
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and fungicidal activity of 3,4-dichloroisothiazole based strobilurins as potent fungicide candidates†
Lai Chena,
Xiao-Feng Guoa,
Zhi-Jin Fan*ab,
Nai-Lou Zhanga,
Yu-Jie Zhua,
Zhi-Ming Zhanga,
Inna Khazhievac,
Morzherin Y. Yurievich c,
Nataliya P. Belskayac and
Vasiliy A. Bakulev*c
c,
Nataliya P. Belskayac and
Vasiliy A. Bakulev*c
aState Key Laboratory and Institute of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: fanzj@nankai.edu.cn; Tel: +86-13920714666
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, P. R. China
cThe Ural Federal University Named after the First President of Russia B. N. Yeltsin, Yeltsin UrFU 620002, Ekaterinburg, Russia. E-mail: v.a.bakulev@urfu.ru
First published on 13th January 2017
Abstract
A series of 3,4-dichloroisothiazole based novel strobilurin analogs were designed and synthesized, and their structures were elucidated by NMR and HRMS, and the typical crystal structure was determined by X-ray diffraction for validation. Results from different biological assays suggested that most target compounds displayed very good fungicidal activity against one or multiple plant pathogens in vitro and in vivo. Among them, compounds 6d, 6g and 8d showed a broad spectrum of fungicidal activity. Further field experiments indicated that compound 8d displayed better efficacy against Sphaerotheca fuliginea than commercial standards azoxystrobin and trifloxystrobin, and better efficacy against Pseudoperonospora cubensis than trifloxystrobin. Overall, a new fungicidal candidate for plant disease management was discovered in this study.
Introduction
A sustainable food supply for an increasing global demand requires new innovations in crop protection technology.1 Continuous global population growth and a shift in consumers' food preference call for an increased crop yield under fixed/shrinking arable lands.2 Agrochemicals have been one of the most effective tools to meet this need. However, pest resistance, pest shifts and an ever evolving regulatory landscape mean a high pressure for current agrochemicals. Innovative agrochemicals that can overcome resistance are always welcome for farmers.3Resistance of plant pathogens to some strobilurin analogs needs innovative products. The strobilurin fungicides are the second largest group of launched fungicides, which act through inhibition of mitochondrial respiration by blocking electron transfer within the respiratory chain, thus in turn causes important cellular biochemical processes to be severely disrupted, and results in cessation of fungal growth.4 Strobilurin fungicides with a broad spectrum are highly efficacious and are suitable for a wide range of crops.5 However, since the first launch of this class of fungicide, widespread applications have led to pathogen resistance.6 Resistant cases of Septoria in European wheat and the U.S. turf market were good examples.7 Therefore, new strobilurins are possibly needed for future markets.
Heterocyclic compounds exhibit wide spectrum of biological activity.8 Isothiazoles constitute a relatively novel class of heterocyclic compounds. As one of their members, 3,4-dichloroisothiazoles possess a broad spectrum of biological activity such as insecticide, fungicide and potential systemic acquired resistance activities, 3,4-dichloroisothiazole-5-carboxylic acid and its' derivatives manifest fungicidal activity;9 isotianil being as its' derivative was developed as a novel fungicide with activating defence responses against a wide range of plant pathogens.10
The purpose of this study was to use strobilurin A as a template to design and synthesize new strobilurin analogs (Fig. 1) with a active substructure of 3,4-dichloroisothiazole. Extensive biological assays demonstrated that newly synthesized compounds displayed good to excellent activity against one or multiple plant pathogens. Field trials suggested that the fungicidal activity of the best candidate 8d was better than commercial standards.
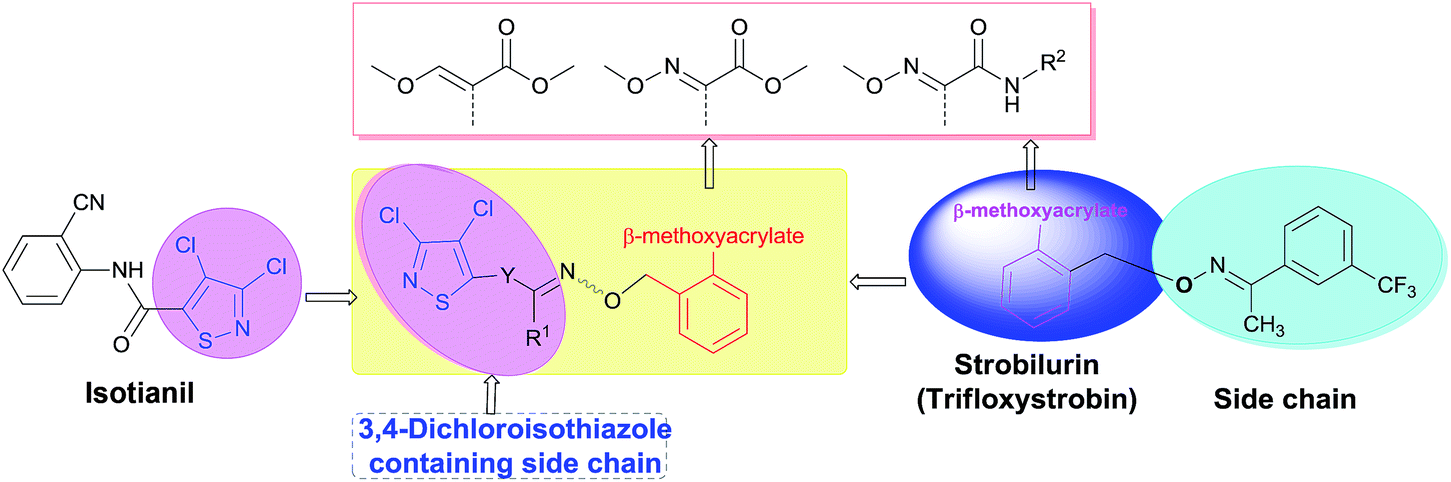 | ||
| Fig. 1 Design of the title compounds. | ||
Results and discussion
Chemistry
The synthetic route of 3a and 3c was outlined in Scheme 1. Compound 2 was obtained in a high yield from compound 1 by ref. 11. Then compound 3a was obtained from compound 2 through substitution reaction. Compound 3b was prepared from the oxidation of 3,4-dichloro-5-hydroxylmethyl isothiazole. Compound 3c was obtained by aldol condensation reaction between compound 3b and acetone.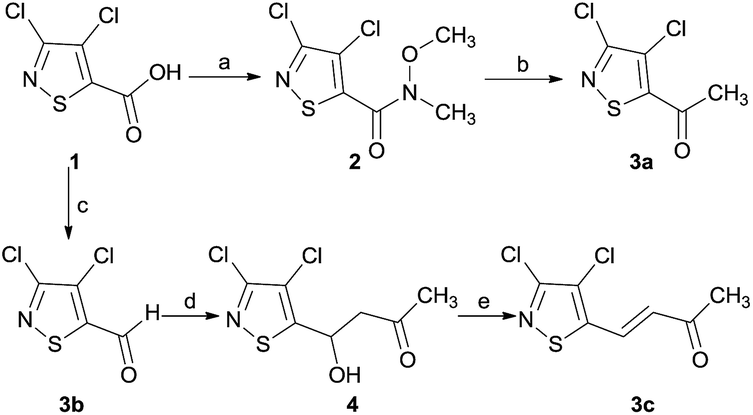 | ||
| Scheme 1 Reagents: (a) EDCI, CH3NHOCH3·HCl, CH2Cl2, rt; (b) CH3MgBr, THF, −30 °C; (c) (i): SOCl2, CH3OH; (ii): NaBH4, CH3OH; (iii): PCC; (d) acetone/H2O, NaOH; (e) SOCl2, DMF, 25 °C. | ||
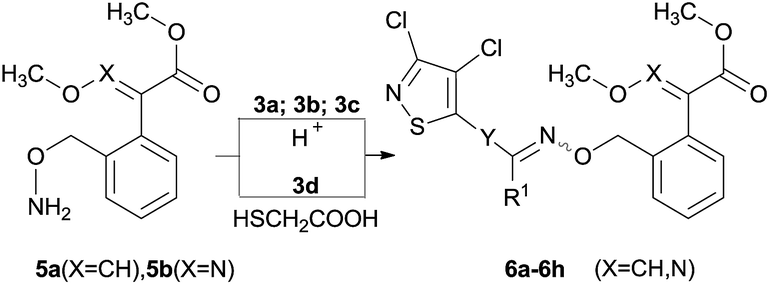
For the target compounds synthesis, compounds 6a–6h were prepared by intermediates 3a–3d reacting with compounds 5a or 5b respectively (Table 1).12 Compounds 8a–8r were synthesized according to the method described in Table 2.
|
|||
|---|---|---|---|
| Compd | X | R1 | Y |
| 6a | CH | ⋯CH3 | Covalent bond |
| 6c | CH | ⋯H | Covalent bond |
| 6e | CH | ⋯NH2 | Covalent bond |
| 6g | CH | ⋯CH3 |  |
| 6b | N | ⋯CH3 | Covalent bond |
| 6d | N | ⋯H | Covalent bond |
| 6f | N | ⋯NH2 | Covalent bond |
| 6h | N | ⋯CH3 |  |
|
|||||||
|---|---|---|---|---|---|---|---|
| Compd | Y | R1 | R2 | Compd | Y | R1 | R2 |
| 8a | Covalent bond | ⋯CH3 |  |
8j |  |
⋯CH3 | ⋯CH3 |
| 8b | Covalent bond | ⋯CH3 |  |
8k |  |
⋯CH3 |  |
| 8c | Covalent bond | ⋯CH3 |  |
8l |  |
⋯CH3 |  |
| 8d | Covalent bond | ⋯CH3 | ⋯CH3 | 8m | Covalent bond | ⋯NH2 |  |
| 8e | Covalent bond | ⋯CH3 |  |
8n | Covalent bond | ⋯NH2 |  |
| 8f | Covalent bond | ⋯CH3 |  |
8o | Covalent bond | ⋯NH2 |  |
| 8g |  |
⋯CH3 |  |
8p | Covalent bond | ⋯NH2 | ⋯CH3 |
| 8h |  |
⋯CH3 |  |
8q | Covalent bond | ⋯NH2 |  |
| 8i |  |
⋯CH3 |  |
8r | Covalent bond | ⋯NH2 |  |

Compounds 7 series were obtained by hydrolytic reaction from the corresponding compounds 6b, 6f or 6h, and then compounds 8 series were obtained by the reaction of the corresponding compound 7 with a corresponding amino compound. The structures of the synthesized compounds were confirmed by 1H NMR, 13C NMR, HRMS or elemental analyses.
Fungicidal activity
The result of in vitro fungicidal activity determination of all synthesized compounds and the positive control azoxystrobin against 9 fungi was assessed at 50 μg mL−1. The results were shown in Table 3.| Compd | AS | CA | GZ | PP | BC | SS | RC | PS | PI |
|---|---|---|---|---|---|---|---|---|---|
| 6a | 43 | 67 | 52 | 78 | 100 | 80 | 100 | 65 | 53 |
| 6b | 47 | 50 | 41 | 53 | 100 | 100 | 85 | 77 | 79 |
| 6c | 57 | 67 | 74 | 78 | 100 | 53 | 100 | 65 | 58 |
| 6d | 100 | 92 | 71 | 62 | 100 | 90 | 98 | 69 | 95 |
| 6e | 57 | 100 | 61 | 87 | 100 | 100 | 100 | 59 | 47 |
| 6f | 53 | 75 | 50 | 71 | 100 | 98 | 89 | 77 | 89 |
| 6g | 57 | 50 | 74 | 74 | 100 | 100 | 100 | 71 | 79 |
| 6h | 36 | 50 | 61 | 48 | 100 | 87 | 81 | 59 | 47 |
| 8a | 37 | 43 | 79 | 40 | 54 | 84 | 88 | 20 | 80 |
| 8b | 29 | 36 | 100 | 49 | 52 | 100 | 87 | 73 | 80 |
| 8c | 28 | 44 | 64 | 61 | 47 | 80 | 61 | 57 | 100 |
| 8d | 48 | 68 | 100 | 72 | 100 | 67 | 100 | 89 | 100 |
| 8e | 27 | 46 | 52 | 43 | 63 | 29 | 76 | 21 | 29 |
| 8f | 16 | 13 | 33 | 33 | 46 | 53 | 76 | 15 | 17 |
| 8g | 34 | 36 | 45 | 30 | 53 | 53 | 85 | 35 | 90 |
| 8h | 41 | 52 | 82 | 65 | 47 | 80 | 100 | 78 | 100 |
| 8i | 48 | 60 | 64 | 72 | 58 | 73 | 71 | 68 | 100 |
| 8j | 54 | 84 | 100 | 65 | 84 | 73 | 85 | 89 | 100 |
| 8k | 5 | 13 | 24 | 40 | 32 | 41 | 58 | 15 | 11 |
| 8l | 13 | 13 | 61 | 58 | 43 | 53 | 62 | 21 | 20 |
| 8m | 13 | 17 | 33 | 40 | 38 | 53 | 62 | 24 | 23 |
| 8n | 8 | 20 | 55 | 19 | 42 | 47 | 66 | 46 | 100 |
| 8o | 21 | 28 | 64 | 23 | 37 | 73 | 85 | 19 | 33 |
| 8p | 34 | 44 | 100 | 40 | 58 | 67 | 100 | 84 | 100 |
| 8q | 16 | 13 | 21 | 28 | 39 | 53 | 62 | 15 | 17 |
| 8r | 9 | 17 | 45 | 11 | 36 | 53 | 54 | 21 | 11 |
| Azoxystrobin | 75 | 81 | 71 | 100 | 91 | 100 | 100 | 81 | 100 |
For each fungus, most of the synthesized compounds were more active than the positive control azoxystrobin under the same condition. Especially for BC, SS and RC, majority of synthesized compounds exhibited better activity than azoxystrobin. For PI, most compounds showed similar or better activity than azoxystrobin except for compounds 8m, 8o and compounds with a sulfur atom in the substitution of R2. Besides, for compounds 8a–8r, it was also found that the compounds with a methyl group at R2 exhibited better activity. Compounds 8d, 8j and 8p exhibited 100% inhibition activity against GZ, while they had higher activity than azoxystrobin against RC. Furthermore, compounds 6d–6g and 8d showed a broad spectrum of fungicidal activity in vitro. Overall, the in vitro fungicidal potency and spectrum are dependent on the individual structures, or substitutions (e.g., X, Y, R1, or R2) in the structures of the compounds 6 or 8 series.
In order to explore the fungicidal potency, precision toxicity determination for the EC50 of compounds 6d–6g, 8d and 8p with a broader fungicidal spectrum described above were further conducted. The results in Table 4 indicated that compound 8d exhibited excellent activity with EC50 of 0.07 μg mL−1 and 0.49 μg mL−1 against RC and PI, respectively; they were at the same level as that of the positive control azoxystrobin. Besides, compound 8d showed higher activity against GZ and BC with much lower EC50 than azoxystrobin. Compound 8d was confirmed with broad spectrum of fungicide activity by this precision toxicity experiments.
| Compd | AS | CA | GZ | BC | SS | RC | PI |
|---|---|---|---|---|---|---|---|
| a Determined values based on the results shown in Table 3.b nd, not detect. | |||||||
| 6d | 33.34a | 14.47 | ndb | 2.94 | 5.21 | 0.13 | 3.10 |
| 6e | nd | 58.25 | nd | 12.15 | 20.06 | 0.21 | nd |
| 6f | nd | nd | nd | 1.62 | 7.53 | nd | nd |
| 6g | nd | nd | nd | 3.93 | 4.47 | 0.11 | nd |
| 8d | nd | nd | 1.75 | 0.15 | nd | 0.07 | 0.49 |
| 8p | nd | nd | 1.81 | nd | nd | 0.69 | 3.84 |
| Azoxystrobin | 185.42 | 2.50 | 6.92 | 6.31 | 4.04 | 0.06 | 0.40 |
The in vivo fungicidal activity of all compounds and positive control (i.e., azoxystrobin) against P. cubensis, E. graminis, P. sorghi Schw and C. lagenarium were further assessed at 400 μg mL−1 and the results were listed in Table 5. Most of the synthesized compounds were more active than azoxystrobin. For P. cubensis, the compounds 6a, 6d, 6f, 8d and 8r exhibited almost 100% activity which were better than azoxystrobin (with only 85% of activity). Besides, the compounds 6a–6g, 8d, 8g, 8h, 8p and azoxystrobin showed the similar 100% activity against E. graminis. Even though azoxystrobin showed no activity against P. sorghi Schw, the compounds 6b, 6d, 6f, 6g, 8a and 8b exhibited 100% activity. Furthermore, almost all of the compounds except for 6b, 6h and 8e had similar to or better activity than azoxystrobin against C. lagenarium. Thus, the compounds 6a–6g, 8b and 8d were confirmed as with broad-spectrum of fungicidal activity in vivo too. Afterwards, 6a, 6d, 6g, 6f, 8b and 8d with good activity were further validated at concentrations of 50 μg mL−1, 12.5 μg mL−1, 3.13 μg mL−1, 0.78 μg mL−1 and 0.20 μg mL−1 in vivo, respectively. The results were shown in Table 6.
| Compd | P. cubensis | E. graminis | P. sorghi Schw | C. lagenarium |
|---|---|---|---|---|
| 6a | 100 | 100 | 0 | 98 |
| 6b | 0 | 100 | 100 | 100 |
| 6c | 85 | 100 | 0 | 98 |
| 6d | 99 | 100 | 100 | 100 |
| 6e | 85 | 100 | 85 | 50 |
| 6f | 98 | 100 | 100 | 80 |
| 6g | 85 | 100 | 100 | 100 |
| 6h | 30 | 0 | 50 | 0 |
| 8a | 75 | 50 | 100 | 98 |
| 8b | 85 | 100 | 100 | 100 |
| 8c | 0 | 30 | 70 | 85 |
| 8d | 100 | 100 | 0 | 80 |
| 8e | 70 | 80 | 40 | 0 |
| 8f | 40 | 0 | 0 | 85 |
| 8g | 0 | 100 | 40 | 85 |
| 8h | 0 | 100 | 40 | 100 |
| 8i | 0 | 0 | 60 | 98 |
| 8j | 0 | 60 | 0 | 100 |
| 8k | 85 | 0 | 0 | 80 |
| 8l | 60 | 0 | 60 | 100 |
| 8m | 80 | 0 | 0 | 98 |
| 8n | 60 | 0 | 80 | 100 |
| 8o | 85 | 0 | 40 | 80 |
| 8p | 70 | 100 | 80 | 85 |
| 8q | 85 | 0 | 0 | 100 |
| 8r | 100 | 0 | 0 | 80 |
| Azoxystrobin | 85 | 100 | 0 | 80 |
| Compd | C (μg mL−1) | P. cubensis | E. graminis | P. sorghi Schw | C. lagenarium |
|---|---|---|---|---|---|
| 6a | 0.2 | 0 | 100 | 95 | 65 |
| 0.78 | 0 | 100 | 95 | 70 | |
| 3.13 | 0 | 100 | 98 | 80 | |
| 12.5 | 0 | 100 | 100 | 90 | |
| 50 | 0 | 100 | 100 | 98 | |
| 6d | 0.2 | 0 | 100 | 0 | 0 |
| 0.78 | 0 | 100 | 0 | 0 | |
| 3.13 | 0 | 100 | 30 | 0 | |
| 12.5 | 0 | 100 | 45 | 15 | |
| 50 | 0 | 100 | 70 | 40 | |
| 6g | 0.2 | 0 | 30 | 0 | 0 |
| 0.78 | 0 | 45 | 0 | 0 | |
| 3.13 | 0 | 65 | 0 | 0 | |
| 12.5 | 0 | 90 | 0 | 0 | |
| 50 | 0 | 100 | 30 | 0 | |
| 6f | 0.2 | 0 | 100 | 0 | 0 |
| 0.78 | 0 | 100 | 0 | 0 | |
| 3.13 | 0 | 100 | 20 | 0 | |
| 12.5 | 0 | 100 | 55 | 0 | |
| 50 | 0 | 100 | 80 | 0 | |
| 8b | 0.2 | 0 | 95 | 0 | 0 |
| 0.78 | 0 | 100 | 0 | 0 | |
| 3.13 | 0 | 100 | 0 | 0 | |
| 12.5 | 0 | 100 | 0 | 0 | |
| 50 | 0 | 100 | 0 | 0 | |
| 8d | 0.2 | 20 | 90 | 10 | 60 |
| 0.78 | 30 | 95 | 45 | 70 | |
| 3.13 | 45 | 100 | 85 | 85 | |
| 12.5 | 55 | 100 | 90 | 95 | |
| 50 | 65 | 100 | 95 | 100 | |
| Trifloxystrobin | 0.2 | 0 | 95 | 95 | 65 |
| 0.78 | 0 | 100 | 98 | 85 | |
| 3.13 | 0 | 100 | 100 | 98 | |
| 12.5 | 0 | 100 | 100 | 98 | |
| 50 | 20 | 100 | 100 | 100 | |
| Enestroburin | 0.2 | 0 | 30 | 30 | 65 |
| 0.78 | 0 | 65 | 75 | 85 | |
| 3.13 | 0 | 95 | 98 | 95 | |
| 12.5 | 0 | 98 | 100 | 98 | |
| 50 | 0 | 100 | 100 | 100 |
As can be seen from Table 6, compounds 6a, 6d, 6f, 8b and 8d exhibited 90–100% inhibition activity against E. graminis even at 0.20 μg mL−1, which were similar to that of positive control trifloxystrobin (with inhibition of 95%) and were much better than enestroburin (with inhibition of 30%). The compound 6a exhibited best activity against P. sorghi Schw as the same as positive control trifloxystrobin, they both had 95% of fungicidal activity at 0.2 μg mL−1; their activities were much better than that of enestroburin (only 30% at 0.20 μg mL−1). The compounds 6a and 8d exhibited best activity against C. lagenarium as the same as positive control trifloxystrobin and enestroburin, they all had about 65% of activity even at 0.20 μg mL−1. Most importantly, the compound 8d also exhibited very good activity against P. cubensis with 20% of inhibition at 0.20 μg mL−1, while the two positive controls trifloxystrobin and enestroburin had no activity against P. cubensis even at 12.5 μg mL−1. Our in vivo confirmation studies indicated, most of the active compounds discovered not only kept its' highly activity, but also acted to a broad spectrum of fungi tested. Compound 8d deserved for further novel fungicide development studies as a candidate.
Field efficacy evaluation of compound 8d
The results of the field experiments were shown in Table 7. Studies indicated that the compound 8d exhibited significantly better efficacy (78.62%) against cucumber S. fuliginea than commercial standards (i.e., azoxystrobin and trifloxystrobin) at the same application rate of 37.5 g ai per hm2 at 21 days after spraying application. At an application rate of 75 g ai per hm2, the compound 8d showed similar activity against P. cubensis as one commercial standard pyraclostrobin, but significantly better than another commercial standard trifloxystrobin. These results suggested that the compound 8d could be considered as an alternative to control S. fuliginea and P. cubensis, it is under further novel pesticide development studies.| Disease | Compd | Amount (g ai per hm2) | Base DIa | After DIb | Efficacy (%) | DDd | |
|---|---|---|---|---|---|---|---|
| 5% | 1% | ||||||
| a Base DI, base disease index.b After DI, disease index after compounds application.c nd, not need to detect.d DD, distinct difference. | |||||||
| S. fuliginea | 9.60% 8d EC | 37.5 | 1.98 | 8.01 | 78.62 ± 4.38 | a | A |
| 250 g L−1 azoxystrobin SC | 37.5 | 1.66 | 9.59 | 70.19 ± 2.91 | b | B | |
| 50% trifloxystrobin WG | 37.5 | 1.64 | 10.04 | 68.02 ± 5.58 | b | B | |
| CK | ndc | 1.07 | 20.63 | nd | nd | nd | |
| P. cubensis | 9.60% 8d EC | 75 | 3.71 | 5.39 | 79.18 ± 1.09 | a | A |
| 50% trifloxystrobin WG | 75 | 3.73 | 7.24 | 72.02 ± 2.15 | b | B | |
| 250 g L−1 pyraclostrobin EC | 75 | 3.57 | 5.78 | 77.02 ± 1.77 | a | AB | |
| CK | ndc | 3.58 | 24.96 | nd | nd | nd | |
Experimental
Equipment and materials
Melting points of all compounds were determined on an X-4 binocular microscope (Gongyi Tech. Instrument Co., Henan, China) and the thermometer was not corrected. Proton NMR spectra were obtained using a Bruker AVANCE-400 MHz spectrometer and chemical shift values (δ) were reported in ppm with deutero-chloroform (CDCl3) as a solvent and tetramethylsilane (TMS) as an internal standard. High resolution mass spectrometry (HRMS) data were obtained on an FTICR-MS Varian 7.0T FTICR-MS instrument. Elemental analyses were taken on a Vario EL III elemental analysis instrument. Crystal structure was recorded by Bruker SMART 1000 CCD diffraction meter. All solvents and reagents were an analytical reagent grade. Column chromatography purification was carried out on silica gel.General procedure for the synthesis of compound 3a
3,4-Dichloro-N-methoxy-N-methylisothiazole-5-carboxamide 2 can be prepared from 3,4-dichloroisothiazole-5-carboxylic acid 1 according to the revising procedures of ref. 11. To a solution of the compound 2 (3.00 g, 12.44 mmol) in anhydrous tetrahydrofuran (45 mL) at −30 °C under N2 atmosphere was added dropwise with a solution of methyl magnesium bromide in Et2O (3 mol L−1, 5.81 mL, 17.42 mmol). Then the mixture was allowed to stir at −30 °C for 1 h and room temperature for another 1 h. When the reaction completed, the reaction mixture was worked-up by the sat. aq. NH4Cl (50 mL). After removal of the tetrahydrofuran under vacuum, the aqueous phase was extracted with ethyl acetate (3 × 50 mL). The organic layers were combined, washed with water (50 mL) and saturated brine (50 mL), and then dried over anhydrous sodium sulfate. After filtration, the solvent was evaporated. The residue was then purified by column chromatography on silica gel using ethyl acetate and petroleum ether (60–90 °C) with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 of υ/υ as an eluent to obtain yellow oil 3a (1.56 g) in a yield of 63.8%. 1H NMR (400 MHz, CDCl3) δ 2.65 (s, 3H, O
:9 of υ/υ as an eluent to obtain yellow oil 3a (1.56 g) in a yield of 63.8%. 1H NMR (400 MHz, CDCl3) δ 2.65 (s, 3H, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH3).
C–CH3).
General procedure for the synthesis of compound 3b
A solution of (3,4-dichloroisothiazol-5-yl)methanol (1.37 g, 7.44 mmol), which can be prepared from 3,4-dichloroisothiazole-5-carboxylic acid, in 50 mL CH2Cl2 was added to the suspension of PCC (2.40 g, 11.16 mmol) and diatomite (2.40 g) in 50 mL CH2Cl2 at 0 °C, then the reaction mixture was allowed to stir at room temperature for overnight. The reaction mixture was filtrated and the solvent was evaporated. The residue was then purified by column chromatography on silica gel using ethyl acetate and petroleum ether (60–90 °C) with 1:10 of υ/υ as an eluent to afford the white solid 3b (0.89 g) in a yield of 65.9%. 1H NMR (400 MHz, CDCl3) δ 10.09 (s, OCH).
General procedure for the synthesis of compound 3c
To a solution of 3b (0.10 g, 0.05 mmol) in 9 mL acetone was added 1 mL water, then 0.15 mL of aqueous NaOH (2%) was added dropwise within 30 s at ice bath. The reaction mixture was stirred at this temperature for about 2 min and then was quenched with 0.50 mL dilute hydrochloric acid (1 mol L−1). After removal of the acetone under vacuum, water (20 mL) was added and the aqueous phase was extracted with ethyl acetate (2 × 10 mL). The organic layers were combined and washed with saturated brine (50 mL), dried over anhydrous sodium sulfate. After filtration, the solvent was evaporated. The residue was then purified by column chromatography on silica gel using ethyl acetate and petroleum ether (60–90 °C) with 1:10 of υ/υ as an eluent to afford the white solid intermediate 4 (0.10 g) in a yield of 75.9%. 1H NMR (400 MHz, CDCl3) δ 5.43 (d, J = 8.8 Hz, 1H, O–CH), 4.00 (s, 1H, OH), 3.14 (d, J = 17.7 Hz, 1H, OC–CH2), 2.81 (dd, J = 18.1, 9.6 Hz, 1H, OC–CH2), 2.24 (s, 3H, OC–CH3).
To a solution of intermediate 4 (0.10 g, 0.42 mmol) in anhydrous CH2Cl2 (20 mL), and SOCl2 (0.1 g, 0.84 mmol) was added and followed by a catalytic amount of DMF. The reaction mixture was allowed to stir at 25 °C for 24 h. The solvent was evaporated under reduced pressure and the residue was then purified by column chromatography on silica gel using ethyl acetate and petroleum ether (60–90 °C) with 1:5 of υ/υ as an eluent to afford the white solid 3c (0.08 g) in a yield of 88.9%. 1H NMR (400 MHz, CDCl3) δ 7.49 (d, J = 16.4 Hz, 1H, CHCH), 6.71 (d, J = 16.3 Hz, 1H, CHCH), 2.39 (s, 3H, CC–CH3).
General procedure for the synthesis of compounds 6a–6d and 6g–6h
Compounds 5a and 5b can be prepared following the procedure reported by ref. 13. A solution of compound 5a or 5b (1.12 mmol) in 15 mL ethanol was add to the solution of intermediates 3a, 3b or 3c (1.02 mmol) in ethanol (15 mL), and the reaction mixture was added the catalytic amount of 2 mol L−1 hydrochloric acid and stirred at room temperature for overnight. The solvent was evaporated under reduced pressure and the residue was then purified by recrystallization in ethanol or column chromatography on silica gel using ethyl acetate and petroleum ether (60–90 °C) with 1:9 to 1:4 of υ/υ as an eluent to obtain the desired derivatives 6 (Table 1).
General procedure for the synthesis of compounds 6e and 6f
Compounds 6e and 6f can be prepared following the procedure reported by ref. 13. A solution of compounds 5a or 5b (1.22 mmol) in 15 mL ethanol was added to the solution of commercially available 3,4-dichloroisothiazole-5-carbonitrile 3d (0.20 g, 1.11 mmol) in ethanol (15 mL), 2-mercaptoacetic acid (0.10 g, 1.09 mmol) was added to the reaction mixture for stirring at room temperature for overnight. The solvent was evaporated under reduced pressure and the residue was then purified by recrystallization in ethyl acetate or column chromatography on silica gel using ethyl acetate and petroleum ether (60–90 °C) with 1:4 to 1:9 of υ/υ as an eluent to give the desired derivatives (Table 1).
General procedure for the synthesis of compounds 7
Intermediates 6b, 6f or 6h (2.49 mmol) was dissolved in 15 mL methanol, the solution of sodium hydroxide (0.30 g, 7.47 mmol) in 15 mL methanol was added to the reaction mixture for 30 min of refluxing. After completing of the reaction, the solvent was evaporated under reduced pressure and the residue was added 15 mL water. The aqueous phase was adjusted to pH 2–3 with dilute hydrochloric acid (3 mol L−1). The aqueous layer was extracted with ethyl acetate (2 × 15 mL). The organic layers were combined and washed with saturated brine (50 mL), dried over sodium sulfate. After filtration, the solvent was evaporated to obtain the compounds 7 as a white solid.General procedure for the synthesis of compounds 8a–8r
The reaction mixture of the compound 7 (0.75 mmol), EDCI (0.17 g, 0.90 mmol), HOBT (0.11 g, 0.77 mmol) in dichloromethane (25 mL) was stirred for 15 min in ice bath. A solution of amine in dichloromethane (25 mL) was added and followed by Et3N (0.09 g, 0.90 mmol), the reaction mixture was stirred for further 16 h. After completion of the reaction, the organic layer was successively washed with water (2 × 30 mL) and saturated brine (40 mL), dried over MgSO4 and concentrated under vacuum. After filtration, the solvent was evaporated. The residue was then purified by column chromatography on silica gel using ethyl acetate and petroleum ether (60–90 °C) with 1:2 to 1:5 of υ/υ as an eluent to give the desired derivatives (Table 2).
Crystal structure determination for compound 6d
The crystal of compound 6d was obtained by recrystallization from ethanol (Fig. 2). X-ray intensity data were recorded on a Bruker SMART 1000 CCD diffraction meter using graphite-monochromatic Mo Kα radiation (λ = 0.71073 Å). A total of 13757 reflections were measured, of which 3004 were unique (Rint = 0.0635) in the range of 3.06° ≤ θ ≤ 25.02° (h, −13 to 13; k, −9 to 9; l, −22 to 19), and 2884 observed reflections with I > 2σ(I) were used in the refinement on F2. The structure was solved by direct methods with the SHELXS-97 program. All of the non-H atoms were refined anisotropically by full-matrix least-squares to give the final R = 0.0698 and wR = 0.2049 (w = 1/[σ2((Fo2) + (0.1200P))2 + 6.9197P], where P = (Fo2 + 2Fc2)/3 with (Δ/σ)max = 0.981 and S = 1.061 by using the SHELXL program. The hydrogen atoms were located from a difference Fourier map and refined isotropically.
 | ||
| Fig. 2 X-ray diffraction structure of the title compound 6d. | ||
Fungicidal assay
The preliminary in vitro fungicidal activities of newly synthesized compounds against Alternaria solani (AS), Botrytis cinerea (BC), Cercospora arachidicola (CA), Gibberella zeae (GZ), Phytophthora infestans (Mont) de Bary (PI), Physalospora piricola (PP), Pellicularia sasakii (PS), Sclerotinia sclerotiorum (SS), and Rhizoctonia cerealis (RC) were tested according to ref. 14. Precision toxicity studies were conducted for the median effective concentration (EC50) calculation according to ref. 14.The preventive in vivo activities of the target compounds against Pseudoperonospora cubensis, Erysiphe graminis, Puccinia sorghi Schw and Colletotrichum lagenarium were tested in green house according to the ref. 15. For the active compounds, different lower concentration tests were also conducted for validation.
Evaluation of field efficacy of compound 8d
Due to well laboratory performance against fungi in vitro and in vivo, the compound 8d was employed to evaluate its efficacy in the cucumber field. A 9.60% EC of 8d was prepared for the efficacy against S. fuliginea and P. cubensis in Wuqing County, Tianjin, P. R. China. Commercial available products of 250 g L−1 azoxystrobin SC, 50% trifloxystrobin WG, 250 g L−1 pyraclostrobin EC were chosen as positive control. An application dosages of 8d and positive standards was 37.5 and 75 g. ai per ha for S. fuliginea and P. cubensis, respectively. Disease index was evaluated by formula as DI = ∑(A × B) × 100/(C × 9); A-means the number of disease leaf; B-means the corresponding grade of A; C-means the total number of investigation leaf. Prevention efficacy was calculated with a formula efficacy (%) = [1 − CK0 × PT1/(CK1 × PT0)] × 100%; CK0-means DI of control group before applying water; PT0 means DI of treatment group before applying compound; CK1 means DI of control group after applying water; PT1 means DI of treatment group after applying compound. Duncan's new multiple range (DMRT) statistic method was used for data analysis.Conclusion
A series of strobilurin analogues with a substructure of 3,4-dichloroisothiazole were synthesized and characterized by NMR, HRMS and X-ray diffraction. These compounds were assessed for biological activity against a few important plant fungi in vitro and in vivo. The fungicidal potency and spectrum varied with compounds and fungi. Most of synthesized compounds displayed good to excellent fungicidal activity against one or multiple plant fungi, some of these compounds showed better activity against one or multiple plant fungi than commercial standard such as azoxystrobin. Further field experiment suggested that compound 8d showed better efficacy against cucumber S. fuliginea at than two commercial standards azoxystrobin and trifloxystrobin at the same application rate. Moreover, the compound 8d showed similar efficacy against P. cubensis as that of pyraclostrobin, but significantly better than that of trifloxystrobin. In summary, an excellent fungicide candidate which reached commercially biological levels against some plant pathogen was developed in this study for further development.Acknowledgements
This research was in part financial supported by the International Science & Technology Cooperation Program of China (Grant no. 2014DFR41030), the National Natural Science Foundation of China (No. 21372132 and 31571991), the "111" Project of Ministry of Education of China (Project No. B06005) and Tianjin Natural Science Foundation (No.: 14JCYBJC20400). V. A. Bakulev thanks The Ministry of Education and Science of the Russian Federation (State task 4.1626.2014/K).Notes and references
- B. A. Chalmers, H. Xing, S. Houston, C. Clark, S. Ghassabian, A. Kuo, B. Cao, A. Reitsma, C. E. P. Murray and J. E. Stok, Angew. Chem., Int. Ed., 2016, 55, 3580–3585 CrossRef CAS PubMed.
- Anonymous, Nature, 2010, 466, 531–532 CrossRef PubMed.
- P. Jeschke, Pest Manage. Sci., 2016, 72, 433–455 CrossRef CAS PubMed.
- (a) D. W. Bartlett, J. M. Clough, J. R. Godwin, A. A. Hall, M. Hamer and B. Parr-Dobrzanski, Pest Manage. Sci., 2002, 58, 649–662 CrossRef CAS PubMed; (b) P. M. Wood and D. W. Hollomon, Pest Manage. Sci., 2003, 59, 499–511 CrossRef CAS PubMed; (c) J. O. Obuya and G. D. Franc, Eur. J. Plant Pathol., 2016, 146, 817–827 CrossRef CAS.
- (a) H. B. Christensen and K. Granby, Food Addit. Contam., 2001, 18, 866–874 CrossRef CAS PubMed; (b) S. Tu, L.-H. Xu, L.-Y. Ye, X. Wang, Y. Sha and Z.-Y. Xiao, J. Agric. Food Chem., 2008, 56, 5247–5253 CrossRef CAS PubMed; (c) Y. Wang, Y.-B. Duan and M.-G. Zhou, Can. J. Plant Pathol., 2014, 36, 354–359 CrossRef CAS; (d) L. Li, M. Li, H. Chi, J. Yang, Z. Li and C. Liu, J. Fluorine Chem., 2016, 185, 173–180 CrossRef CAS.
- (a) E. Flampouri, S. Mavrikou, A.-C. Mouzaki-Paxinou and S. Kintzios, Biochem. Pharmacol., 2016, 113, 97–109 CrossRef CAS PubMed; (b) J. Leiminger, B. Adolf and H. Hausladen, Plant Pathol., 2014, 63, 640–650 CrossRef CAS.
- U. Gisi, H. Sierotzki, A. Cook and A. McCaffery, Pest Manage. Sci., 2002, 58, 859–867 CrossRef CAS PubMed.
- (a) Y. J. Zhu, X. F. Guo, Z. J. Fan, L. Chen, L. Y. Ma, H. X. Wang, Y. Wei, X. M. Xu, J. P. Lin and V. A. Bakulev, RSC Adv., 2016, 6, 112704–112711 RSC; (b) S. Pulya, Y. Kommagalla, D. G. Sant, S. U. Jorwekar, S. G. Tupe, M. V. Deshpande and C. V. Ramana, RSC Adv., 2016, 6, 11691–11701 RSC; (c) B. Varun, G. Divya, A. Vikrant, D. Saurabh and S. Poonam, RSC Adv., 2015, 5, 15233–15266 RSC; (d) N. P. Belskaya, V. A. Bakulev and Z. J. Fan, Chem. Heterocycl. Compd., 2016, 52, 627–636 CrossRef CAS; (e) Z. Jin, Nat. Prod. Rep., 2006, 23, 464–496 RSC.
- (a) F. Clerici, M. L. Gelmi, S. Pellegrino and D. Pocar, Top. Heterocycl. Chem., 2007, 9, 179–264 CAS; (b) R. V. Kaberdin and V. I. Potkin, Russ. Chem. Rev., 2002, 71, 673–694 CrossRef CAS; (c) X. Y. Chen, L. Y. Dai, Y. D. Li, W. T. Mao, Z. Fang, J. J. Li, D. Wang, K. Tatiana and Z. J. Fan, Chin. J. Pestic. Sci., 2013, 15, 140–144 CAS.
- (a) Y. Bektas and T. Eulgem, Front. Plant Sci., 2015, 5, 804 Search PubMed; (b) X. Y. Chen, D. Wang, J. Huang, Y. Huang and Z. J. Fan, J. China Agrochem., 2012, 56, 31–34 Search PubMed.
- D. K. Friel, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 9942–9951 CrossRef CAS PubMed.
- F. Al-Saffar, S. Berlin, T. Musil and S. Sivadasan, WO Pat., 2006/090153, 2006.
- P. Strazzolini and A. Pavsler, Ind. Eng. Chem. Res., 2005, 44, 1625–1626 CrossRef CAS.
- Z. J. Fan, Z. K. Yang, H. K. Zhang, N. Mi, H. Wang, F. Cai, X. Zuo, Q. X. Zheng and H. B. Song, J. Agric. Food Chem., 2010, 58, 2630–2636 CrossRef CAS PubMed.
- Y. Q. Xie, Y. B. Huang, J. S. Liu, L. Y. Ye, L. M. Che, S. Tu and C. L. Liu, Pest Manage. Sci., 2015, 71, 404–414 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystal structure data of compound 6d, characterization and copies of NMR spectra for all target compounds 6 and 8. CCDC 1509531. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra25520e |
| This journal is © The Royal Society of Chemistry 2017 |