
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermal oxidative etching method derived graphitic C3N4: highly efficient metal-free catalyst in the selective epoxidation of styrene†
Jia Ren‡
a,
Xin Liu‡a,
Lu Zhanga,
Qianqian Liua,
Ruihua Gao*b and
Wei-Lin Dai*a
aDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Material, Fudan University, Shanghai 200433, P. R. China. E-mail: wldai@fudan.edu.cn; Fax: +86 021 5566 5572; Tel: +86 021 5566 4678
bState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai, 200433, P. R. China. E-mail: ruihuagao@fudan.edu.cn; Tel: +86 021 5566 5626
First published on 17th January 2017
Abstract
A series of g-C3N4 nanosheets were synthesized by thermal oxidation with long time heating and etching. With an increase in heating time from 1.0 to 3.0 h, the g-C3N4 nanosheet was obtained with a thinner layer thickness, larger BET surface area and higher graphic nitrogen ratio. As a metal-free heterogeneous catalyst, the g-C3N4 NS-3 h nanosheets show a superior catalytic performance for the epoxidation of styrene to styrene oxide than that of the bulk g-C3N4 and other recently reported metal-free materials. The higher activity of the g-C3N4 NS-3 h synthesized by long time thermal oxidative etching might be ascribed to the enlarged specific surface, pore volume and higher graphic nitrogen ratio with the loose and soft laminar morphology. The graphitic nitrogen species play a key role in the catalytic reaction based on a good linear correlation between the content of these species and the activity results. The g-C3N4 nanosheets are very stable and can be reused 5 times without obvious loss of catalytic activity.
1. Introduction
Alkene epoxidation reaction is a first and important step in industrial processes because the products are versatile intermediates in the synthesis of fine chemicals such as plasticizers, perfumes, epoxy resin, pharmaceuticals, etc. Traditionally, epoxidation reaction was carried out by directly using stoichiometric peracids as the oxidants,1 which produced a large amount of by-products. Even worse, the peracids were expensive, unsafe and hazardous. Due to the increasing attention paid to the environmentally sustainable growth, much more of an attempt has been made to develop various new processes for alkene epoxidation reaction. The current research is being concentrated on the metal-related catalysts, such as metal-embedded molecular sieves, heteropolyacid modified complexes and noble metal supported catalysts which are usually synthesized though complicated process.2–4 For these catalytic systems, the metals play an irreplaceable role in the adsorption and activation of the alkenes and oxidants. Particularly, titanium substituted silicalite (TS-1) has been demonstrated to be very effective for styrene epoxidation, and Ti species dispersed in this molecular sieve was identified as the key active center of the catalyst.5 Wang and coworkers found that g-C3N4 could promote the catalytic epoxidation of styrene using molecular oxygen as the primary oxidant after modification with Co and Fe.6 In our previous work,7 the catalytic performance of the Zr–CeO2 is mainly attributed to the oxygen vacancy and the Ce3+.As sustainable development was hindered by the issue of metal-based catalysts, such as their scarcity in nature, deactivation, high cost in disposal of waste catalysts, and secondary pollution,8 it is potential to develop metal-free catalysts for alkene epoxidation reactions. Recently, graphene-based non-metal materials have gained more and more attention in the realm of catalysis. Garcia and co-workers achieved a breakthrough by synthesizing nitrogen- and boron-doped graphene, which can efficiently activate di-oxygen in the epoxidation of benzylic styrene compounds.9 In addition, Ma and co-workers discovered that nitrogen-doped graphene can effectively catalyze the C–C double bond epoxidation reaction, and the nitrogen dopants at the quaternary site were proven to be the active species.10 However, there are still few studies reported on the application of non-metal catalysts in the alkene epoxidation reaction. These interesting results motivate us to pioneer green nitrogen-rich carbon materials applying in the alkene epoxidation.
It is well known that graphitic carbon nitride materials are a kind of nitrogen rich material which has similar sheet-like structure with graphene. In addition, polymeric graphitic carbon nitride materials (g-C3N4) have attracted much attention owing to their similarity to graphene.11 Meanwhile, the g-C3N4 materials are especially promising catalysts for introducing N into carbon materials, due to their high N content, high thermal, chemical stability, low price, and easily tailorable structure.12 The catalytic activity of g-C3N4 has recently been reported for hydrogen evolution,12 oxidation of alkanes, including benzene,14 cyclohexane,15 toluene,16 and alcohols,17 and photodegradation of pollutants,18 etc. However, it is worth noting that bulk g-C3N4 shows a low catalytic activity by far. It is necessary to optimize the structure of g-C3N4 for getting a better catalytic performance. For example, exfoliating bulk g-C3N4 into nanosheets have been proved to be able to enhance the efficiency of hydrogen evolution by one order of magnitude,13 offer novel activity in photocatalytic disinfection19 and bio-imaging application.20 Currently, the g-C3N4 materials can also be used as photodegradation catalysts in the field of photocatalysis. However, scarce efforts have been taken to employ g-C3N4 as the metal-free catalysts for the epoxidation of olefins.
Herein, the g-C3N4 nanosheets with varying thickness of layer, surface chemistries and surface areas were obtained by a simple method, namely, thermal oxidative etching of bulk g-C3N4 in air with various time, leading to different catalytic performances. The g-C3N4 was firstly used as a metal-free catalyst in the epoxidation of styrene with a stable and excellent catalytic performance. The g-C3N4 nanosheets demonstrate morphology-dependent activity in the epoxidation of styrene, which is much higher than that of bulk g-C3N4. By detailed characterizations, the graphitic nitrogen rich species and high surface areas are responsible for the epoxidation reaction. The excellent catalytic performances as well as the wide applicability make g-C3N4 nanosheets a promising catalyst for “green” oxidation of styrene.
2. Experimental
2.1 Preparation and activity test
The g-C3N4 nanosheets with different morphologies and sizes can be directly synthesized via thermal oxidative etching method (denoted as g-C3N4 NS-t h, where t represents the thermal oxidative etching time). Typically, a certain amount of as-prepared bulk g-C3N4 was placed in a crucible and heated for 2 h in a muffle furnace at 500 °C with a ramping rate of 5 °C min−1. After cooling to room temperature, a light yellow powder of g-C3N4 NS-2 h was finally obtained. In addition, thermal oxidative etching time is a key factor in controlling the formation and mass yield of the obtained g-C3N4 nanosheets (Scheme 1).
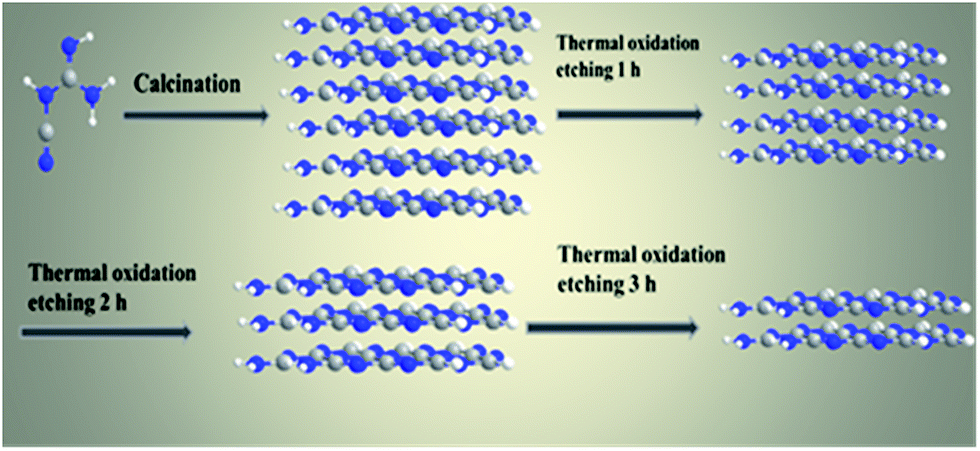 | ||
| Scheme 1 Schematic diagram of the synthesis process of bulk g-C3N4 and the thermal etching process. | ||
2.2 Catalyst characterization
The samples were characterized by wide-angle XRD patterns on a Bruker D8 Advance X-ray diffractometer in a scanning angle (2θ) range of 10–90° at a scanning speed of 8° min−1, operated at 40 kV and 40 mA with nickel-filtered Cu Kα radiation (λ = 0.15406 nm).The Brunauer–Emmett–Teller (BET) specific surface areas and pore volume of the catalysts were characterized by nitrogen adsorption–desorption (Micromeritics Tristar ASAP 3000) employing BET method. The samples were pre-treated at 250 °C in vacuum for 3 h before test to desorb moisture adsorbed. Transmission electron micrographs (TEM) images were obtained on a JOEL JEM 2010 transmission electron microscope. The samples were supported on carbon-coated copper grids. Fourier transform infrared spectra (FT-IR) were gained from Nicolet Avatar-360 FT-IR spectrometer. Scanning electron micrographs (SEM) were obtained using a PHILIPS XL 30 microscope operating at an accelerating voltage of 20 kV.
Tapping-mode atomic force microscopy (AFM, Nanoscope Multimode 8) with Si-tip cantilever was used to evaluate the morphology and thickness of the obtained nanosheets on the mica substrate.
The X-ray photoelectron spectroscopy (XPS) were determined on a RBD-147 upgraded Perkin-Elmer PHI 5000C ESCA system equipped with a dual X-ray source and a hemispherical electron energy analyzer. The Mg Kα (1253.6 eV) anode was operated at 14 kV and 20 mA. The spectra were performed in the constant pass energy mode with a value of 46.95 eV, and all binding energies were calibrated referenced to the carbonaceous C1s at 284.6 eV.
2.3 Activity measurements
The activity measurement for styrene epoxidation was carried out in a closed 25 mL regular glass reactor with a reflux condenser using styrene as a substrate, 70 wt% aqueous t-butyl hydroperoxide (TBHP) as the oxidant and acetonitrile as solvent. In a typical oxidation reaction, 50 mg of catalysts, 10 mL of acetonitrile and 8.7 mmol of TBHP (70 wt% aqueous) were introduced into the regular glass reactor at 80 °C with magnetic stirring. After the reaction, the catalyst was separated and the quantitative analysis of the reaction products was carried out by gas chromatography (GC), and the determination of different products in the reaction mixture was performed by GC-mass spectroscopy (GC-MS).| Substrate conversion = moles of substrate converted/moles of substrate used |
| Product selectivity = moles of product formed/moles of substrate converted |
The stability was tested as follows: the re-used catalyst was separated by centrifuge, washed by distilled water and ethanol for several times, and dried under 80 °C for 12 h for the next run.
3. Results and discussion
3.1 Characterization of the as-prepared samples
The XRD profiles of the as-prepared samples before and after heat treatment are shown in Fig. 1. All the samples show two obvious diffraction peaks at 2θ = 12.9° and 27.3°, respectively. The strong peak at 27.3° is ascribed to the (002) plane with d = 0.326 nm, which is assigned to a characteristic interlayer spacing of aromatic systems.11 With increasing the thermal oxidative etching time, the diffraction intensity of this (002) peak remarkably decreases and the diffraction peak shifts to the higher angle end, which demonstrates that the increased heating time during thermal oxidation process may lead to the exfoliation of bulk g-C3N4 to nanosheets and a denser packing between the basic sheets.11,21–23 Additionally, Antonietti et al. reported that undulated single layers in bulk g-C3N4 could be planarized by further heating, resulting in a denser stacking.21,24 Furthermore, the low-angle reflection peaks at 13.1°, stemmed from in-planar tris-s-triazine structural packing, are not changed with different etching time, which indicated that the planar size of the layers changed little during the thermal oxidative etching of bulk g-C3N4.13,22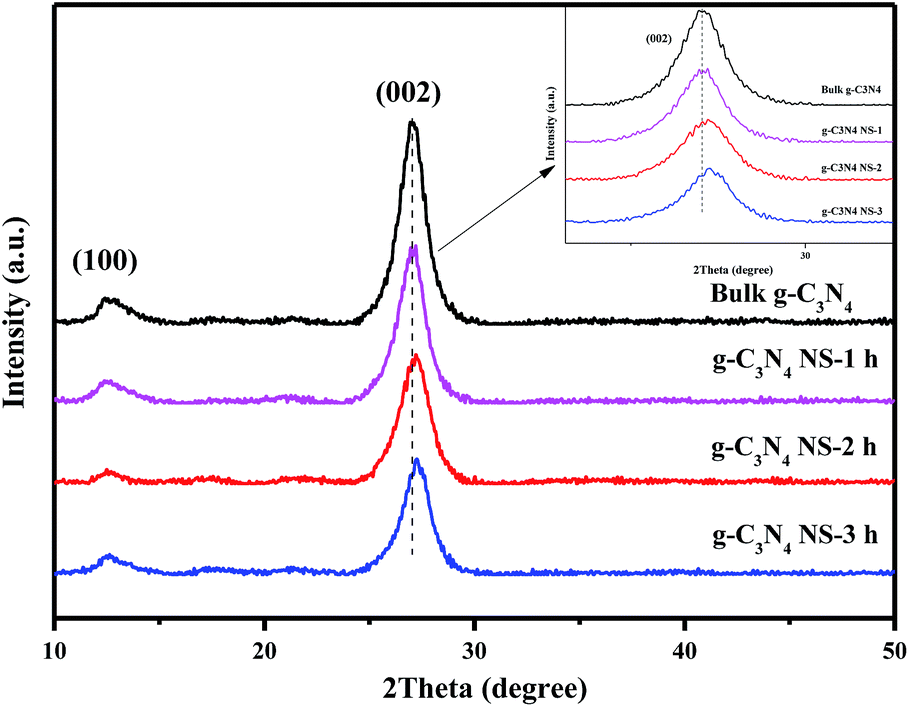 | ||
| Fig. 1 XRD patterns of bulk g-C3N4, g-C3N4 NS-1 h, g-C3N4 NS-2 h, and g-C3N4 NS-3 h. | ||
The morphology and microstructure of the as-prepared g-C3N4 NS-3 h and bulk C3N4 were investigated via SEM and TEM. As shown in Fig. 2b, the as-prepared g-C3N4 nanosheets are transparent to electron beams owing to their thinner nature compared with bulk g-C3N4 (Fig. 2a). Furthermore, the basic sheet edges roll up in order to minimize the surface-energy of the sheet. Simultaneously, very similar trend was observed by Niu et al.22 To a certain extent, the appearance of g-C3N4 nanosheets in Fig. 2b is quite similar to that of graphene or graphene oxide. Fig. 2c shows the bulk g-C3N4 display in the form of irregular blocky agglomerates, and the size distribution is mainly in several micrometers. While upon thermal treatment at 500 °C for 3 h, the resulting sample g-C3N4 NS-3 h (Fig. 2d) appeared as loose and soft agglomerates consisting of laminar morphology like petals. Obviously, great numbers of small gaps between these petals indicate the formation of mesoporous structure and the exfoliation of bulk g-C3N4, which is in a good agreement with the XRD and BET results.
 | ||
| Fig. 2 TEM and SEM images of bulk g-C3N4 (a and c), and g-C3N4 NS-3 h (b and d). | ||
To gain insight into the structural features of g-C3N4 nanosheets, atomic cross-sectional AFM was conducted. As shown in Fig. 3, the representative AFM image and thick analyses of the g-C3N4 NS-3 h show average thickness of ca. 1.301 nm, revealing the same laminar morphology appeared in the observations from SEM and TEM. It is well known that the theoretical thickness of monolayer is 0.326 nm.25,26 Besides, Tang et al. conclude that the thickness analysis of 1-, 2- and 4-layer g-C3N4 nanosheet by AFM revealed an average thickness of ca. 0.4, 0.7 and 1.3 nm, respectively.27 From this point of view, it could be believed that herein the g-C3N4 NS-3 h fabricated by a simple thermal oxidative etching process was in form of four C–N layers.
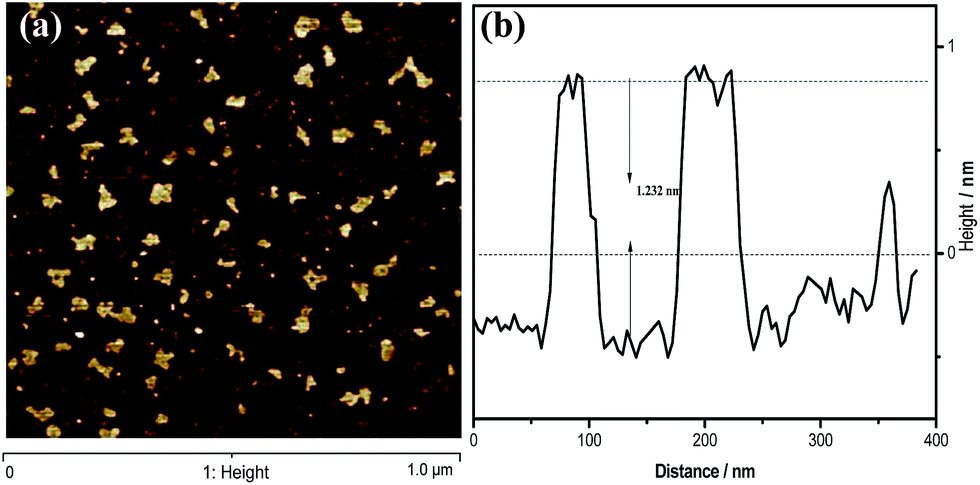 | ||
| Fig. 3 (a) The typical AFM images of the g-C3N4 NS-3 h, (b) height analysis of the nanosheets corresponding to the write curves in the AFM images. | ||
The change of chemical structure of g-C3N4 with prolonging heat treatment time was further confirmed by the FTIR spectra shown in Fig. S1.† All samples display similar characteristic IR absorption spectrum. The sharp peak at ca. 810 cm−1 is originated from the characteristic breathing mode of heptazine units.28 The peaks in the region from ca. 1200 to 1480 cm−1 are assigned to the typical stretching vibrations of either trigonal C–N(–C)–C or bridging C–N(–H)–C heterocycles associated with skeletal stretching vibrations of aromatic rings,29 and these bands become sharper compared with bulk C3N4, probably due to the more ordered packing of hydrogen-bond cohered long stands of polymeric melon units survived after thermal oxidative etching in the layer of nanosheets.22 The absorption bands at ca. 1633 and 1570 cm−1 is due primarily to the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in the s-triazine. The broad peaks between 3000 and 3600 cm−1 are originated from the O–H stretches, suggesting there are adsorbed hydroxyl species in the surface of the catalysts. The similarity between all the bands indicates that the heat-treatment of the C3N4 sample does not lead to significant changes in the structure of the g-C3N4 nanosheets. Besides, the most prominent change of the IR is the band in the ca. 3600–3000 cm−1 region, corresponding to the surface-bonded H2O molecules, which is much stronger after the thermal treatment, implying the enlarger BET surface area.30 The nitrogen adsorption–desorption isotherms were used to further characterize the microstructural change of obtained C3N4 with increasing the thermal etching time. According to IUPAC classification, all the g-C3N4-t h samples and bulk-C3N4 can be identified as Type IV adsorption–desorption isotherms with H3 hysteresis loop in the relative pressure range of 0.6–1.0 (Fig. 4).23,31 The obtained specific BET surface areas are 22, 52, 151 and 245 m2 g−1, respectively, for bulk-C3N4, g-C3N4-1 h, g-C3N4-2 h, g-C3N4-3 h, suggesting the increased specific BET surface area with thermal etching time. The BET surface area of the g-C3N4-3 h is about 11 times higher than that of bulk g-C3N4. And the large surface area could provide more reactive sites and adsorb more reactants. Furthermore, as seen from the BJH pore-size distribution curve (inset), the distribution of pores centered between 25 and 50 nm may attribute to the mesopores originated from the stacking of C3N4 nanosheets, which also increased with the thermal etching time. These variation tendencies further confirm that the laminar structure bulk g-C3N4 can be effectively exfoliated by extending the thermal etching time. This result supports those from SEM, TEM and AFM. Most importantly, g-C3N4 material with mesopores and large surface area can be obtained by using this simple etching method even though the final mass yields decrease at much longer thermal etching time (as shown in Table S1†). Therefore, according to the results from SEM, TEM, AFM and N2 adsorption, we can conclude that the increased etching time in our study gives rise to significant changes in the textural properties of carbon nitride. The bulk g-C3N4 in the form of irregular blocky agglomerates was divided into loose and soft agglomerates, consisting of laminar morphology like petals after long time heating and etching process.
N in the s-triazine. The broad peaks between 3000 and 3600 cm−1 are originated from the O–H stretches, suggesting there are adsorbed hydroxyl species in the surface of the catalysts. The similarity between all the bands indicates that the heat-treatment of the C3N4 sample does not lead to significant changes in the structure of the g-C3N4 nanosheets. Besides, the most prominent change of the IR is the band in the ca. 3600–3000 cm−1 region, corresponding to the surface-bonded H2O molecules, which is much stronger after the thermal treatment, implying the enlarger BET surface area.30 The nitrogen adsorption–desorption isotherms were used to further characterize the microstructural change of obtained C3N4 with increasing the thermal etching time. According to IUPAC classification, all the g-C3N4-t h samples and bulk-C3N4 can be identified as Type IV adsorption–desorption isotherms with H3 hysteresis loop in the relative pressure range of 0.6–1.0 (Fig. 4).23,31 The obtained specific BET surface areas are 22, 52, 151 and 245 m2 g−1, respectively, for bulk-C3N4, g-C3N4-1 h, g-C3N4-2 h, g-C3N4-3 h, suggesting the increased specific BET surface area with thermal etching time. The BET surface area of the g-C3N4-3 h is about 11 times higher than that of bulk g-C3N4. And the large surface area could provide more reactive sites and adsorb more reactants. Furthermore, as seen from the BJH pore-size distribution curve (inset), the distribution of pores centered between 25 and 50 nm may attribute to the mesopores originated from the stacking of C3N4 nanosheets, which also increased with the thermal etching time. These variation tendencies further confirm that the laminar structure bulk g-C3N4 can be effectively exfoliated by extending the thermal etching time. This result supports those from SEM, TEM and AFM. Most importantly, g-C3N4 material with mesopores and large surface area can be obtained by using this simple etching method even though the final mass yields decrease at much longer thermal etching time (as shown in Table S1†). Therefore, according to the results from SEM, TEM, AFM and N2 adsorption, we can conclude that the increased etching time in our study gives rise to significant changes in the textural properties of carbon nitride. The bulk g-C3N4 in the form of irregular blocky agglomerates was divided into loose and soft agglomerates, consisting of laminar morphology like petals after long time heating and etching process.
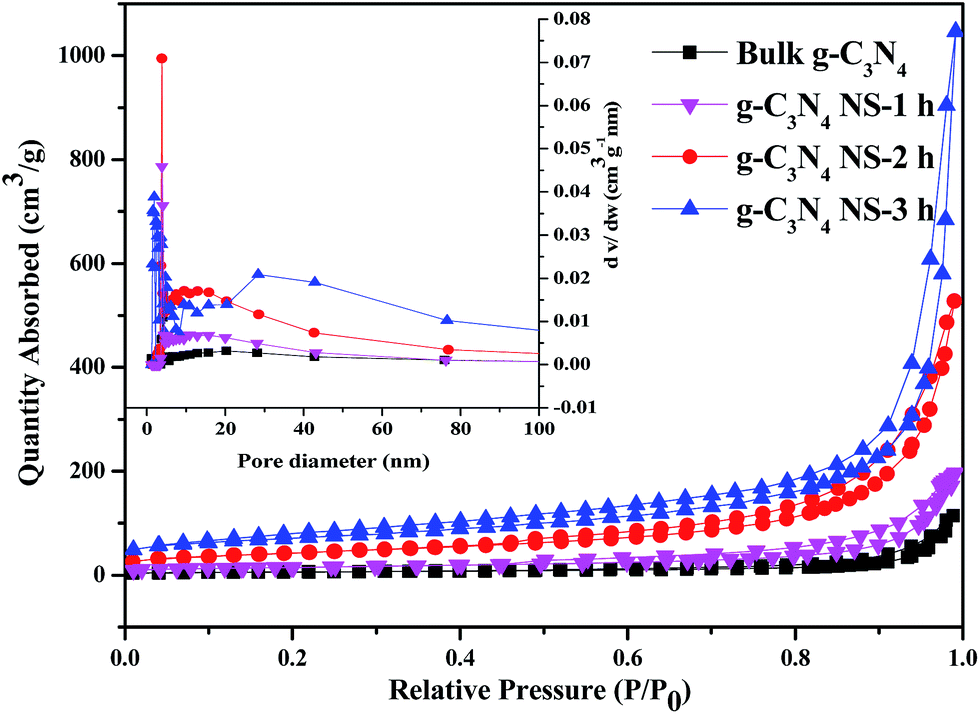 | ||
| Fig. 4 The N2 adsorption–desorption isotherm and the corresponding BJH pore-size distribution curve (inset) of bulk g-C3N4, g-C3N4 NS-1 h, g-C3N4 NS-2 h, and g-C3N4 NS-3 h. | ||
To further investigate the compositions and chemical state of the samples, the XPS measurements were carried out. As shown in Fig. S2,† only three non-metal elements (C, N, O) are detected in the XPS spectra. The O1s peak is assigned to the adsorbed H2O on the sample surface, which appeared at about 532.5 eV for all samples.29 In order to gain insight into the chemical bonding between nitrogen and carbon atoms in the samples, the high resolution C1s and N1s spectra of g-C3N4 were further deconvoluted into Gaussian–Lorentzian peaks, respectively. As shown in Fig. S3,† the C1s spectra shows three peaks centered at ca. 284.6, 287.2 and 287.8 eV. The peak at 287.2 eV is identified as sp2 hybridized C atoms bonded to N inside the aromatic structure, while the peak at 287.8 eV is attributed to the sp2 hybridized C atoms bonded to N inside the aromatic structure.12,31,32 The C1s peak at ca. 284.6 eV is typically originated from surface adventitious carbon.22
Meanwhile, the high-resolution N1s spectra can also be deconvoluted into four Gaussian–Lorentzian peaks. The four peaks centered at about 398.8, 400.3, 400.9 and 404.5 eV are assigned to N1, N2, N3 and N4, respectively (Fig. 5).33 The dominate N1 peak is typically assigned to sp2-bonded N(C–NC) involved in the triazine rings.34,35 While the N2 is attributed to the graphite-like N in the form of N–(C)3, and the peak at 400.9 eV (N3) is assigned to bridging N atoms [exocyclic: NH(C3N3)2, NH2(C3N3)], originating from the defective condensation of heptazine substructures.36–38 The very weak peak N4 at 404.1 eV can be indicative of the charging effects or positive charge localization in the heterocycles.37
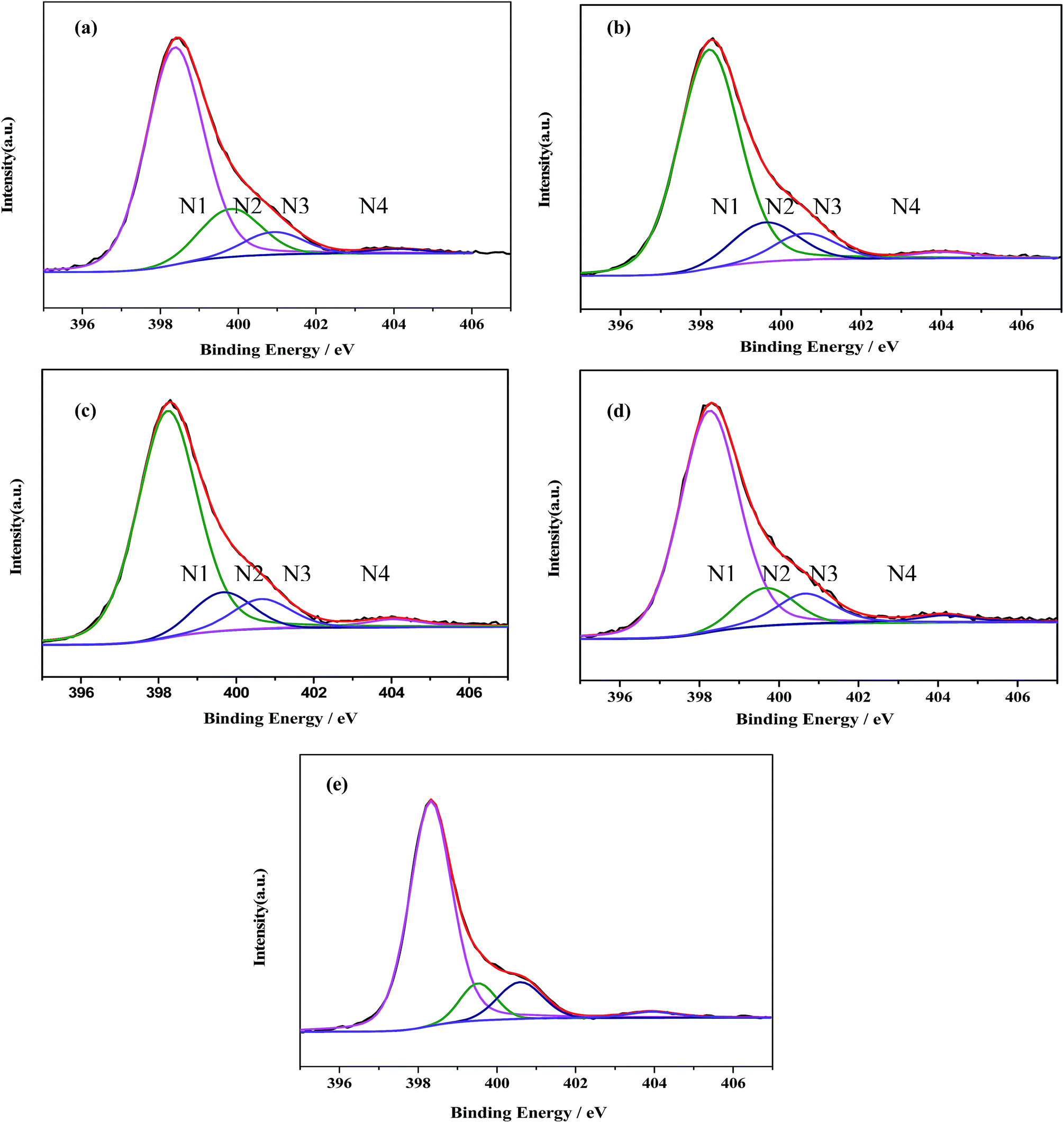 | ||
| Fig. 5 Deconvolution of the XPS N1s peaks for (a) g-C3N4 NS-3 h; (b) g-C3N4 NS-2 h; (c) g-C3N4 NS-1 h; (d) bulk g-C3N4; (e) U-g-C3N4. | ||
The distributions of the four kinds of nitrogen in the samples were estimated according to their percent in total XPS peak area. As shown in Table 1, the N1 ratio keeps substantially constant (ca. 76 at%), while the ratios of N2 and N3 change with different thermal oxidative etching time. With the increase of thermal oxidative etching time, the N2 ratio increases along the decrease of the N3 ratio, which suggests that thermal oxidative etching promote the poly-condensation and the structural rearrangement of small oligomeric fragments. These variation trends are similar with those found in the process of vacuum heat-treatment of carbon nitride.39 As well known, reaction path for the formation of graphitic C3N4 starting from di-cyanodiamide is a combination of a poly-addition and poly-condensation scheme, which consist of several processes, such as the condensation of precursors towards melamine, the rearrangements of melamine to form tris-s-triazine, and the condensation of this tris-s-triazine to polymers.40 The final polymeric C3N4 is composed of a series of different degrees of polymerization of the polymer, so poly-condensation and the structural rearrangement of small oligomeric fragments occur in the thermal etching process where N3 is eliminated.
| Samples | N states | N1 | N2 | N3 | N4 |
|---|---|---|---|---|---|
| g-C3N4 NS-3 h | B.E/eV | 398.4 | 399.8 | 400.9 | 403.9 |
| Atom content/% | 76.73 | 11.04 | 8.97 | 3.26 | |
| g-C3N4 NS-2 h | B.E/eV | 398.2 | 399.6 | 400.7 | 404 |
| Atom content/% | 76.66 | 9.71 | 9.75 | 3.88 | |
| g-C3N4 NS-1 h | B.E/eV | 398.2 | 399.7 | 400.7 | 404.1 |
| Atom content/% | 76.83 | 9.18 | 9.99 | 4.00 | |
| Bulk g-C3N4 | B.E/eV | 398.3 | 399.7 | 400.7 | 404.1 |
| Atom content/% | 76.25 | 8.81 | 11.03 | 3.91 | |
| Bulk U-g-C3N4 | B.E/eV | 398.3 | 399.5 | 400.6 | 403.9 |
| Atom content/% | 75.27 | 8.52 | 12.60 | 3.61 |
3.2 Reaction behavior
The reaction behavior of epoxidation of styrene over g-C3N4 NS-2 h under different reaction conditions was investigated (Fig. S4†). Fig. S4b† depicts the reaction pathway where the conversion of (mol%) and selectivity (mol%) of the products are plotted against reaction time. As expected, styrene conversion increases steadily in the first 12 h, and then grows slowly. The selectivity of epoxidation products dropped with the progress of the reaction, and the selectivity of benzaldehyde changes in the opposite trend simultaneously. This finding is mainly because of the further oxidation of primarily formed epoxide into benzaldehyde by TBHP.Fig. S4d† illustrates the effect of reaction temperature on the conversion (mol%) of styrene and the epoxide selectivity (mol%). As expected, styrene conversion increased from 30% to 76% by increasing the reaction temperature from 333 K to 363 K. Further, selectivity to styrene oxide also increased mainly at the cost of benzaldehyde. This result indicates that the epoxidation process is a thermodynamics controlled reaction, while the oxidative CC bond cleavage is a kinetics controlled reaction. The reusability of a heterogeneous catalyst is a key role for its practical application. For this reason, the practical reusability of the g-C3N4 nanosheet in the epoxidation of styrene was tested to evaluate the stability of the catalyst. The recycling test was performed for continuous five reaction cycles under the identical reaction as mentioned above. The re-used g-C3N4 nanosheet catalyst was separated by centrifugation from the reaction solution, washed with distilled water and ethanol several times, and then dried at 353 K for 12 h. It is interesting to find that the g-C3N4 nanosheet catalyst also shows desired recyclability. As illustrated in Fig. S5,† during the six reaction cycles, there is almost no difference in the conversion and epoxide selectivity of the g-C3N4 NS-3 h on the epoxidation of styrene. In addition, the used catalyst kept the same structure and morphology with fresh g-C3N4 NS-3 h, confirmed by the determination of TEM and XRD. These results strongly indicate that g-C3N4 nanosheets are very stable and active inexpensive catalysts for the epoxidation reaction, meaning that it can be used for further industrial applications.
3.3 Catalyst activities of the g-C3N4 nanosheets
For the first time, the C3N4 nanosheets were used as metal-free catalyst for the epoxidation of styrene to styrene oxide by using TBHP (70 wt% in water) as the oxidant and CH3CN as the solvent. As shown in Table 2, without any catalyst (Entry 5, Table 2), autooxidation of styrene into styrene oxide and benzaldehyde occurs, whereby the conversion of the substrate is very low (∼5%). This value is much lower than that of bulk C3N4 with the conversion of 51.6% and the selectivity of 82.1% (Entry 1, Table 2). It should be noted that the g-C3N4 nanosheet samples apparently exhibit high styrene oxide yield than the bulk C3N4. The thermal etching of bulk C3N4 improves the catalytic performance significantly. Among all the catalysts, g-C3N4 NS-3 h exhibits an optimal conversion of 81.2% with the highest yield of 66.7% (Entry 4, Table 2). This result is superior to those recently reported metal-free catalysts (Entry 10–12, Table 2) under similar conditions.9 In order to exclude the self-polymerization of styrene in the strong oxidizing environment, tributyl phosphate is added to the reaction system employed as an effective polymerization inhibitor.41 The result (Entry 9, Table 2) reveals that high conversion for styrene arises from catalysis rather than polymerization.| Entry | Catal. | Styrene conversion (%) | Selectivity (%) | Yield of styrene oxide (%) | ||
|---|---|---|---|---|---|---|
| Styrene oxide | Benzaldehyde | Others | ||||
| a Reaction condition: 4.35 mmol styrene, 50 mg catalyst, 5 mL solvent (CH3CN), 8.7 mmol TBHP (70% aqueous solution), 80 °C, 12 h.b Without Catal.c Without TBHP.d H2O2 as oxidant.e 2,6-Di-tert-butyl-4-methylphenol was added as a free-radical quencher.f Tributyl phosphate is added as an polymerization inhibitor.g Ref. 10.h Ref. 36.i Ref. 9 (the error range is below ±0.5%).j The bulk g-C3N4 from urea as the catalyst. | ||||||
| 1 | Bulk-g-C3N4 | 51.6 | 82.1 | 14.6 | 3.3 | 42.4 |
| 2 | g-C3N4-NS-1 h | 56.8 | 81.9 | 12.6 | 5.5 | 46.5 |
| 3 | g-C3N4-NS-2 h | 64.0 | 82.7 | 13.9 | 3.4 | 52.9 |
| 4 | g-C3N4-NS-3 h | 81.2 | 82.2 | 12.9 | 4.9 | 66.7 |
| 5b | — | 5.4 | 81.0 | 19.0 | — | 4.37 |
| 6c | g-C3N4-NS-3 h | — | — | — | — | — |
| 7d | g-C3N4-NS-3 h | 13.0 | 7.8 | 88.6 | 3.6 | 1.0 |
| 8e | g-C3N4-NS-3 h | 12.1 | 60.9 | 25.9 | 13.2 | 7.4 |
| 9f | g-C3N4-NS-3 h | 80.5 | 80.7 | 11.3 | 8.0 | 65.0 |
| 10g | N-OLC-3 | 92.9 | 24.2 | 75.8 | — | 22.5 |
| 11h | N-Graphene | 58.0 | 40.0 | 60.0 | — | 23.2 |
| 12i | NG-800 | 87.6 | 72.8 | 27.2 | — | 63.8 |
| 13j | U-Bulk-g-C3N4 | 36.1 | 85.6 | 10.1 | 4.3 | 30.9 |
The remarkably improved catalytic activities of g-C3N4 nanosheets caused by thermal oxidative etching can be mainly attributed to the thin lamella conjugate structure with the larger surface area and porous structures, which provide more surface active sites for promoting the adsorption and more channels for mass transfer. In addition to the BET area affecting the catalytic activity of g-C3N4, the graphitic nitrogen species N2 is one of the main factors. As shown in Fig. 6, the catalytic performance is in line with the N2 ratio which increases with the thermal oxidative etching time. Similarly, the linear relationship between the catalytic activity and the concentration of graphitic nitrogen is also reported by Su and coworkers.10
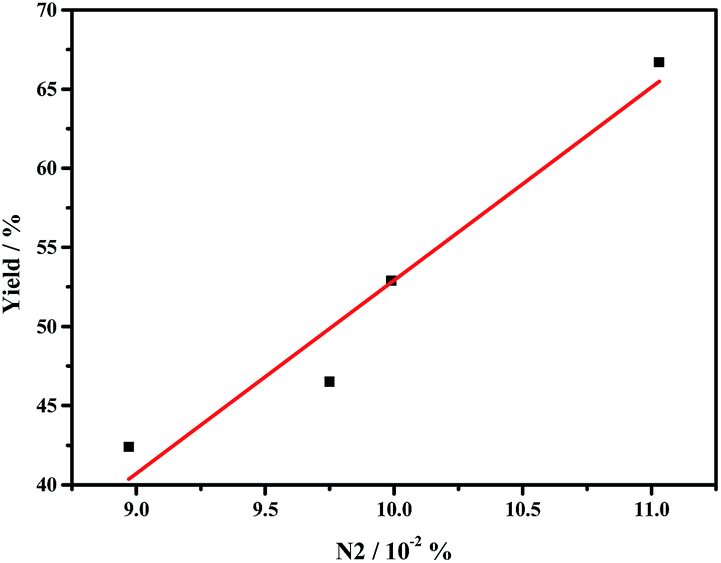 | ||
| Fig. 6 The relationship between the catalytic performance (yield of styrene oxide) and the concentration of graphic nitrogen (N2) in g-C3N4 nanosheet. | ||
Ma et al. reported that graphitic nitrogen modulated the electronic structure of the adjacent sp2 carbon atoms, finally confers the nitrogen-neighboring carbon a metal-like d-band electronic phenomenon.42,43 These unique electronic structure of carbon atoms make them more capable of adsorbing the peroxide-like species such as –OOH.10,42,44,45 To confirm that the catalytic performance of the g-C3N4 is not only decided by the BET area but also the concentration of graphitic nitrogen, we prepared the bulk g-C3N4 using urea as precursor, where the BET area (84.5 m2 g−1) is about 4 times of that bulk g-C3N4 derived from di-cyanodiamine. Compared with the sample using di-cyanodiamine as precursor which goes through a direct poly-addition process to melamine, the urea precursor undergoes a series of gas-releasing stages to form melamine, leading to the formation of a less condensed and porous g-C3N4 with more edge excited amino groups,46 namely higher N3 ratio and lower N2 ratio. As shown in Table 2 (Entry 13), the conversion of styrene on the g-C3N4 from urea is lower that of g-C3N4 derived from di-cyanodiamine. This finding implies that graphitic nitrogen species N2 is critical for the epoxidation activity.
Based on the discussion above and the reported literature,10,43,47 the possible catalytic reaction mechanism of the g-C3N4 nanosheets were proposed and illustrated in Scheme 2. Catalytic reactions traditionally using TBHP have been reported to proceed via a free radical mechanism, which was indirectly verified in this reaction system by using 2,6-di-tert-butyl-4-methylphenol as a free-radical quencher (Entry 8, Table 2). Due to the formation of a metal-like d band electronic structure, the nitrogen-neighboring carbon atom in g-C3N4 nanosheets (Cat) may be more able to interact with TBHP to form Cat-OH species, which can be further converted to Cat-OOH species under the oxidation conditions. In addition, based on the big π system of g-C3N4, styrene with π orbitals are able to adsorb on g-C3N4 surface via a strong π–π interaction, subsequently, on account of the specific properties of free radicals,48 carbon–carbon double bond of styrene is attacked by the α-oxygen of the Cat-OOH species, broke up to carbon–carbon single bond and then formed a transition state with a new C–O bond and C6H5−˙CH–CH2 intermediate species. Subsequently, since the ring-closure barrier is much lower than the C–O bond formation barrier, a ring-closure process essentially leads to the formation of the target epoxide product. In the final step, the surface reactive oxygen species can be recovered by the reaction between tBuOOH, which completed the reaction cycle. In addition, the abundant existence of tBuOH after reaction by using gas chromatography-mass spectrometry verified indirectly the rationality of the proposed mechanism process.
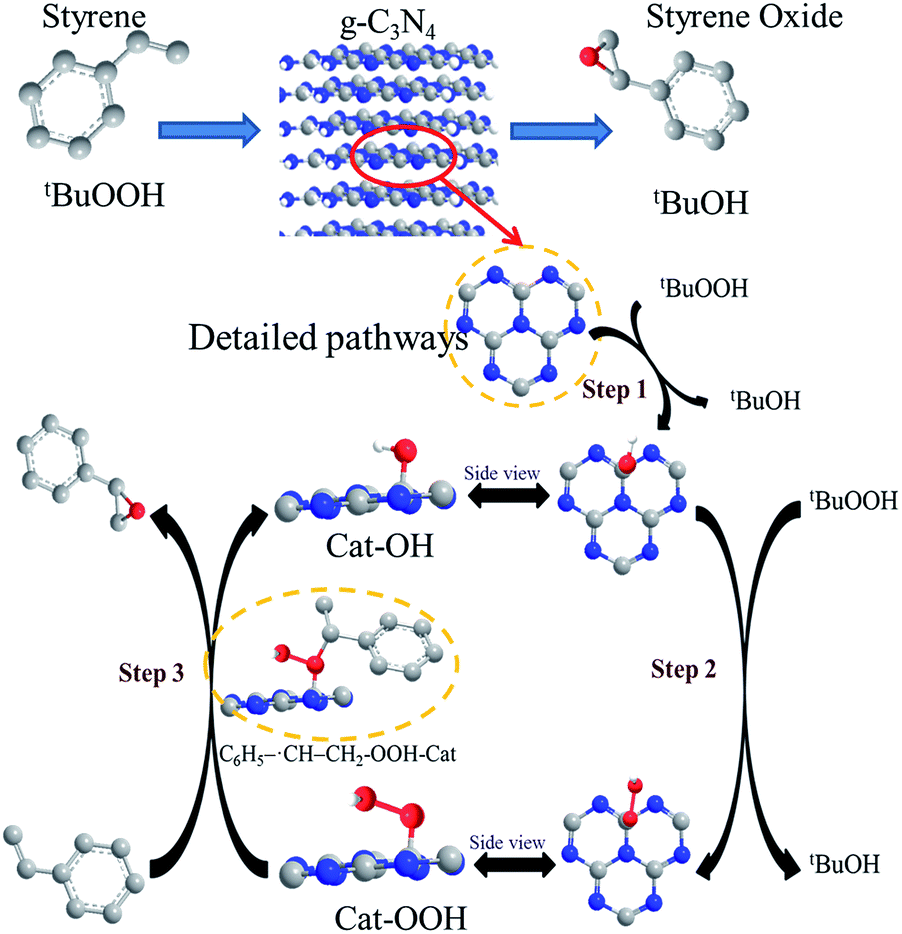 | ||
| Scheme 2 The possible reaction mechanism for the epoxidation of styrene on g-C3N4 nanosheet. | ||
4. Conclusions
In summary, the g-C3N4 nanosheets were prepared by a simple thermal oxidative etching process. Thermal oxidative etching time has a significant effect on the exfoliation process of g-C3N4, lamellar thickness, morphology evolutions and chemical state of nitrogen on the g-C3N4. As a metal-free heterogeneous catalyst, the g-C3N4 NS-3 h nanosheet shows more superior catalytic performance for the epoxidation of styrene to styrene oxide than that of the bulk g-C3N4 and other recently reported metal-free materials. The thin lamellar structure with large specific surface area and pore volume, graphitic nitrogen in g-C3N4 nanosheets contribute to the superior catalytic performance. The optimal styrene conversion and epoxide selectivity reach 81.2% and 82.2%, respectively. The possible reaction mechanism is proposed on the basis of the unique properties of graphitic nitrogen and the free radical theory. This finding put forward a new and eco-friendly methodology for olefin epoxidation using the metal-free catalysts.Acknowledgements
We would like to thank financial support by the Major State Basic Resource Development Program (Grant No. 2012CB224804), NSFC (Project 21373054, 21303023, 21173052), the Natural Science Foundation of Shanghai Science and Technology Committee (08DZ2270500).Notes and references
- A. Murphy, G. Dubois and T. D. P. Stack, J. Am. Chem. Soc., 2003, 125, 5250–5251 CrossRef CAS PubMed
.
- J. M. Thomas and G. Sankar, Acc. Chem. Res., 2001, 34, 571–581 CrossRef CAS PubMed
- G. D. Yadav and A. A. Pujari, Org. Process Res. Dev., 2000, 4, 88–93 CrossRef CAS
- X. Deng and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 17178–17179 CrossRef CAS PubMed
- S. B. Kumar, S. P. Mirajkar, G. C. G. Pais, P. Kumar and R. Kumar, J. Catal., 1995, 156, 163–169 CrossRef CAS
- Z. X. Ding, X. F. Chen, M. Antonietti and X. C. Wang, ChemSusChem, 2011, 4, 274–281 CAS
- X. Liu, J. Ding, X. Lin, R. H. Gao, Z. H. Li and W. L. Dai, Appl. Catal., A, 2015, 503, 117–123 CrossRef CAS
- X. Duan, H. Sun, Y. Wang, J. Kang and S. Wang, ACS Catal., 2015, 5, 553–559 CrossRef CAS
- A. Dhakshinamoorthy, A. Primo, P. Concepcion, M. Alvaro and H. Garcia, Chem.–Eur. J., 2013, 19, 7547–7554 CrossRef CAS PubMed
- W. Li, Y. Gao, W. Chen, P. Tang, W. Z. Li, Z. J. Shi, D. S. Su, J. G. Wang and D. Ma, ACS Catal., 2014, 4, 1261–1266 CrossRef CAS
- Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 40, 4893–4908 RSC
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed
- X. F. Chen, J. S. Zhang, X. Z. Fu, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed
- Y. Wang, J. S. Zhang, X. C. Wang, M. Antonietti and H. R. Li, Angew. Chem., Int. Ed., 2010, 122, 3428–3431 CrossRef
- X. H. Li, X. C. Wang and M. Antonietti, ACS Catal., 2012, 2, 2082–2086 CrossRef CAS
- F. Z. Su, S. C. Mathew, G. Lipner, X. Z. Fu, M. Antonietti, S. Blechert and X. C. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed
- H. Zhao, H. Yu, X. Quan, S. Chen, Y. Zhang, H. Zhao and H. Wang, Appl. Catal., B, 2014, 152–153, 46–50 CrossRef CAS
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed
- P. Niu, L. L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS
- Q. Gu, Z. Gao, H. Zhao, Z. Lou, Y. Liao and C. Xue, RSC Adv., 2015, 5, 49317–49325 RSC
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS
- H. Zhao, H. Yu, X. Quan, S. Chen, H. Zhao and H. Wang, RSC Adv., 2014, 4, 624–628 RSC
- K. Schwinghammer, B. Tuffy, M. B. Mesch, E. Wirnhier, C. Martineau, F. Taulelle, W. Schnick, J. Senker and B. V. Lotsch, Angew. Chem., Int. Ed., 2013, 52, 2435–2439 CrossRef CAS PubMed
- J. Jiang, L. Yang, L. Zhu, A. Zheng, J. Zou, X. Yi and H. Tang, Carbon, 2014, 80, 213–221 CrossRef CAS
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS
- F. Dong, Z. Zhao, T. Xiong, Z.L. Ni, W.D. Zhang, Y.J. Sun and W.K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CAS
- Q. Han, B. Wang, J. Gao, Z. Cheng, Y. Zhao, Z. Zhang and L. Qu, ACS Nano, 2016, 10, 2745–2751 CrossRef CAS PubMed
- C. Yuan, X. Zhang, L. Su, B. Gao and L. Shen, J. Mater. Chem., 2009, 19, 5772–5777 RSC
- J. Zhang, M. Zhang, G. Zhang and Z. Wang, ACS Catal., 2012, 2, 940–948 CrossRef CAS
- J. Xu, L. Zhang, R. Shi and Y. Zhu, J. Mater. Chem. A, 2013, 1, 14766–14772 CAS
- A. Vinu, Adv. Funct. Mater., 2008, 18, 816–827 CrossRef CAS
- Y. J. Cui, J. S. Zhang, G. G. Zhang, J. H. Huang, P. Liu, M. Antonietti and X. C. Wang, J. Mater. Chem., 2011, 21, 13032–13039 RSC
- W. K. Ho, Z. Z. Zhang, W. Lin, S. Huang, X.W. Zhang, X.X. Wang and Y. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 5497–5505 CAS
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed
- Y. Cao, Z. Zhang, J. Long, J. Liang, H. Lin, H. Lin and X. Wang, J. Mater. Chem. A, 2014, 2, 17797–17807 CAS
- B. Jürgens, E. Irran, J. Senker, P. Kroll, H. Müller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288–10300 CrossRef PubMed
- J. Li, G. Zhao, S. Gao, Y. Lv, J. Li and Z. Xi, Org. Process Res. Dev., 2006, 10, 876–880 CrossRef CAS
- Y. J. Gao, G. Hu, J. Zhong, Z. J. Shi, Y. S. Zhu, D. S. Su, J. G. Wang, X. H. Bao and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 2109–2133 CrossRef CAS PubMed
- J. Ding, Q. Q. Liu, Z. Y. Zhang, X. Liu, J. Q. Zhao, S. B. Cheng, B. N. Zong and W. L. Dai, Appl. Catal., B, 2015, 165, 511–518 CrossRef CAS
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110–3116 CrossRef CAS PubMed
- X. Zhong, H. Y. Yu, G. L. Zhuang, Q. Li, X. D. Wang, Y. S. Zhu, L. Liu, X. N. Li, M. D. Dong and J. G. Wang, J. Mater. Chem. A, 2014, 2, 897–901 CAS
- Q. Su, J. Sun, J. Wang, Z. Yang, W. Cheng and S. Zhang, Catal. Sci. Technol., 2014, 4, 1556–1562 CAS
- Y. M. Lin, X. L. Pan, W. Qi, B. S. Zhang and D. S. Su, J. Mater. Chem. A, 2014, 2, 12475–12483 CAS
- A. J. Catino, R. E. Forslund and M. P. Doyle, J. Am. Chem. Soc., 2004, 126, 13622–13623 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26137j |
| ‡ These authors contribute equally to the paper. |
| This journal is © The Royal Society of Chemistry 2017 |