O–CH2CH3 group. Due to the addition of CaH2, the produced H2O was efficiently removed. In the second step, the triazine compounds and phenol were reacted with one another via Mannich reaction to form the benzoxazine heterocyclic compound.34 The chemical structure of BS was confirmed using 1H, 13C and 29Si NMR, FTIR and EA. Fig. 1a shows the 1H NMR spectrum of BS. The resonances appearing at 3.91 ppm and 4.82 ppm were assigned to the methylene protons of Ar–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–N and O–C2–N in the benzoxazine group, respectively. The characteristic resonance peaks of BS appear at 3.77 ppm for O–C2–CH3, 2.71 ppm for N–C2–CH2, 1.64 ppm for CH2–C2–CH2, 1.55 ppm for C–C3, 1.19 ppm for CH2–C3 and 0.64 ppm for Si–C2. The aromatic protons display multiple characteristic resonance peaks at 6.92–6.88, 6.79–6.76, and 6.64–6.60 ppm. In the corresponding 13C NMR spectrum shown in Fig. 1b, the resonances appearing at 50.5 ppm and 82.5 ppm were assigned to the methylene carbons in the benzoxazine group. The characteristic resonance peaks appear at 58.3 ppm for O–
2–N and O–C2–N in the benzoxazine group, respectively. The characteristic resonance peaks of BS appear at 3.77 ppm for O–C2–CH3, 2.71 ppm for N–C2–CH2, 1.64 ppm for CH2–C2–CH2, 1.55 ppm for C–C3, 1.19 ppm for CH2–C3 and 0.64 ppm for Si–C2. The aromatic protons display multiple characteristic resonance peaks at 6.92–6.88, 6.79–6.76, and 6.64–6.60 ppm. In the corresponding 13C NMR spectrum shown in Fig. 1b, the resonances appearing at 50.5 ppm and 82.5 ppm were assigned to the methylene carbons in the benzoxazine group. The characteristic resonance peaks appear at 58.3 ppm for O–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2–CH3, 54.2 ppm for N–H2–CH2, 21.6 ppm for CH2–H2–CH2, 41.5 ppm for CH3––CH3, 31.1 ppm for C–H3, 18.3 ppm for CH2–H3 and 7.64 ppm for Si–CH2. The characteristic resonance peaks at 115.7, 119.5, 125.2, 126.2, 142.7, and 151.7 ppm were assigned to the aromatic carbons. The appearance of the characteristic resonance peaks for the protons and carbons in the benzoxazine group indicate that the structure contains a benzoxazine ring. If the ring is not formed, characteristic resonance peaks at chemical shifts of about 3.0–3.5 ppm are observed. There are no characteristic resonance peaks appearing at about 3.0–3.5 ppm, which also provides evidence for the formation of the benzoxazine ring. Moreover, BS displays only one resonance peak at about −45.1 ppm in the 29Si NMR spectrum (see Fig. 1c), suggesting the high purity of BS and also the absence of the hydrolysis reaction of Si–O–CH2CH3 in the preparation process of BS.
H2–CH3, 54.2 ppm for N–H2–CH2, 21.6 ppm for CH2–H2–CH2, 41.5 ppm for CH3––CH3, 31.1 ppm for C–H3, 18.3 ppm for CH2–H3 and 7.64 ppm for Si–CH2. The characteristic resonance peaks at 115.7, 119.5, 125.2, 126.2, 142.7, and 151.7 ppm were assigned to the aromatic carbons. The appearance of the characteristic resonance peaks for the protons and carbons in the benzoxazine group indicate that the structure contains a benzoxazine ring. If the ring is not formed, characteristic resonance peaks at chemical shifts of about 3.0–3.5 ppm are observed. There are no characteristic resonance peaks appearing at about 3.0–3.5 ppm, which also provides evidence for the formation of the benzoxazine ring. Moreover, BS displays only one resonance peak at about −45.1 ppm in the 29Si NMR spectrum (see Fig. 1c), suggesting the high purity of BS and also the absence of the hydrolysis reaction of Si–O–CH2CH3 in the preparation process of BS.
 |
| | Fig. 1 The NMR spectra of BS (a) 1H NMR; (b) 13C NMR and (c) 29Si NMR and the NMR spectrum of HBS8 (d) 29Si NMR. | |
The FTIR spectrum of BS is shown in Fig. 2. The characteristic absorption at 1171 cm−1 was assigned to the asymmetric stretching of C–N–C. The characteristic absorptions at 1068 and 1238 cm−1 are attributed to the symmetric and asymmetric stretching of C–O–C. The bond at 1367 cm−1 suggests the wagging mode of CH2 groups in the benzoxazine structure. The absorptions at 956 cm−1, 1463 and 1491 cm−1 were attributed to benzene with an attached oxazine ring. The peak located at 1108 cm−1 is the characteristic absorption of Si–O–C. The absorption at 1600 cm−1 is assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of benzene ring. The absorptions at 1463 and 1491 cm−1 were attributed to the stretching vibrations of the benzene ring attached to the oxazine ring. In addition, no peaks were observed at 3000–3500 cm−1, which also demonstrates that the structure contains a benzoxazine ring.
C stretching of benzene ring. The absorptions at 1463 and 1491 cm−1 were attributed to the stretching vibrations of the benzene ring attached to the oxazine ring. In addition, no peaks were observed at 3000–3500 cm−1, which also demonstrates that the structure contains a benzoxazine ring.
 |
| | Fig. 2 The FTIR spectra of BS, HBS2, HBS4 and HBS8. | |
The elemental analysis results obtained for BS show that the elements' content values (C, 61.63%; H, 8.54%; N, 4.17%; O, 17.92% and Si, 7.62%) are in reasonable agreement with those of the theoretical values (C, 61.8%; H, 8.69%; N, 3.9%; O, 17.8% and Si, 7.81%).
3.1.2 Chemical structure characterization of HBSn. In this study, the non-polar 1,4-dioxane was chosen as the hydrolysis condensation reaction solvent during the formation process of HBSn. The changes in the chemical structures of these benzoxazine-bridged polysilsesquioxanes precursors were characterized using FTIR spectroscopy (see Fig. 2) and could be used to track the progress of the hydrolysis condensation reaction. By comparing the characteristic absorptions of Si–O–C during the preparation of BS and HBSn, it was found that the doublet peaks at 1078 and 1120 cm−1 appearing in the FTIR spectrum of BS showed a tendency to change into one broad peak at about 1100 cm−1 in HBSn. This result suggests the presence of the hydrolysis condensation reaction of Si–O–CH2CH3 in the preparation process of HBSn. Due to the occurrence of the hydrolysis condensation reaction, the content of –CH2CH3 was decreased, which probably led to the absorption intensity at 1100 cm−1 for –CH2 possessing a weakening trend and the change in the absorption peak shape. Furthermore, the appearance of a broad absorption peak at 3440 cm−1 for HBS4 and HBS8 probably demonstrates the presence of Si–OH. However, it is noted that the intensities at 956 and 1238 cm−1 assigned to the benzoxazine ring show a weakening trend upon increasing the hydrolysis condensation reaction time. The interpretation of these results in terms of the benzoxazine ring-opening polymerization is, however, very difficult because the benzoxazine ring is poorly activated and opened in this system.35 We proposed that the hydrolysis condensation reaction of silsesquioxanes probably results in the formation of some cage structures,36–39 which can enwrap the benzoxazine rings and generate some shield effects.40 Based on our experimental method, the incomplete hydrolysis of silsesquioxanes will not be conducive to the formation of regular cages. In order to further confirm the occurrence of the hydrolysis condensation reaction, Si elemental analysis of HBSn was conducted. The content of Si was 8.59% for HBS2, 9.73% for HBS4 and 10.56% for HBS8. The content of Si for HBS8 is close to the theoretical value (10.56%) for a completely hydrolyzed product. For the 29Si NMR spectrum (see Fig. 1d) of HBS8, the resonance peak appearing at about −71.6 ppm was essentially in agreement with that of completely condensed silsesquioxanes.
3.1.3 Characterization of the thermal homopolymerization. The hybrid materials containing the structures of polybenzoxazines and polysilsesquioxanes can be prepared via the thermal polymerization of the series of hydrolysis products of BS. Fig. 3 shows the non-isothermal homopolymerization DSC thermograms of the benzoxazine-containing precursors (for BS, HBS2, HBS4, and HBS8). The homopolymerization curves obtained at the heating rate of 10 °C min−1 exhibit one broad exothermic peak ranging from 150 °C to 280 °C. The peak temperature, Tp, and polymerization enthalpy, ΔH, values for each exothermic peak obtained from Fig. 3 are summarized in Table 1. As can be seen, the Tps value for HBSn was higher than that found for BS. With an increase in the degree of hydrolysis of BS, the values of Tp gradually increase, which can be explained by the steric hindrance effect of the bulky polysilsesquioxanes cage.41–43 As described in previous sections, the cage or partial cage structures were formed during the hydrolysis condensation of the silsesquioxanes contained in BS. The steric hindrance of the polysilsesquioxanes cages may play an important role in preventing the homopolymerization of the benzoxazine-bridged polysilsesquioxanes precursors, which can cause an increase in the Tps value. Intriguingly, the values obtained for the polymerization enthalpy did not exhibit any influence on the thermal homopolymerization (see Table 1). From the view-point of the polymerization reaction kinetics, the curing degree was dependent on the curing time. It was noted that during the non-isothermal polymerization process, the effect of the chemically-controlled process is stronger than that of the diffusion-controlled process, which makes the non-reacted benzoxazine groups involved in the polymerization stage of the diffusion-controlled process. Hence, one post-cure process with a prolonged curing time at a correspondingly high temperature is necessary.
 |
| | Fig. 3 The non-isothermal DSC curves obtained for the benzoxazine-bridged polysilsesquioxanes precursors (BS, HBS2, HBS4 and HBS8). | |
Table 1 Thermogravimetric data for the non-isothermal DSC at the heating rate of 10 °C min−1 obtained for the benzoxazine-bridged polysilsesquioxanes precursors
| Sample |
Tp (°C) |
ΔH (J g−1) |
| BS |
222 |
−246 |
| HBS2 |
237 |
−282 |
| HBS4 |
236 |
−237 |
| HBS8 |
246 |
−243 |
The FTIR spectra of the hybrid polybenzoxazine-bridged polysilsesquioxanes materials are displayed in Fig. 4, which further prove the bulky polysilsesquioxanes cages' steric hindrance influence on the thermal homopolymerization. After post-curing at 250 °C for 2 h, the absorptions for 1,2,3-trisubstituted benzene obtained via the ring-opening thermal homopolymerization were observed at about 1610 cm−1 (1618 cm−1 for PBS, 1620 cm−1 for PHBS2, 1636 cm−1 for PHBS4, and 1642 cm−1 for PHBS8) and 1496 cm−1. The intensities of the characteristic absorptions for the benzoxazine ring at 1232 cm−1 (for Ar–O–C), 939 cm−1 (for oxazine absorption), and 1600 and 1498 cm−1 (for the ortho-substituted benzene absorption) exhibit a distinct weakening. It was noted that these characteristic absorption peaks of the benzoxazine ring in PHBSn did not disappear completely except those in PBS. Hence, we can speculate that a handful of benzoxazine rings were probably wrapped in the polysilsesquioxanes during the hydrolysis condensation process, which could not be involved into the ring-opening thermal homopolymerization reaction during the polymerization stage of the diffusion-controlled process.
 |
| | Fig. 4 The FTIR spectra of the benzoxazine-bridged polysilsesquioxanes precursors (PBS, PHBS2, PHBS4 and PHBS8). | |
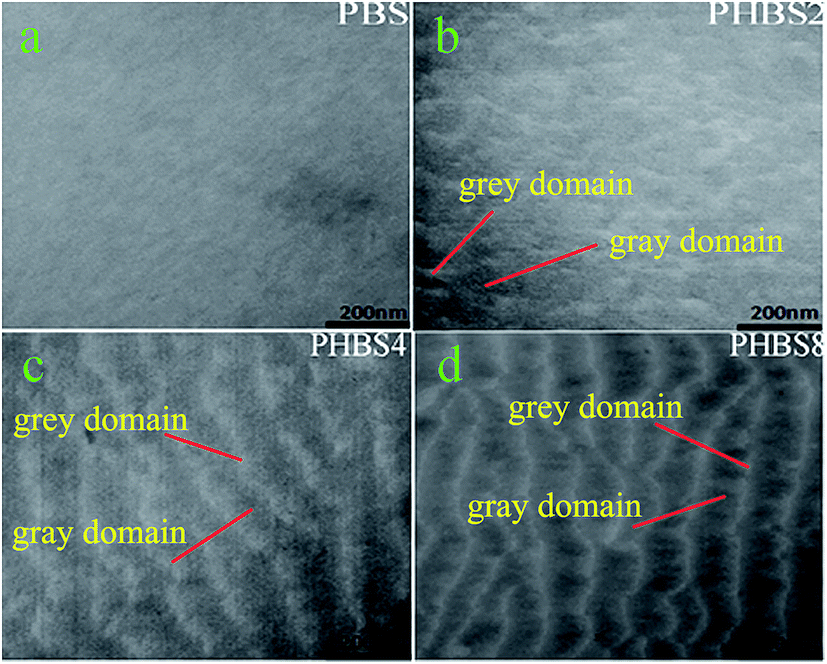
3.1.4 Characterization of controllable morphologies for the hybrid materials. By investigating the characteristic morphologies and chemical components of the materials we can further understand the mechanism of the formation of the characteristic morphologies. As a novel method used for the investigation of the morphology transition behavior of materials, TEM/EDS observation provides detailed imaging information on the morphology and chemical components of a material. Fig. 5 displays the morphological changes in PBS, PHBS2, PHBS4 and PHBS8. PBS possesses a cloud-layer morphology and does not show any evident characteristic morphology (see Fig. 5a). For PHBS2, some hierarchical morphologies are observed (see Fig. 5b). We can see that the lamellar domains possessed by PHBS4 and PHBS8 are clearly observed. Fig. 5c and 6d display the clear layer-by-layer morphologies of these two hybrid materials. Evidently, the degree of this characteristic lamellar structure was found to increase upon an increase in the degree of hydrolysis. With the help of EDS, the chemical components of the different lamellar domains were detected (see Table 2). Due to the small size of the domain areas, the morphology of PBS was not analyzed using EDS. Based on the chemical component analysis of the different domains, there is a striking discrepancy between the gray domain and the grey domain for the contents of Si and C. As shown in Table 2, the Si content found for all of samples in the gray domains was much higher than that found in the grey domains; the C content for all of the samples in the gray domains was much less than that found in the grey domains. Accordingly, we suggest that the nano-size lamellar structures of these hybrid materials are comprised of the silicon-rich (Si-rich) and carbon-rich (C-rich) layers. In the Si-rich layer domain, the content of Si increases upon increasing the degree of hydrolysis according to the following order: PHBS2 < PHBS4 < PHBS8. This phenomenon is absent in the C-rich layer domain. In the C-rich layer domain, the content of C increases upon increasing the degree of hydrolysis according to the following order: PHBS2 < PHBS4 < PHBS8. This phenomenon is absent in the Si-rich layer domain. From the view-point of changes in the morphology and chemical components, phase separation behavior with nanometer dimensions (about 20–50 nm) occurred during the thermal homopolymerization of the benzoxazine-bridged polysilsesquioxanes precursors. As for the mechanism of the phase separation, first, the presence of the strong self-organization ability of the polysilsesquioxanes provides indispensable power for the formation of the lamellar structure during the homopolymerization of the benzoxazine-bridged polysilsesquioxanes precursors.44 For the mechanism of self-assembly, the precursors of benzoxazine act as selective solvents for the polysilsesquioxanes and self-organized nanophases are formed prior to the homopolymerization. It is judged that the size of the self-organized nanophases depends on the degree of hydrolysis. When more silsesquioxanes were involved into the hydrolysis condensation, the self-organized nanophases of the polysilsesquioxanes are assembled favorably and miscible in the benzoxazine precursors. Second, the homopolymerization of benzoxazine possesses an induction effect for phase separation, which should be attributed to the heterocyclic structure of benzoxazine that forms strong intermolecular hydrogen bonds and π–π stacking interactions.45 The strong molecular forces between the benzoxazine moieties are perceived as favorable factors for the ordered self-organization of BS. From the perspective of polarity, BS consists of two different polar groups: the polar benzoxazine moiety and the lower polar triethoxysilane moiety. Two different polar groups possessed in one molecule should also be another favorable factor for the self-assembly of BS. As the reaction proceeds, the self-organized nanophases of the polysilsesquioxanes are further locked in the subsequent curing reaction and gradually become non-miscible in the benzoxazine precursors. Accordingly, with the driving power of the intermolecular forces and polarity guidance, the occurrence of phase separation becomes possible. The independent aggregation of the self-organized nanophases of the polysilsesquioxanes and the cross-linked polybenzoxazine networks promote the formation of the lamellar structures. Due to the chemical bridges between the polysilsesquioxanes and polybenzoxazine, phase separation is nano-dimensional and well controllable. Based on our experimental method, by taking advantage of the controllable preparation of benzoxazine-bridged polysilsesquioxanes precursors we can obtain hybrid materials with controllable morphology.
 |
| | Fig. 5 TEM images of the hybrid materials (a) PBS; (b) PHBS2; (c) PHBS4 and (d) PHBS8. | |
 |
| | Fig. 6 Photoluminescent spectrum of the hybrid materials under excitation at 365 nm (PBS, PHBS2, PHBS4 and PHBS8). | |
Table 2 The chemical composition content of each domain in PHBS2, PHBS4 and PHBS8
| Sample |
Chemical composition content (%) |
| Grey domains |
Gray domains |
| C |
Si |
C |
Si |
| PHBS2 |
18.7 |
30.2 |
68.6 |
6.9 |
| PHBS4 |
11.3 |
38.6 |
72.5 |
4.7 |
| PHBS8 |
6.5 |
48.3 |
75.8 |
1.8 |
3.1.5 Characterization of the photoluminescence (PL) properties. Bridged polysilsesquioxanes inorganic–organic hybrid materials displaying photoluminescence (PL) properties have been well studied over recent years.13,46 Emission spectroscopy is characterized by the presence of bands arising from processes occurring in both inorganic and organic domains. Herein, we also investigated the PL properties of the polysilsesquioxanes hybrid materials, PBS, PBS2, PBS4 and PHBS8, under excitation at 365 nm. As shown in Fig. 6, the absorbance bands around 400–450 nm are distinctly observed in the spectra obtained from the samples. Interestingly, there are two closely-spaced shoulders, one around 400 nm and the other around 425 nm, in the spectra of all the samples. Following the order, from PBS, PHBS2, PHBS4 to PHBS8, the shoulder around 425 nm presents a weakening tendency. The intensities of the PL are sharply decreased, according to the following order: PBS > PHBS2 > PHBS4 > PHBS8. To the best of our knowledge, there have been few literature articles reporting this phenomenon. In view of the present situation, we propose that the two closely-spaced shoulders may be ascribed to two types of spectral absorption. For one type, the shoulder around 425 nm may be ascribed to the spectral absorption of silsesquioxane. Zaitoun et al.47 reported the mechanism of electron–hole carriers that may originate from the defects in a silica xerogel matrix system. In our research, the different degrees of hydrolysis condensation may lead to the formation of defects. Hence, due to the presence of the electron–hole carriers generated from the defects, the PL properties of the polysilsesquioxanes may result in the spectral absorption around 425 nm. When the degree of hydrolysis condensation of BS was increased, the decrease in the number of defects contributes to the weakening tendency of the shoulder peak around 425 nm. For the other type, the shoulder around 400 nm can be ascribed to the spectral absorption of the benzoxazine rings enwrapped by the cage structures of the polysilsesquioxanes. As we know, regular benzoxazine rings possess great π–π conjugate interactions, which produce spectral absorption. Upon increasing the degree of hydrolysis condensation of BS, more and more regular benzoxazine rings can be enwrapped in the polysilsesquioxane cages. In combination with the discussion in the previous section, the enrichment of Si separated from the organic phase can generate some shield effects, which may depress the spectral absorption intensities in the PL spectra. Moreover, the phenomenon of reducing radiation efficiency of emission spectrum will occur when the stacking of the molecular chains of the materials becomes tighter.48 Hence, it is probably the independent aggregations of the self-organized nanophases of the polysilsesquioxanes and the cross-linked polybenzoxazine networks that result in the stacking of the molecular chains that depresses the spectral absorption intensities in the PL spectra.
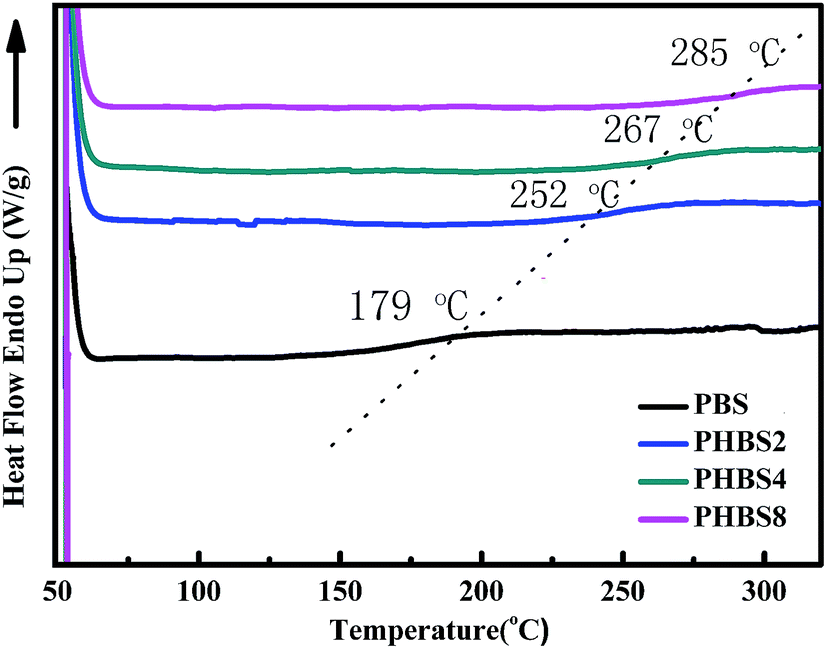
3.1.6 Characterization of thermal properties. As shown in Fig. 7, the Tg values (179 °C for PBS, 252 °C for PHBS2, 267 °C for PHBS4 and 285 °C for PHBS8) were obtained using DSC. It can be seen that as the degree of hydrolysis condensation of BS was increased, the Tgs values increased, which can be attributed to the incorporation of the polysilsesquioxanes. As discussed above, due to the steric hindrance effect of the bulky polysilsesquioxanes cage, the polybenzoxazine molecular chains are stacked more compactly and find it difficult to move upon increasing the amount of polysilsesquioxanes, which could be the major reason for the increase in the Tgs value.41,49 For secondary reasons, the strong molecular forces between the polybenzoxazine moieties and the high cross-linking density are also factors that increase Tgs. Furthermore, in combination with the performance of the characteristic morphologies for these samples, the enrichment of Si separated from the organic phase may reduce the plasticization effect of the inorganic particles on the polybenzoxazine phase.
 |
| | Fig. 7 The Tgs of the hybrid materials (PBS, PHBS2, PHBS4 and PHBS8). | |
The thermal degradation stabilities of the hybrid materials have been studied using TGA and the weight loss curves obtained for PBS, PHBS2, PHBS4 and PHBS8 are shown in Fig. 8. From the TG and DTG curves, the parameters for all of the samples, two onset degradation temperatures (Tds) evaluated by the temperature with 5% weight loss (T5%) and the temperature with 10% weight loss (T10%) and one maximum weight loss rate (Tmax) temperature are shown in Fig. 8b, and the residual char at 800 °C (Rc800) is listed in Table 3. The values of Tds, Tmax and Rc800, important parameters for evaluating the thermal degradation stability of polymer materials, for PBS are all lower than those obtained for PHBSn. For the three samples of PHBSn, the Tds, Tmax and Rc800 values obtained for PHBS8 are higher than those observed for PHBS2 and PHBS4. Between PHBS2 and PHBS4, there are no evident differences in the T5% values; however, the T10%, Tmax and Rc800 values obtained for PHBS2 are lower than those observed for PHBS4. According to the complete results, it can be proposed that the incorporation of polysilsesquioxanes can promote the enhancement of the thermal stability of these hybrid materials. Ishida et al.50 have acknowledged that the thermal stabilities of polybenzoxazine with secondary cross-linking mechanisms are expected since no new chemical functionalities have been incorporated in this series of compounds. To further significantly increase the char yield and thermal degradation stability, the possibility of forming more thermally stable cyclic structures should be introduced. Polysilsesquioxane possessing excellent thermal stability will play a significant role. One insight into the role played by the polysilsesquioxanes inorganic phase at T10% is its impact on the reticulated structure of the hybrid material.51,52 This structure restricts the movement of the polybenzoxazine chains, and hence retarding the onset of thermal decomposition improves the residual char properties of the hybrid materials. Due to the presence of the char layer with relatively high pyrolysis resistance, further degradation of the materials is retarded.
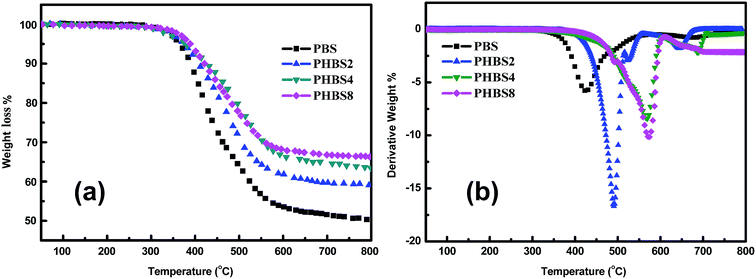 |
| | Fig. 8 TG (a) and DTG (b) thermograms of the hybrid materials (PBS, PHBS2, PHBS4 and PHBS8) under nitrogen at a heating rate of 10 °C min−1. | |
Table 3 The thermal degradation stabilities of the hybrid materials obtained from TGA
| Sample |
Specific temperature of weight loss (°C) |
Residual char at 800 °C (%) |
| T5% |
T10% |
Tmax |
| PBS |
355 |
386 |
421 |
50.1 |
| PHBS2 |
372 |
411 |
499 |
58.5 |
| PHBS4 |
378 |
440 |
568 |
63.7 |
| PHBS8 |
391 |
441 |
571 |
66.2 |
4. Conclusions
A series of inorganic–organic hybrid materials has been prepared via the thermal homopolymerization of benzoxazine-bridged polysilsesquioxanes precursors obtained from the controllable degree of hydrolysis condensation of silsesquioxanes. It was found that because of the application of this chemical controllable preparation method, the degree of the characteristic lamellar structure was increased upon increasing the degree of hydrolysis. Due to the chemical bridging between the polysilsesquioxanes and polybenzoxazine, phase separation is nanodimensional and well controlled. This significant result can be explained by the phase separation due to the synergistic effect of the polymerization-induced enrichment of the benzoxazine organic fragments and the self-organization enrichment of the inorganic polysilsesquioxanes. Upon increasing the degree of hydrolysis, the PL properties of the hybrid materials exhibited a weakening tendency due to the presence of the polysilsesquioxane cage effects. Furthermore, due to the incorporation of polysilsesquioxanes, the thermal stabilities of the hybrid materials were also improved. As expected, the nanodimensional inorganic phase layer was embedded in the organic phase layer and plays a key role in the structure–property relationships, which has been demonstrated by the changes in the optical and thermal properties of the hybrid materials. Hence, in view of the present situation, we propose that this research provides an effective approach to preparing hybrid materials with controllable morphology.
Acknowledgements
The authors gratefully acknowledge the support of the National Natural Science Foundation of China (21406058).
References
- M.-h. Sun, S.-z. Huang, L.-h. Chen, Y. Li, X.-y. Yang, Z.-y. Yuan and B.-l. Su, Chem. Soc. Rev., 2016, 45, 3479–3563 RSC.
- H.-y. Hsueh, C.-t. Yao and R.-m. Ho, Chem. Soc. Rev., 2015, 44, 1974–2018 RSC.
- Y.-h. Deng, Y. Cai, Z.-k. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D.-y. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS PubMed.
- Y.-f. Shen, J. Mater. Chem. A, 2015, 3, 13114–13188 CAS.
- Z.-q. Hu and G.-m. Chen, Adv. Mater., 2014, 26, 5950–5956 CrossRef CAS PubMed.
- X. Ji, K.-t. Lee and L.-f. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- T.-h. Kim, J. Hwang, W.-s. Hwang, J. Huh, H.-c. Kim, S.-h. Kim, J.-m. Hong, E.-l. Thomas and C. Park, Adv. Mater., 2008, 20, 522–527 CrossRef CAS.
- X.-m. Zhu, L.-q. Wang, J.-p. Lin and L.-s. Zhang, ACS Nano, 2010, 4, 4979–4988 CrossRef CAS PubMed.
- F. Liu, Y. Zhang, L. Xu and W.-a. Zhang, Chem.–Eur. J., 2015, 21, 5540–5547 CrossRef CAS PubMed.
- W.-a. Zhang and A. H. E. Müller, Prog. Polym. Sci., 2013, 38, 1121–1162 CrossRef CAS.
- C.-y. Gao and G.-m. Chen, Compos. Sci. Technol., 2016, 124, 52–70 CrossRef CAS.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- H.-c. Liu, W.-c. Su and Y.-l. Liu, J. Mater. Chem., 2011, 21, 7182–7187 RSC.
- T. Kishida, N. Fujita, K. Sada and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 7298–7299 CrossRef CAS PubMed.
- A. Shimojima, Z. Liu, T. Ohsuna, O. Terasaki and K. Kuroda, J. Am. Chem. Soc., 2005, 127, 14108–14116 CrossRef CAS PubMed.
- X.-f. Zhou, S. Yang, C.-z. Yu, Z.-h. Li, X.-x. Yan, Y. Cao and D.-y. Zhao, Chemistry, 2006, 12, 8484–8490 CrossRef CAS PubMed.
- K. Yamamoto, Y. Nohara, Y. Domon, Y. Takahashi, Y. Sakata, J. Plévert and T. Tatsumi, Chem. Mater., 2005, 17, 3913–3920 CrossRef CAS.
- S. Yun, H.-j. Luo and Y.-f. Gao, J. Mater. Chem. A, 2015, 3, 3390–3398 CAS.
- L. Xu, V. R. Manda, L. E. McNamara, M. P. Jahan, H. Rathnayake and N. I. Hammer, RSC Adv., 2014, 4, 30172–30179 RSC.
- M. A. Wahab and J. N. Beltramini, RSC Adv., 2015, 5, 79129–79151 RSC.
- D. A. Loy and K. J. Shea, Chem. Rev., 1995, 95, 1431–1442 CrossRef CAS.
- U. Díaz, T. García, A. Velty and A. Corma, Chem.–Eur. J., 2012, 18, 8659–8672 CrossRef PubMed.
- D.-x. Wang, W.-y. Yang, L.-g. Li, X. Zhao, S.-y. Feng and H.-z. Liu, J. Mater. Chem. A, 2013, 1, 13549–13558 CAS.
- S.-m. Wu, T. Hayakawa, R. Kikuchi, S. J. Grunzinger and M.-a. Kakimoto, Macromolecules, 2008, 41, 3481–3487 CrossRef CAS.
- Y. Kaneko, H. Imamura, T. Sugioka and Y. Sumida, Polymer, 2016, 92, 250–255 CrossRef CAS.
- Y.-c. Gan, X.-s. Jiang and J. Yin, J. Mater. Chem. C, 2014, 2, 5533–5539 RSC.
- H. E. Romeo, M. A. Fanovich, R. J. J. Williams, L. Matějka, J. Pleštil and J. Brus, Macromolecules, 2007, 40, 1435–1443 CrossRef CAS.
- J. P. Randall, M. A. B. Meador and S. C. Jana, ASC Appl. Mater. Interfaces, 2011, 3, 613–626 CrossRef CAS PubMed.
- Y. Fujimoto, M. Heishi, A. Shimojima and K. Kuroda, J. Mater. Chem., 2005, 15, 5151–5157 RSC.
- Z.-e. Fu, K. Xu, X. Liu, J. Wu, C.-j. Tan and M.-c. Chen, Macromol. Chem. Phys., 2013, 214, 1122–1130 CrossRef CAS.
- Z.-e. Fu, H. Liu, H.-l. Cai, X. Liu, Y. Guo, K. Xu and M.-c. Chen, Polym. Eng. Sci., 2012, 52, 2473–2481 CAS.
- P. Velez-Herrera, K. Doyama, H. Abe and H. Ishida, Macromolecules, 2008, 41, 9704–9714 CrossRef CAS.
- R. J. P. Corriu, J. J. E. Moreau, P. Thepot and M. W. C. Man, Chem. Mater., 1996, 8, 100–106 CrossRef CAS.
- H. Ishida and Y. L. Hong, J. Appl. Polym. Sci., 1998, 69, 2559–2567 CrossRef CAS.
- X. Ning and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1121–1129 CrossRef CAS.
- B. P. Pichon, M. Wong Chi Man, P. Dieudonné, J.-L. Bantignies, C. Bied, J.-L. Sauvajol and J. J. E. Moreau, Adv. Funct. Mater., 2007, 17, 2349–2355 CrossRef CAS.
- J. Peng, K. Xu, H.-l. Cai, J.-c. Wu, W.-h. Lin, Z.-w. Yu and M.-c. Chen, RSC Adv., 2014, 4, 7124–7131 RSC.
- Y. Kaneko, M. Shoirikia and T. Mizumob, J. Mater. Chem., 2012, 22, 14475–14478 RSC.
- H. Ghanbari, B. G. Cousins and A. M. Seifalian, Macromol. Rapid Commun., 2011, 32, 1032–1046 CrossRef CAS PubMed.
- C.-h. Chou, S.-L. Hsu, K. Dinakaran, M.-y. Chiu and K.-h. Wei, Macromolecules, 2005, 38, 745–751 CrossRef CAS.
- Y. Gao, C.-l. He, Y.-g. Huang and F.-l. Qing, Polymer, 2010, 51, 5997–6004 CrossRef CAS.
- L.-g. Li, R.-d. Liang, Y.-j. Li, H.-z. Liu and S.-y. Feng, J. Colloid Interface Sci., 2013, 406, 30–36 CrossRef CAS PubMed.
- Y.-w. Li, K. Guo, H. Su, X.-p. Li, X.-y. Feng, Z. Wang, W. Zhang, S.-s. Zhu, C. Wesdemiotis, S. Z. D. Cheng and W.-b. Zhang, Chem. Sci., 2014, 5, 1046–1053 RSC.
- C.-x. Liu, Z.-q. Li and Y.-g. Wang, J. Sol-Gel Sci. Technol., 2016 DOI:10.1007/s10971-016-4219-5.
- P. Velez-Herrera, K. Doyama, H. Abe and H. Ishida, Macromolecules, 2008, 41, 9704–9714 CrossRef CAS.
- M. L. Gómez, D. P. Fasce, R. J. J. Williams, H. A. Montejano and C. M. Previtali, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 289–296 CrossRef.
- M. A. Zaitoun, T. Kim and C. T. Lin, J. Phys. Chem. B, 1998, 102, 1122–1125 CrossRef CAS.
- X. Yang, L. Liu, C. He, W.-s. Chin and T. Lin, J. Mater. Chem., 2006, 16, 829–836 RSC.
- P. M. And and S. L. Wunder, Chem. Mater., 2002, 14, 4494–4497 CrossRef.
- H. Ishida and D. P. Sanders, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 3289–3301 CrossRef CAS.
- H. W. P. Carvalho, A. F. Suzana, C. V. Santilli and S. H. Pulcinelli, Polym. Degrad. Stab., 2014, 104, 112–119 CrossRef CAS.
- N. Katsikis, F. Zahradnik, A. Helmschrott, H. Munstedt and A. Vital, Polym. Degrad. Stab., 2007, 92, 1966–1976 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Zi-en Fu*c,
Fei Songab,
Qingquan Liuab and
Lijuan Chenab
ab,
Zi-en Fu*c,
Fei Songab,
Qingquan Liuab and
Lijuan Chenab