
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fluorescent carbon nitride nanofibrous hydrogel for selective sensing of Cu2+
Zhijun Huang ,
Feng-Wen Yan* and
Guoqing Yuan*
,
Feng-Wen Yan* and
Guoqing Yuan*
Beijing National Laboratory of Molecular Sciences, Key Laboratory of Green Printing, Institute of Chemistry, Chinese Academy of Sciences, Beijing, PR China. E-mail: yanfw@iccas.ac.cn; yuangq@iccas.ac.cn; Fax: +86 10 62559373
First published on 5th January 2017
Abstract
A carbon nitride nanofibrous hydrogel was fabricated by self-assembly/gelation of carbon nitride aqueous solution derived from thermal evaporation of bulk graphitic carbon nitride. The structure, chemical composition and optical properties of the as-prepared nanofibers were well investigated. The π–π interactions between nitrogen containing aromatic rings and hydrogen bonds between hydroxyl and amino groups were the driving forces for the formation of nanofibers and the hydrogel. A fluorescent nanofiber suspension can be obtained by dilution of the hydrogel with water. Due to the excellent fluorescence properties and the high dispersion stability in water, the carbon nitride nanofibers can be employed as a fluorescent probe for the selective sensing of Cu2+.
1 Introduction
Carbon materials have long been used in adsorption, separation, catalysis and electrochemistry. Heteroatom-doping is an effective method to improve the performance and expand the application of carbon based materials.1 By far, nitrogen is the most widely investigated heteroatom.2 Both the chemical reactivity and optical and electronic properties could be improved by nitrogen-doping.3,4Graphitic carbon nitride (g-C3N4), a nitrogen-rich carbon material, has received much interest due to its unusual optical and electronic properties and surprisingly high thermal and chemical stability.5,6 It is generally prepared by directly condensation of nitrogen and carbon containing precursors, such as s-triazine based compounds,7,8 urea9 and guanidine salts.10 This in situ preparation technique provides g-C3N4 with a homogeneously nitrogen-doped carbon backbone. It is composed of tri-s-triazine units connected via trigonal nitrogen atoms. Over the past decade, g-C3N4 has been widely used in metal-free (photo)catalysis,11 catalyst supports,12 sensing,13 and bioimaging.14 It shows high fluorescence quantum yield, high photostability, and good water solubility.15 Various metal ions, including Hg2+,13,16,17 Fe3+,18,19 Cr5+,20 Cu2+,21–23 and Ag+,24,25 can be detected by g-C3N4 through fluorescence sensing.
Low-dimensional nanomaterials have received significant attention in the past two decades. The superiority of such materials over traditional materials is due to the remarkable surface, optical, and electronic properties. To date, low-dimensional nanomaterials have showed great potential applications in the fields of energy storage and conversion,26 catalysis,27 engineering materials,28 environmental science29 and sensors.30 The most widely investigated low-dimensional g-C3N4 nanomaterial is nanosheet. Similar to graphite, bulk g-C3N4 are stacked arrays of two-dimensional (2D) layers. g-C3N4 nanosheets with the thickness of single or several atomic layers can be facially prepared through ultrasonic assistant liquid exfoliation,31 thermal oxidation,32 chemical exfoliation33 and self-assembly.34
A few studies have focused on the one-dimensional (1D) g-C3N4 nanomaterials. The strong π–π stacking of tri-s-triazine units is beneficial to fabricate 1D g-C3N4 nanofiber/nanobelt/nanotube/nanoribbon. Microfibrous g-C3N4 with a mean diameter of about 0.8 μm was synthesized on a large scale by a thermal evaporation method via a vapor-solid process.35 A solvothermal approach was employed to synthesize g-C3N4 nanobelts about 50–60 nm in width by copolymerizing cyanuric chloride and melamine.36 Nanotubular g-C3N4 with a diameter of 300–500 nm was prepared by heating precursors synthesized via self-assembly of protonated melamine in glycol.37 g-C3N4 nanoribbons were synthesized by a one-step hydrothermal method with the existence of graphene oxide.38 Although a number of techniques were developed to prepare 1D g-C3N4 nanomaterials, the fabrication of 1D g-C3N4 with small size and large length–diameter ratio via environmentally friendly strategies is still a challenge.18
Herein, we report a thermal evaporation combined self-assembly method for the preparation of carbon nitride nanofibers. Self-assembly/gelation of the carbon nitride aqueous solution derived from thermal evaporation of bulk g-C3N4 resulted in carbon nitride nanofibrous hydrogel. Preparation of carbon nitride nanofibrous hydrogel was recently reported through hydrolyzing bulk g-C3N4 in 3 M NaOH aqueous solution.39 Our novel method successfully avoids using a large amount of corrosive and harmful reagents. The as-prepared nanofibers with diameter of 10–15 nm and length of 0.5–3 μm show excellent dispersion stability in water. Additionally, the g-C3N4 nanofibrous suspension exhibits strong fluorescence and can be employed as a fluorosensor for the selective detection of Cu2+.
2 Experimental
2.1 Reagents and materials
Melamine was purchased from Sinopharm Chemical Reagent Co., Ltd. Cu(NO3)2, HgCl2, AgNO3, Cr(NO3)3, Fe(NO3)3, Ni(NO3)2, Pb(NO3)2, Cd(NO3)2, Mn(NO3)2, NaNO3, Ca(NO3)2 and quinine sulphate were purchased from Aladdin Reagent Co., Ltd. All reagents were analytical reagent grade and were used as received without further purification.2.2 Synthesis of materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min, and the obtained CNNF was washed with ethanol and dried at 60 °C under vacuum over night.
000 rpm for 10 min, and the obtained CNNF was washed with ethanol and dried at 60 °C under vacuum over night.
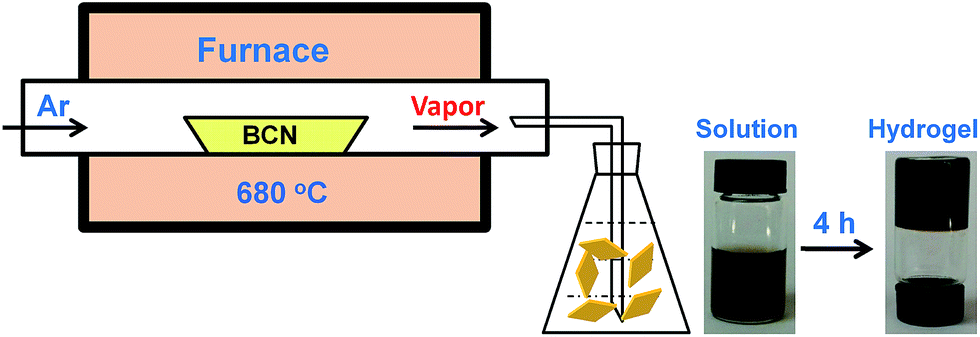 | ||
| Scheme 1 Illustration of carbon nitride hydrogel formation via self-assembly/gelation of carbon nitride aqueous solution. | ||
2.3 Characterizations
Scanning electron microscopy (SEM) images were collected on a Hitachi S-4800 instrument. Transmission electron microscopy (TEM) images were collected on a JEOL JSM-2100 instrument with an accelerating voltage of 200 kV. Powder X-ray diffraction (XRD) dates were obtained on a Rigaku Rotaex diffractometer equipped with a Cu Kα radiation source (40 kV, 200 mA; λ = 1.54056 Å). Fourier transform infrared (FT-IR) spectra were recorded on a Bruker Tensor 27 by using KBr pellets. X-ray photoelectron spectroscopy (XPS) data were recorded on a Thermo Scientific ESCALab 250Xi using 200 W monochromated Al Kα radiation. Elemental analysis experiments were performed on a Flash EA 1112. Thermogravimetric analyses (TGA) were performed on a Pyris 1 TGA (Perkin Elmer) at a heating rate of 10 °C min−1 in air flux. UV-visible (UV-vis) absorption spectra were collected on a Shimadzu UV-2600 UV-visible spectrometer. Fluorescence spectra were recorded on a Hitachi F-4500 fluorescence spectrometer.2.4 Fluorescence sensing of Cu2+
Typically, 20 μL of CNNF suspension (0.5 mg mL−1) was mixed with 10 mL of 25 mM phosphate buffer solution (pH 7.0) containing different concentrations of Cu2+ or other metal ions. The fluorescence emission spectra with an excitation wavelength of 335 nm were recorded at room temperature after 10 min of reactions.3 Results and discussion
3.1 Characterization of materials
It was reported that g-C3N4 obtained by direct heating melamine are bulk morphology with several micrometers in size.41 The thermal stability of BCN is so high that the sublimation and pyrolysis only can occur above 600 °C.42 When BCN was heated at 680 °C under argon flux, it was sublimed slowly and carbon nitride vapor was generated continuously. A hydrogel was formed via self-assembly/gelation of carbon nitride aqueous solution (Scheme 1). After dilution of the hydrogel with deionized water to 0.5 mg mL−1, a fluorescent suspension can be obtained. This suspension is very stable and no precipitates can be found within several months. SEM images of CNNF in Fig. 1a clearly indicate the formation of a large amount of nanofibers with homogeneous diameters. Most of them are 0.5–3 μm long. TEM images (Fig. 1b) show that the diameters of the nanofibers are in the range of 10–15 nm. | ||
| Fig. 1 SEM (a) and TEM (b) images of CNNF. | ||
The crystal structures of the samples were characterized by XRD. As presented in Fig. 2, BCN shows two obvious characteristic diffraction peaks. The strong peak located at 27.4° corresponds to the typical interlayer-stacking structures (002). The weak one located at 13.0° belongs to the in-planar repeating tri-s-triazine units (100).43 CNNF exhibits a similar (002) peak, suggesting the interlayer-stacking structures have not been destroyed. The crystallinity of CNNF decreased slightly, which can be explained by the weakened intensity of the (002) peak. No (100) peak for CNNF was observed. This may be caused by the partial pyrolysis and structural deform of tri-s-triazine units. The pyrolysis of tri-s-triazine units induced an increased interlayer distance, which is identified by the shift of (002) peak from 27.4 to 27.2°.
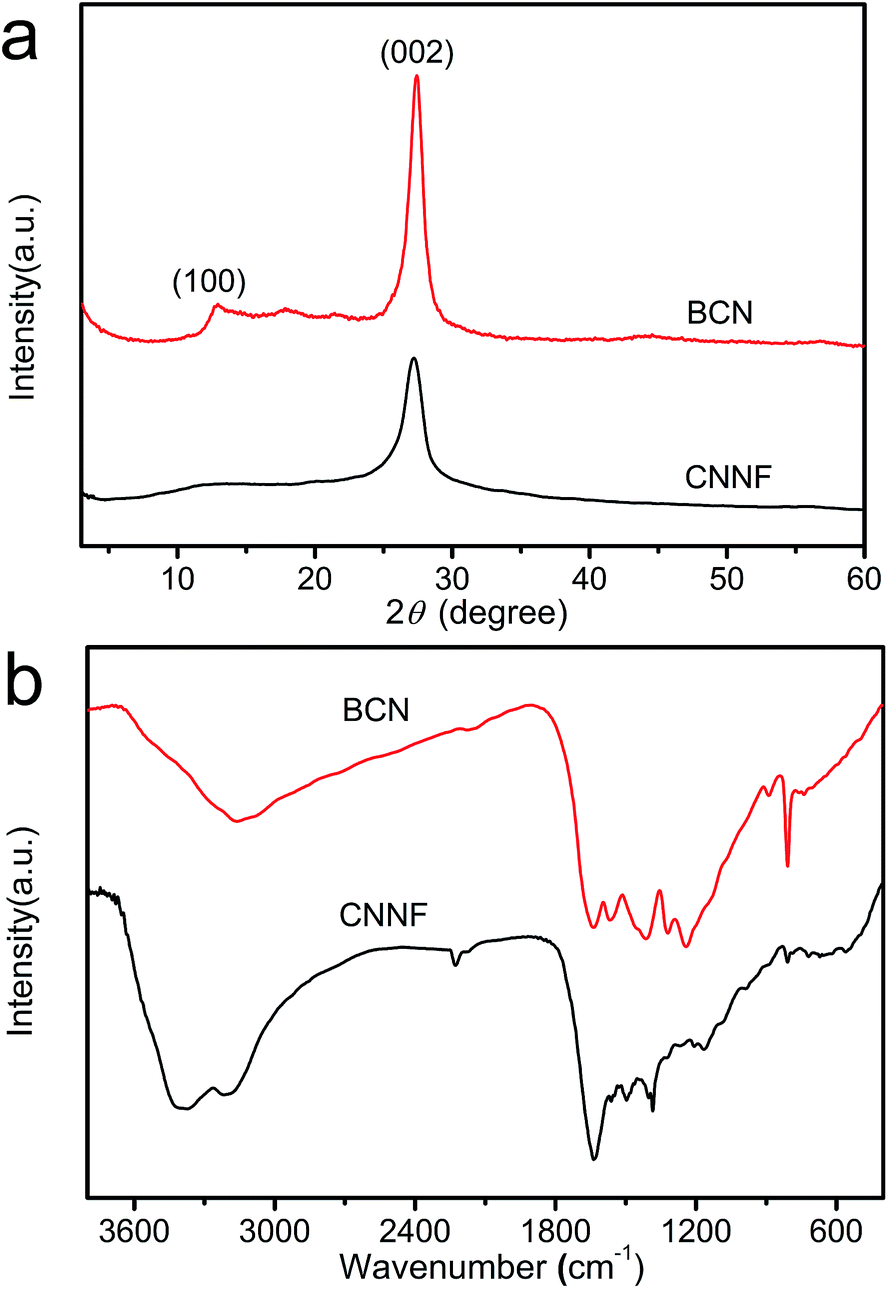 | ||
| Fig. 2 XRD patterns (a) and FT-IR spectra (b) of BCN and CNNF. | ||
The partial pyrolysis of tri-s-triazine units was further investigated by FT-IR spectra (Fig. 2b). BCN exhibits a sharp absorption peak at 806 cm−1. This peak can be assigned to the breathing mode of tri-s-triazine units.44 As can be seen, it becomes weaker and shows a little shift to 810 cm−1 for CNNF, which results from the partial decomposition of tri-s-triazine units. This is also confirmed by the obviously changed absorption peaks in 1250–1650 cm−1 region. The signal at 1384 cm−1, which belongs to the secondary (2C–N) and tertiary (3C–N) amine fragments, becomes stronger, indicating the breaking of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and the generation of C–N.35 During the decomposition of tri-s-triazine units, a small amount of cyano group is generated, which can be identified by the obvious absorption peak at 2226 cm−1. After thermal evaporation, the intensities of the signals corresponding to the N–H bond at 3180 cm−1 and the O–H bond at 3370 cm−1 are enhanced compared to that of BCN. This indicates that some hydroxyl and amino groups (–NH2 or NH groups) were formed during thermal evaporation and subsequent self-assembly/gelation in aqueous solution.45 Hydroxyl and amino groups provide CNNF with excellent dispersion stability in water.
N and the generation of C–N.35 During the decomposition of tri-s-triazine units, a small amount of cyano group is generated, which can be identified by the obvious absorption peak at 2226 cm−1. After thermal evaporation, the intensities of the signals corresponding to the N–H bond at 3180 cm−1 and the O–H bond at 3370 cm−1 are enhanced compared to that of BCN. This indicates that some hydroxyl and amino groups (–NH2 or NH groups) were formed during thermal evaporation and subsequent self-assembly/gelation in aqueous solution.45 Hydroxyl and amino groups provide CNNF with excellent dispersion stability in water.
EA and XPS were employed to investigate the chemical compositions. As shown in Table 1, the C/N atomic ratio of BCN determined by EA is 0.66, which is much lower than the theoretical value (0.75). This can be explained by the incomplete polymerization of nitrogen-rich intermediates during condensation.46 The C/N atomic ratio of CNNF increases to 1.00. Clearly, compared to carbon, nitrogen is preferentially lost during thermal evaporation due to the lower thermal stability. XPS measurements present a similar result. As shown in Fig. 3a, both BCN and CNNF are mainly composed of carbon, nitrogen and oxygen elements. The C/N atomic ratio is increased largely from 0.95 for BCN to 1.47 for CNNF. To further clarify the chemical structure of CNNF, the high-resolution of C 1s XPS spectra were investigated (Fig. 3b). The C 1s XPS spectrum of BCN was fitted into two peaks. The peak at a binding energy of 288.1 eV is ascribed to carbon in tri-s-triazine units, and the peak at 284.8 eV is ascribed to graphite carbon.47 The content of graphite carbon for CNNF is comparable to that of BCN. However, the carbon in tri-s-triazine units decreased sharply from 71.2% for BCN to 20.3% for CNNF. A new chemical state of carbon at 286.5 eV was found in the C 1s XPS spectrum of CNNF. This can be attributed to C–N and C–O species, which is accordance with the FT-IR results.48 The existence of C–OH species was also confirmed by O 1s XPS spectrum. As shown in Fig. 3c, the O 1s XPS spectrum was fitted into two peaks at 532.8 and 531.8 eV. These signals could be ascribed to the adsorbed water and C–OH species respectively.49
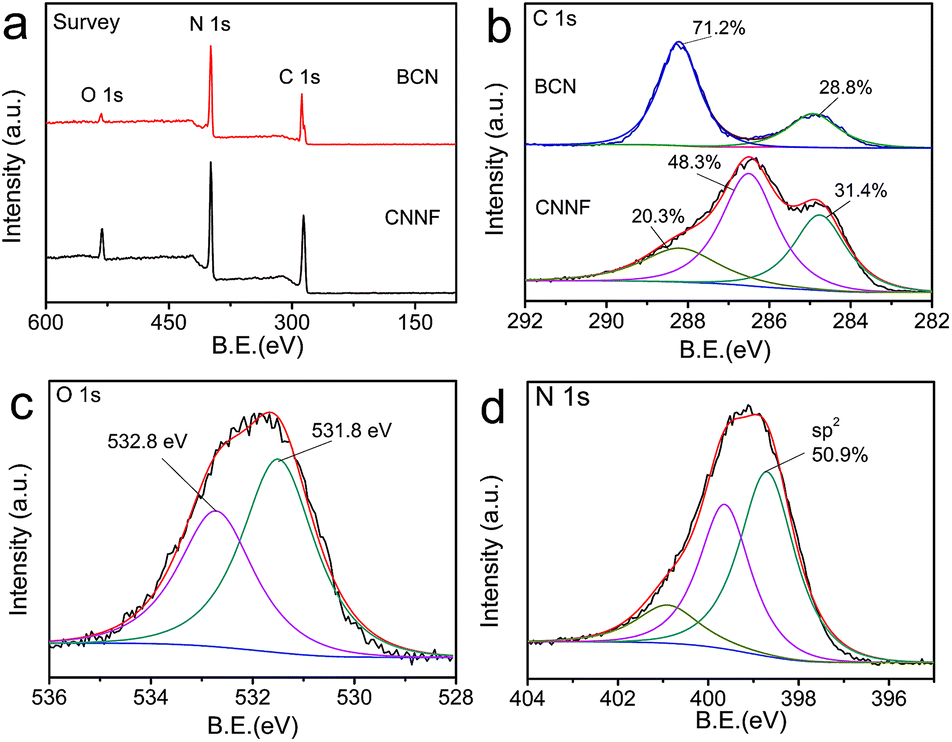 | ||
| Fig. 3 XPS survey spectra (a) and C 1s XPS spectra (b) of BCN and CNNF. O 1s XPS spectrum (c) and N 1s XPS spectrum (d) of CNNF. | ||
Due to the high thermal stability, tri-s-triazine units cannot be decomposed completely. This was already demonstrated by the FT-IR and C 1s XPS measurements. To further clarify the chemical states of nitrogen atoms, N 1s XPS spectrum was fitted into three peaks with the binding energies of 398.7, 399.6 and 400.9 eV, corresponding to the sp2-hybridized nitrogen within aromatic rings, tertiary nitrogen connected with aromatic rings and terminal amino groups, respectively.50 The content of the sp2 nitrogen is 50.9% (Fig. 3d), suggesting that about half of the nitrogen atoms exist in the form of tri-s-triazine or s-triazine. These nitrogen containing aromatic rings provide CNNF with π–π stacked structures and specific optical properties.
The thermal behaviour of CNNF was measured by TGA analysis under air flux. For comparison, the result for BCN was also presented. As shown in Fig. 4, BCN shows an obvious weight loss above 600 °C, related to the sublimation and pyrolysis of the tri-s-triazine units. CNNF shows a markedly different thermal behavior. The TGA curve exhibits a moderate weight loss between 100 and 500 °C due to the loss of amino, hydroxyl and s-triazine groups. Above 500 °C, the weight loss accelerates, which is ascribed to the polymerization of s-triazine units and pyrolysis of tri-s-triazine units.
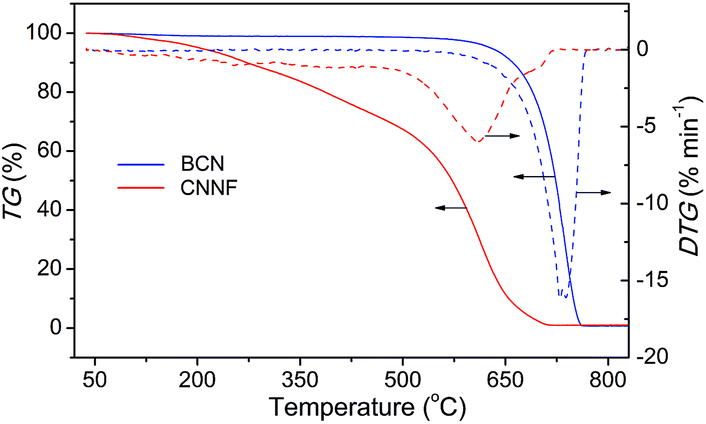 | ||
| Fig. 4 TGA curves of BCN and CNNF at a heating rate of 10 °C min−1 under air flux. | ||
3.2 Mechanism for the formation of nanofibers and hydrogels
In order to investigate the mechanism for the formation of nanofibers and hydrogels, time-dependent TEM images of the self-assembled products were provided in Fig. 5. Nanoparticles with 10–15 nm in diameters and a small amount of short nanorods with the same diameters are found in the immediately obtained carbon nitride aqueous solution (Scheme 1). After maintained at room temperature for 0.5 h, the colour of the solution becomes darker gradually. The nanoparticles are disappeared and nanorods with lengths of tens to hundreds of nanometers are obtained. The lengths of the nanorods increase continuously with the increase in time. Finally, nanofibers with the lengths of 0.5–3 μm are formed after 4 h. At the same time, the viscosity of the solution increases continuously, a black hydrogel is obtained (Scheme 1). These observations as well as the conclusions made from the above analyses of the crystal structures and chemical compositions allow proposing a mechanism for the formation of nanofibers and hydrogels. Firstly, BCN is sublimed at 680 °C to form carbon nitride vapor. Some amino groups are generated during the partial decomposition. Carbon nitride vapor is then dissolved in water and carbon nitride aqueous solution is obtained. Simultaneously, some carbon nitride moleculars react with water to form hydroxyl groups. The existence of amino and hydroxyl groups means that extensive hydrogen bonding may take place, which leads to the self-assembly and generation of nanoparticles. Due to the high thermal stability of BCN, some aromatic nucleuses in the form of s-triazine or tri-s-triazine are left in the carbon nitride moleculars. The π–π stacking of these aromatic nucleuses as well as the hydrogen bonding serves as the driving force for the further growth of nanoparticles to nanofibers.51 Finally, hydrogel is formed via self-organization of the obtained fibrous materials.52 | ||
| Fig. 5 TEM images of the self-assembled products obtained at different times. | ||
3.3 Optical properties of CNNF suspension
The optical properties of CNNF suspensions were investigated by UV-vis absorption spectrum and fluorescence excitation and emission spectra. As shown in Fig. 6a, BCN gives a strong UV-vis absorption with an absorption edge of about 450 nm. Due to the partial decomposition of tri-s-triazine units, CNNF demonstrates a different UV-vis absorption. A absorption peak cantered at around 295 nm is observed, which may be ascribed to the π–π* electronic transitions.53 The terminal groups provide CNNF with an obvious absorption shoulder between 400 and 600 nm.54 Strong fluorescence excitation peak is presented at around 335 nm (Fig. 6b). Under the excitation wavelength of 335 nm, CNNF exhibits an emission peak cantered at around 435 nm. Excitation wavelength affects the fluorescence emission largely (Fig. 6c). When the excitation wavelength increases from 280 to 320 nm, the emission peak of CNNF shows no obvious shift but the fluorescence intensity increases significantly. Further increase in the excitation wavelengths, the emission peak shows an obvious red shift, and the intensity decreases gradually. The excitation-dependent emission feature indicates that CNNF contains more than one emitter. This can be ascribed to the abundant surface functional groups in CNNF.19 The relative fluorescence quantum yield of CNNF suspension was determined to be 7.26% in reference to quinine sulphate. The effect of the solution pH on the fluorescence intensity of the CNNF suspension was also investigated. It can be seen from Fig. 6d that no obvious fluctuation in the fluorescence intensity was observed within the pH range from 3 to 10.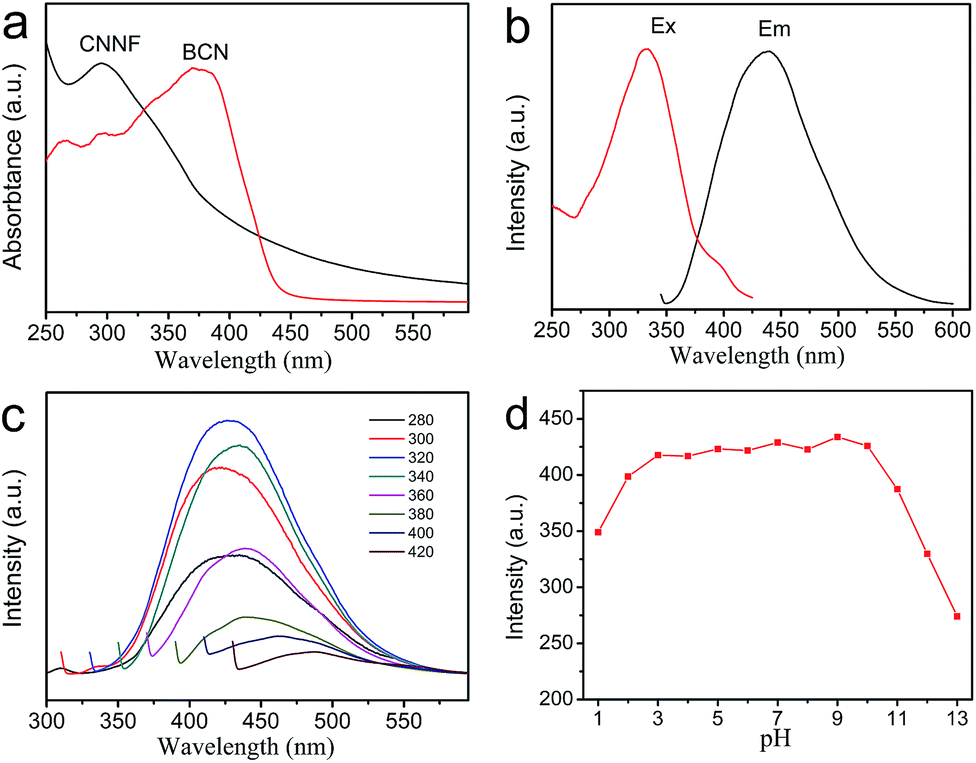 | ||
| Fig. 6 (a) UV-vis absorption spectra of BCN and CNNF suspension. (b) Fluorescence excitation and emission spectra of CNNF suspension. (c) Fluorescence emission spectra of CNNF suspension corresponding to different excitation wavelengths from 280–420 nm. (d) Effect of pH on the fluorescence intensity of CNNF suspension (λex = 335 nm). | ||
3.4 Fluorescence sensing of Cu2+
The excellent fluorescence properties make CNNF a potential fluorescent sensor for detection of metal ion. As shown in Fig. 7a, fluorescence responses of CNNF are different in the presence of different metal ions (50 μM). The fluorescence intensity was greatly quenched by adding Cu2+. It is reported that the fluorescence responses of g-C3N4 based nanomaterials is sensitive to Hg2+,55 Ag+,24 and Fe3+.18 For CNNF, these ions as well as other ions (Cr3+, Ni2+, Pb2+, Cd2+, Mn2+, Na+ and Ca2+) tested under the same conditions show no obvious effects on the fluorescence intensities. The interference of these metal ions was further investigated. As shown in Fig. 7b, no significant differences of the fluorescence intensities between CNNF + Cu2+ and CNNF + Cu2+ + Mn+ systems are observed, indicating that CNNF is an effective sensing material for the selective detection of Cu2+.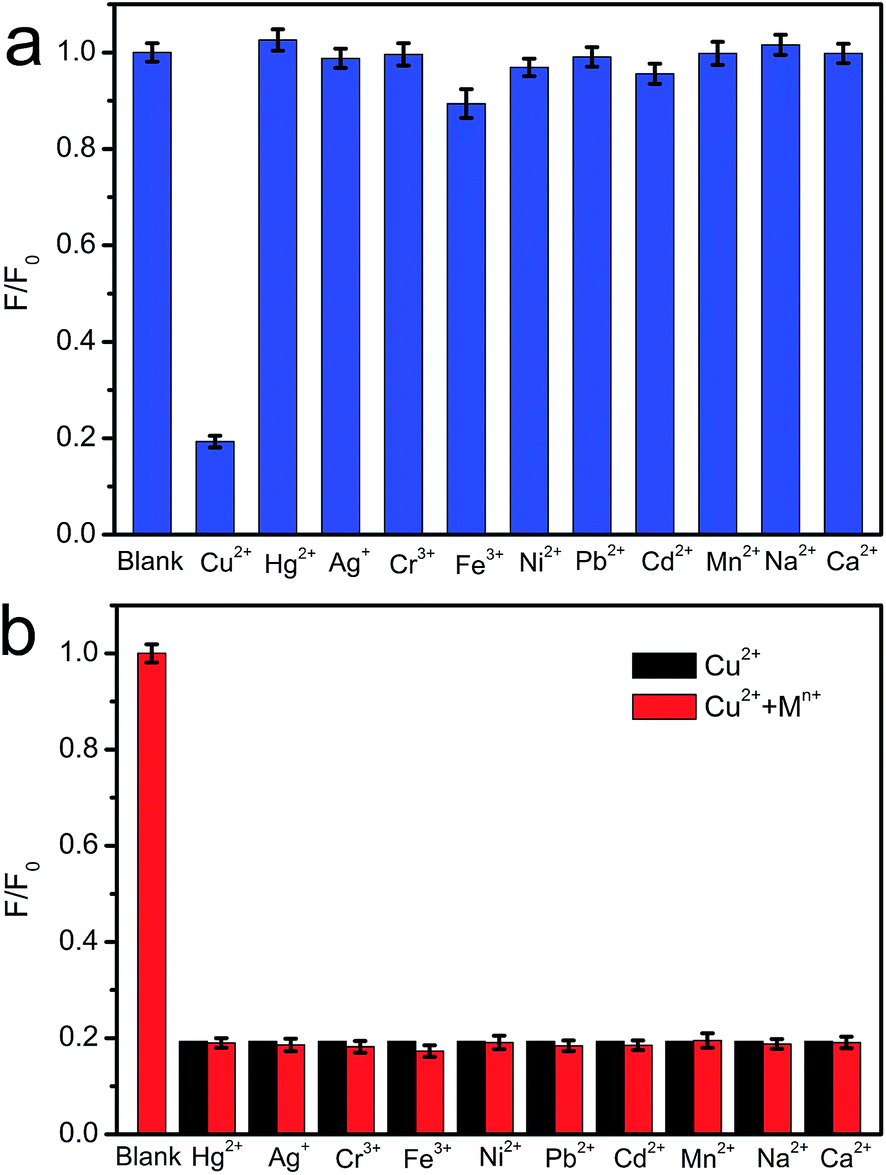 | ||
| Fig. 7 (a) Fluorescence selectivity of CNNF suspension in the presence of 50 μM of various metal ions. (b) Fluorescence response of CNNF suspension towards 50 μM Cu2+ and interference of 50 μM of other metal ions with 50 μM Cu2+. | ||
The quenching mechanism was investigated by XPS spectrum. Fig. 8 shows the Cu 2p3/2 spectrum of CNNF treated with Cu2+. The spectrum is fitted into two peaks. One at 932.8 eV is derived from Cu2+. The other at 934.8 eV is ascribed to Cu+. The Cu+ species are stemmed from the reduction of Cu2+. The redox potential of Cu2+/Cu+ lies between the conduction band (CB) and valence band (VB) of CNNF. The photoinduced electron is transferred from the CB of CNNF to Cu2+, which leads to the reduction of Cu2+ to Cu+. Meanwhile, the fluorescence of CNNF is quenched.24
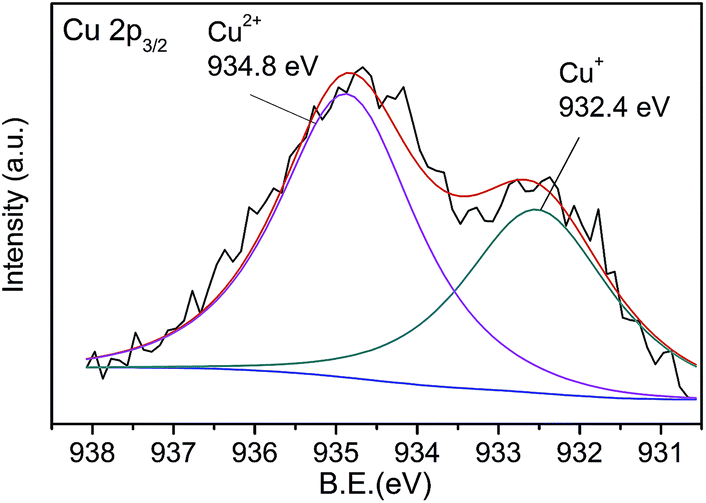 | ||
| Fig. 8 Cu 2p3/2 XPS spectrum of CNNF treated with Cu2+. | ||
The dependence of fluorescence intensity on the concentration of Cu2+ was studied. As shown in Fig. 9a, the fluorescence intensity decreases gradually with the increase in Cu2+ concentration. Two good linearities between the fluorescence intensity and the Cu2+ concentration are observed in 0–0.8 μM and 0.8–15 μM region, respectively. Two different interaction sites for Cu2+ may exist in CNNF.16 At the concentration of 0–0.8 μM, Cu2+ interacts preferentially with the stronger sites. At the concentration of 0.8–15 μM, Cu2+ interacts with the weaker sites. The time-dependent fluorescence spectra of the CNNF suspension treated with Cu2+ in Fig. 9b indicate a similar result. The fluorescence intensity quenches quickly within the first 1 minute, which is ascribed to the interaction of Cu2+ with the stronger sites. While the stronger sites are depleted, Cu2+ interacts with the weaker sites, leading to a slower fluorescence quenching. The content of the stronger sites is lower than that of the weaker sites, which can be explained by the relatively narrow concentration range of Cu2+ for the first linearity. The detection limit was calculated to be 0.06 μM.
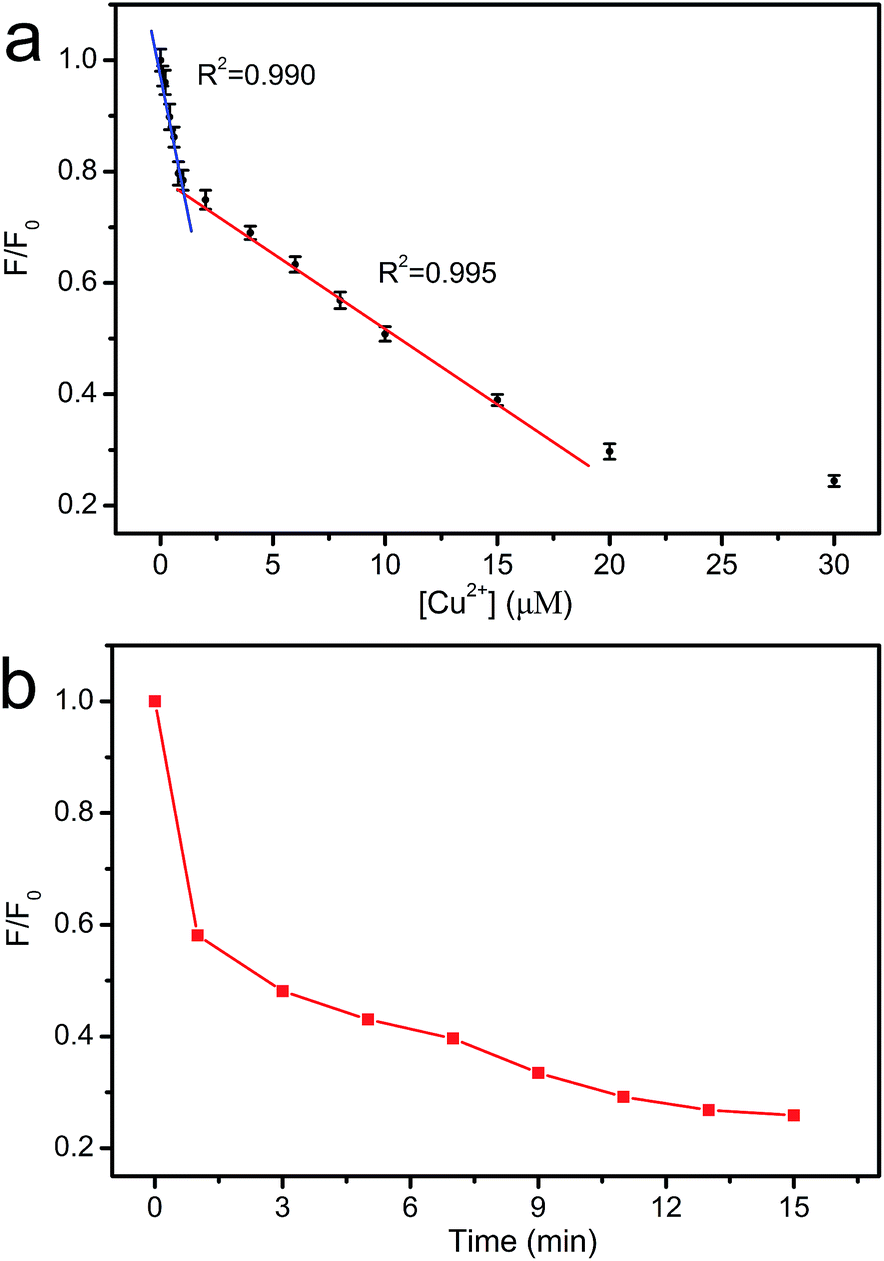 | ||
| Fig. 9 (a) The relationship between F/F0 and the concentration of Cu2+. (b) Time-dependent fluorescence intensity of CNNF suspension (λex = 335 nm). | ||
Tap water was selected as the real sample to investigate the practical application of the fluorescent probe (Fig. 10). CNNF exhibits excellent selectivity to Cu2+. Other metal ions in tap water have little effect on the fluorescence intensities of CNNF. Adding Cu2+ into the tap water led to a significant fluorescence quenching. The fluorescence intensity decreases with the increase of Cu2+ concentration from 0 to 1 μM. Good linear relationship between the fluorescence intensity and the Cu2+ concentration can be observed in the range of 0–0.8 μM. This implies that the fluorescent probe has potential application in real water samples for the detection of Cu2+.
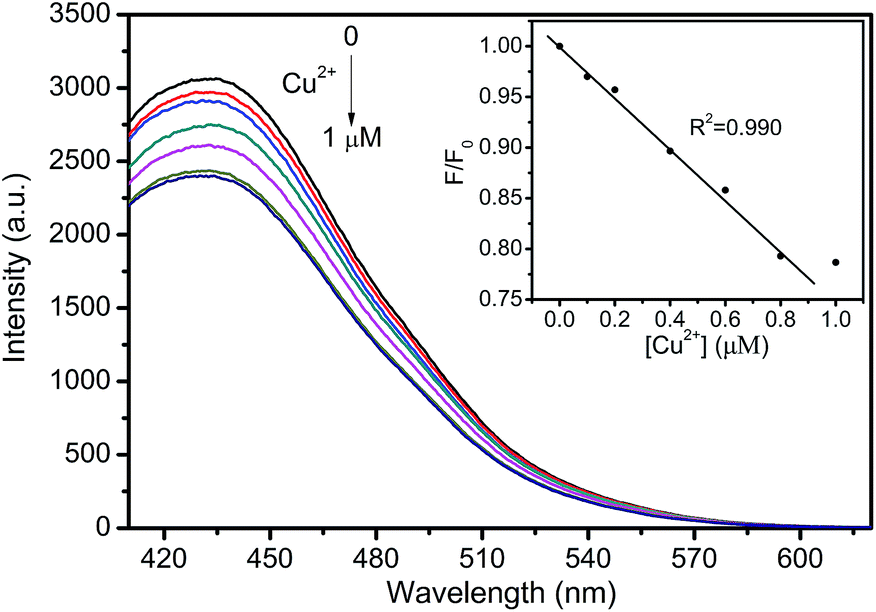 | ||
| Fig. 10 Fluorescence spectra of CNNF dispersion in the presence of different Cu2+ concentrations (0, 0.1, 0.2, 0.4, 0.6, 0.8 and 1 μM) in tap water. Inset: the relationship between F/F0 and the concentration of Cu2+. | ||
4 Conclusions
In conclusion, we reported a fluorescent carbon nitride nanofibrous hydrogel for the selective sensing of Cu2+. Thermal evaporation and subsequent self-assembly/gelation was proved to be an effective method for the preparation of carbon nitride nanofibrous hydrogel. The π–π stacking of the nitrogen containing aromatic rings and the hydrogen bonding serve as the driving forces for the formation of nanofibrous hydrogel. The fluorescent nanofiber suspension was obtained by dilution of the hydrogel with water. The excellent fluorescence properties and the high dispersion stability in water make carbon nitride nanofiber promising for the sensing application in the selective detection of Cu2+. Therefore, our present study has opened a facile way for the synthesis of 1D g-C3N4 nanomaterials with improved structural and optical properties.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21304101).Notes and references
- L. Song, Z. Wu, H. Liang, F. Zhou, Z. Yu, L. Xu, Z. Pan and S. Yu, Nano Energy, 2016, 19, 117–127 CrossRef CAS.
- Q. Wei, X. Tong, G. Zhang, J. Qiao, Q. Gong and S. Sun, Catalysts, 2015, 5, 1574–1602 CrossRef CAS.
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Gong, M. Li, H. Li and Y. Wang, Green Chem., 2015, 17, 715–736 RSC.
- F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171–15174 RSC.
- Z. Huang, F. Li, B. Chen and G. Yuan, Catal. Sci. Technol., 2014, 4, 4258–4264 CAS.
- J. Zhu, P. Xiao, H. Li and S. A. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CAS.
- Z. Huang, F. Li, B. Chen, F. Xue, G. Chen and G. Yuan, Appl. Catal., A, 2011, 403, 104–111 CrossRef CAS.
- H. Zhang, Y. Huang, S. Hu, Q. Huang, C. Wei, W. Zhang, L. Kang, Z. Huang and A. Hao, J. Mater. Chem. C, 2015, 3, 2093–2100 RSC.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed.
- Z. X. Zhou, Y. F. Shen, Y. Li, A. R. Liu, S. Q. Liu and Y. J. Zhang, ACS Nano, 2015, 9, 12480–12487 CrossRef CAS PubMed.
- J. Ma, B. Guo, X. Cao, Y. Lin, B. Yao, F. Li, W. Weng and L. Huang, Talanta, 2015, 143, 205–211 CrossRef CAS PubMed.
- X. Cao, J. Ma, Y. Lin, B. Yao, F. Li, W. Weng and X. Lin, Spectrochim. Acta, Part A, 2015, 151, 875–880 CrossRef CAS PubMed.
- J. Tian, Q. Liu, A. M. Asiri, X. Sun and Y. He, Sens. Actuators, B, 2015, 216, 453–460 CrossRef CAS.
- S. Zhang, J. Li, M. Zeng, J. Xu, X. Wang and W. Hu, Nanoscale, 2014, 6, 4157–4162 RSC.
- M. Rong, L. Lin, X. Song, Y. Wang, Y. Zhong, J. Yan, Y. Feng, X. Zeng and X. Chen, Biosens. Bioelectron., 2015, 68, 210–217 CrossRef CAS PubMed.
- M. Rong, X. Song, T. Zhao, Q. Yao, Y. Wang and X. Chen, J. Mater. Chem. C, 2015, 3, 10916–10924 RSC.
- J. Tian, Q. Liu, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2013, 85, 5595–5599 CrossRef CAS PubMed.
- B. Xia, M. Chu, S. Wang, W. Wang, S. Yang, C. Liu and S. Luo, Anal. Chim. Acta, 2015, 891, 113–119 CrossRef CAS PubMed.
- H. Huang, R. Chen, J. Ma, L. Yan, Y. Zhao, Y. Wang, W. Zhang, J. Fan and X. Chen, Chem. Commun., 2014, 50, 15415–15418 RSC.
- Y. Cao, W. Wu, S. Wang, H. Peng, X. Hu and Y. Yu, J. Fluoresc., 2016, 26, 739–744 CrossRef CAS PubMed.
- P. Kumar, F. Y. Wu, L. H. Hu, S. Ali Abbas, J. Ming, C. N. Lin, J. Fang, C. W. Chu and L. J. Li, Nanoscale, 2015, 7, 8093–8100 RSC.
- X. L. Pan and X. H. Bao, Acc. Chem. Res., 2011, 44, 553–562 CrossRef CAS PubMed.
- X. Sun, H. Sun, H. Li and H. Peng, Adv. Mater., 2013, 25, 5153–5176 CrossRef CAS PubMed.
- Y. Jin, S. C. Hawkins, C. P. Huynh and S. Su, Energy Environ. Sci., 2013, 6, 2591–2596 CAS.
- Y. Du and S. Guo, Nanoscale, 2016, 8, 2532–2543 RSC.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- J. Xu, L. Zhang, R. Shi and Y. Zhu, J. Mater. Chem. A, 2013, 1, 14766–14772 CAS.
- Z. J. Huang, F. B. Li, B. F. Chen and G. Q. Yuan, ChemSusChem, 2016, 9, 478–484 CrossRef CAS PubMed.
- Y. Zhao, Z. Liu, W. Chu, L. Song, Z. Zhang, D. Yu, Y. Tian, S. Xie and L. Sun, Adv. Mater., 2008, 20, 1777–1781 CrossRef CAS.
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 11814–11818 CrossRef CAS PubMed.
- J. Gao, Y. Zhou, Z. Li, S. Yan, N. Wang and Z. Zou, Nanoscale, 2012, 4, 3687–3692 RSC.
- Y. Zhao, F. Zhao, X. Wang, C. Xu, Z. Zhang, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2014, 53, 13934–13939 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhou, Y. Shen, Q. Zhou, J. Wang, A. Liu, S. Liu and Y. Zhang, ACS Nano, 2016, 10, 9036–9043 CrossRef CAS PubMed.
- Z. Huang, F. Li, B. Chen and G. Yuan, RSC Adv., 2015, 5, 102700–102706 RSC.
- Z. Huang, F. Li, B. Chen and G. Yuan, RSC Adv., 2015, 5, 14027–14033 RSC.
- Z. Yang, Y. Zhang and Z. Schnepp, J. Mater. Chem. A, 2015, 3, 14081–14092 CAS.
- J. Qin, J. Huo, P. Zhang, J. Zeng, T. Wang and H. Zeng, Nanoscale, 2016, 8, 2249–2259 RSC.
- P. Yang, J. Zhao, W. Qiao, L. Li and Z. Zhu, Nanoscale, 2015, 7, 18887–18890 RSC.
- Z. Huang, F. Li, B. Chen and G. Yuan, Catal. Sci. Technol., 2016, 6, 2942–2948 CAS.
- V. W. Lau, M. B. Mesch, V. Duppel, V. Blum, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2015, 137, 1064–1072 CrossRef CAS PubMed.
- Z. Zhou, J. Wang, J. Yu, Y. Shen, Y. Li, A. Liu, S. Liu and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 2179–2182 CrossRef CAS PubMed.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC.
- G. Dong, Z. Ai and L. Zhang, RSC Adv., 2014, 4, 5553–5560 RSC.
- G. Algara-Siller, N. Severin, S. Y. Chong, T. Bjorkman, R. G. Palgrave, A. Laybourn, M. Antonietti, Y. Z. Khimyak, A. V. Krasheninnikov, J. P. Rabe, U. Kaiser, A. I. Cooper, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2014, 53, 7450–7455 CrossRef CAS PubMed.
- M. L. Ma, Y. Kuang, Y. Gao, Y. Zhang, P. Gao and B. Xu, J. Am. Chem. Soc., 2010, 132, 2719–2728 CrossRef CAS PubMed.
- Y. Chen, M. Quan, H. Yu, L. Zhang, H. Yang and Y. Lu, RSC Adv., 2015, 5, 31219–31225 RSC.
- Z. Geng, H. Zhang, Q. Xiong, Y. Zhang, H. Zhao and G. Wang, J. Mater. Chem. A, 2015, 3, 19455–19460 CAS.
- P. Wu, J. Wang, J. Zhao, L. Guo and F. E. Osterloh, J. Mater. Chem. A, 2014, 2, 20338–20344 CAS.
- Y. Lu, J. Chen, A. Wang, N. Bao, J. Feng, W. Wang and L. Shao, J. Mater. Chem. C, 2015, 3, 73–78 RSC.
| This journal is © The Royal Society of Chemistry 2017 |