
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic carbon gel assisted-synthesis of Li1.2Mn0.6Ni0.2O2 for a high-performance cathode material for Li-ion batteries
Errui Wanga,
Chunfeng Shaoa,
Shujun Qiuab,
Hailiang Chu *ab,
Yongjin Zouab,
Cuili Xiangab,
Fen Xuab and
Lixian Sun*ab
*ab,
Yongjin Zouab,
Cuili Xiangab,
Fen Xuab and
Lixian Sun*ab
aSchool of Materials Science and Engineering, Guilin University of Electronic Technology, Guilin 541004, P. R. China
bGuangxi Key Laboratory of Information Materials, Guangxi Collaborative Innovation Center of Structure and Property for New Energy and Materials, Guilin University of Electronic Technology, Guilin 541004, P. R. China. E-mail: chuhailiang@guet.edu.cn; sunlx@guet.edu.cn
First published on 5th January 2017
Abstract
Lithium-rich layered oxide Li1.2Ni0.2Mn0.6O2 with a stable network flake structure has been synthesized through a facile resorcinol–formaldehyde (RF) organic carbon gel-assisted method. The as-prepared sample used as a cathode material in lithium ion batteries (LIBs) was characterized by X-ray powder diffraction (XRD), X-ray photoelectron spectroscopy (XPS), field-emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM) and electrochemical measurements. The stable network flake structure is assembled through a dense stack of nanoparticles with an average size of 50–200 nm. As an active material for LIB cathodes, the Li1.2Ni0.2Mn0.6O2 sample shows excellent rate capacities and cycling stability, and delivers a high initial discharge capacity of 273.3 mA h g−1 at 0.1C (1C = 200 mA g−1) between 2.0 V and 4.8 V. When the discharge rate is increased to 2C, an initial capacity of 196.7 mA h g−1 is obtained. After 150 cycles, a discharge capacity of 183.7 mA h g−1 and a high capacity retention of 93.4% are yielded at a rate of 2C.
1. Introduction
Lithium ion batteries (LIBs) with long cycle life, high energy density, high power density, and decent environmental friendliness have great potential to be widely employed as large-scale power sources for electric vehicles (EVs) and hybrid electric vehicles (HEVs).1–3 The cathode materials have been extensively investigated because they play an important role in LIBs. Commercial LiCoO2, as a typical example, has been initially used as a cathode material since Sony Corporation introduced it into LIBs.4 However, the disadvantages of commercial LiCoO2 such as relatively high cost, low energy density, and toxicity have impeded its widespread application in the field of EVs and HEVs. Thus, the development of cathode materials with high energy/power density, low cost, and environmental friendliness is of great importance.5In recent years, the high-voltage Li-rich and Mn-based layered oxide cathode materials with the chemical composition of xLi2MnO3·(1 – x)LiMO2 (M = Ni, Co, Mn) have attracted an increasing attention owing to the high capacity (>250 mA h g−1) when charged above 4.5 V, and high average operating potential.6–10 Nevertheless, the Li-rich cathode materials suffer from a few of serious problems, which hindered them in the commercial application, such as the high irreversible capacity due to the oxidation of O2− accompanying the extraction of Li+ at high working voltage (>4.5 V) during first cycle, the rapid voltage fading during subsequent cycles, and the inferior rate capability.11–15 These obstacles have been overcome to some extent through some effective methods in the reported results. Reducing particle sizes to the nanoscale level has been considered as one of the most effective ways to improve the rate capability of Li-rich cathode materials due to the shortening of the lithium-ion diffusion length.16–18 Moreover, the electrochemical performance of active material in LIB cathode is closely related to the morphology and particle size.19 Controlling of the morphology via the synthesizing strategies has made great progress, such as solid-state reactions,20,21 co-precipitation,22,23 microwave heating,24 hydrothermal process,25–27 molten salt process,28,29 sol–gel preparation,30,31 and so on. Among them, the sol–gel method has been applied widely in the fabrication process of Li-rich materials with higher purity and smaller particle size. The process is based on the reaction between inorganic reagents (generally acetates) and fuels (e.g., urea, resorcinol–formaldehyde (RF), citric acid), which is an effective technique to synthesize electrode materials with nanoscale particle size.31–33 The formation of organic carbon-gel are used to aid for metallic ions to achieve the homogeneous state during chemical reaction of RF. Gao and coworkers have successfully synthesized LiCoO2 cathode material via a facile RF sol–gel method to improve the rate performance, which exhibited a discharge capacity of 97.4 mA h g−1 at 2C.32 Chen et al. prepared LiNi1/3Co1/3Mn1/3O2 by a carbon gel–combustion process using RF gel. As cathode material, it showed excellent rate capability and cycling performance, i.e., the average discharge capacity of 169.1 and 90.5 mA h g−1 at 0.5 and 50C, respectively.33
In the study, the Li1.2Ni0.2Mn0.6O2 material was synthesized via a facile RF organic carbon gel approach, which is an effective technique to prepare electrode materials with the molecular-level. The initial in situ polymerization of resorcinol, formaldehyde, and metal acetate solution helps to achieve the uniformly mixing of raw materials. After calcination, the Li-rich oxide with well-formed layered structure was obtained, which exhibits much better electrochemical properties.
2. Experimental
2.1 Materials preparation
All the raw materials used were analytical purity grade and used as received without any further treatment. The synthesis process of Li1.2Ni0.2Mn0.6O2 samples through a facile solvothermal method is illustrated in Fig. 1. To start with, the starting materials of resorcinol (3.75 mL), formaldehyde (2.7778 g), lithium acetate (2.5968 g), manganese(II) acetate (2.9707 g), and nickel(II) acetate (1.0157 g) with a stoichiometric amount were dissolved in 40 mL absolute ethanol under magnetic stirring for 1 h at room temperature. Then the solution was transferred and sealed in a 100 mL Teflon-lined stainless steel autoclave with a volume filling ratio of 80%, which was kept at 85 °C for 72 h. After that, the solution was naturally cooled down to room temperature and then RF organic carbon-gel was obtained. Subsequently, RF organic carbon-gel was dried completely at 80 °C for 24 h and ground to be powder by using an agate mortar and pestle. Finally, the dried-gels was first calcined at 500 °C for 5 h and then at 850 °C for another 12 h (ref. 30) in alumina crucibles in air atmosphere before it is naturally cooled down to room temperature in the furnace.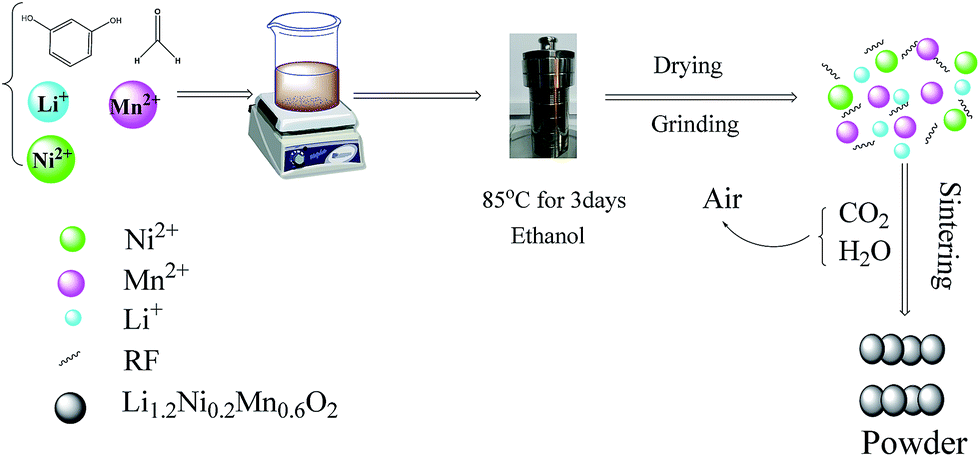 | ||
| Fig. 1 A schematic of RF organic carbon gel-assisted the formation process of Li1.2Mn0.6Ni0.2O2 sample. | ||
2.2 Materials characterization
The crystal structure of the as-prepared powder was characterized through powder X-ray diffraction (XRD, Bruker, D8 Advance, Germany) with Cu Kα radiation operated at 40 kV and 40 mA at a scanning rate of 1° min−1 within 2 theta range of 10–80°. The morphology of materials was observed by field-emission scanning electron microscopy (FESEM, Quanta 200, FEI, USA) and transmission electron microscopy (TEM, Tecnai G2 F20 S-TWIN, Japan). X-ray photoelectron spectroscopy (XPS, Thermo Scientific Escalab 250Xi, USA) was performed to analyze the chemical valence states of Mn and Ni elements. The specific surface area of the samples was determined via Brunauer–Emmett–Teller (BET) procedure from nitrogen sorption isotherms at 77 K using a Quantachrome Autosorb-iQ2 gas sorptometer. The tap density was measured on a Quantachrome Autotap analyzer.2.3 Electrochemical measurements
A two-electrode cell was used in the electrochemical characterization by galvanostatic charge/discharge cycling. The working electrode was fabricated by a slurry coating procedure. The slurry was prepared by mixing 80 wt% active materials, 10 wt% Super P conductive carbon black, 10 wt% polyvinylidene fluoride (PVDF) in N-methyl-2-pyrrolidone (NMP). The electrode slurry was uniformly dispersed and spread on treated aluminum foil as current collector. After drying at 120 °C for 12 h, the aluminum foil with the active materials was pressed under a pressure of 20 MPa. The single-sided cathode has a thickness of 0.033 mm with 3.8–4 mg active material loaded in each electrode. A metallic lithium foil served as the anode. 1 M LiPF6 in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was used as the electrolyte and the Celgard 2400 was the separator. Finally, the cells with CR2025 coin-type half-cells were assembled in an argon-filled glove box with H2O and O2 concentrations below 1 ppm.
:1 was used as the electrolyte and the Celgard 2400 was the separator. Finally, the cells with CR2025 coin-type half-cells were assembled in an argon-filled glove box with H2O and O2 concentrations below 1 ppm.
The charge–discharge performance of the cells was conducted using CT2001A battery testing system (LAND, Wuhan, China) between 2.0 and 4.8 V (vs. Li/Li+) at current densities of 20–400 mA g−1 (1C = 200 mA g−1) at room temperature.
3. Results and discussion
The crystal phase structure of the Li1.2Mn0.6Ni0.2O2 sample was examined by XRD and shown in Fig. 2. The vast majority of the peaks can be well indexed to a hexgonal NaFeO2 layered structure with space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (a = 2.86280(6) Å and c = 14.2608(8) Å) using the TOPAS software with Rwp = 7.242%, except the weak diffraction peaks between 20° and 25°. Two weak peaks at 20.9° and 21.7° can be indexed by (020) and (110) plane diffraction of the monoclinic Li2MnO3 (Li[Li1/3Mn2/3]O2) structure corresponding to space group C2/m. These two weak peaks are caused by the arrangement of LiMn6 cation, always occurring in the transition metal layers of Li2MnO3 nano-domains, which is known to the characteristic peaks of Li-rich phase.5,34–37 No diffraction peaks of any other impurity phases are detectable in the whole XRD patterns, indicating a high purity of the as-prepared materials. In addition, the splitting of peaks corresponding to (006)/(012) and (018)/(110) planes are observed clearly, which are the signatures for the well layered hexagonal structure. The ratio of I(003)/I(104) was used to indicate the cation mixing of the layered structure.38,39 In this work, the ratio of I(003)/I(104) for the Li1.2Mn0.6Ni0.2O2 sample is 1.44 and much more than 1.2, indicating a quite low level of Li+/Ni2+ disorder. Similarly, a value of R-factor (R = [I(012) + I(006)]/I(101)) indicates the order degree of the hexagonal lattice.40,41 The R factor is 0.2997 in our case, suggesting better hexagonal ordering for the as-prepared material. This will then benefit the reversible insertion/extraction of lithium ion through the two-dimension channel built by MO6 and enhance the electrochemical performance.5,39
m (a = 2.86280(6) Å and c = 14.2608(8) Å) using the TOPAS software with Rwp = 7.242%, except the weak diffraction peaks between 20° and 25°. Two weak peaks at 20.9° and 21.7° can be indexed by (020) and (110) plane diffraction of the monoclinic Li2MnO3 (Li[Li1/3Mn2/3]O2) structure corresponding to space group C2/m. These two weak peaks are caused by the arrangement of LiMn6 cation, always occurring in the transition metal layers of Li2MnO3 nano-domains, which is known to the characteristic peaks of Li-rich phase.5,34–37 No diffraction peaks of any other impurity phases are detectable in the whole XRD patterns, indicating a high purity of the as-prepared materials. In addition, the splitting of peaks corresponding to (006)/(012) and (018)/(110) planes are observed clearly, which are the signatures for the well layered hexagonal structure. The ratio of I(003)/I(104) was used to indicate the cation mixing of the layered structure.38,39 In this work, the ratio of I(003)/I(104) for the Li1.2Mn0.6Ni0.2O2 sample is 1.44 and much more than 1.2, indicating a quite low level of Li+/Ni2+ disorder. Similarly, a value of R-factor (R = [I(012) + I(006)]/I(101)) indicates the order degree of the hexagonal lattice.40,41 The R factor is 0.2997 in our case, suggesting better hexagonal ordering for the as-prepared material. This will then benefit the reversible insertion/extraction of lithium ion through the two-dimension channel built by MO6 and enhance the electrochemical performance.5,39
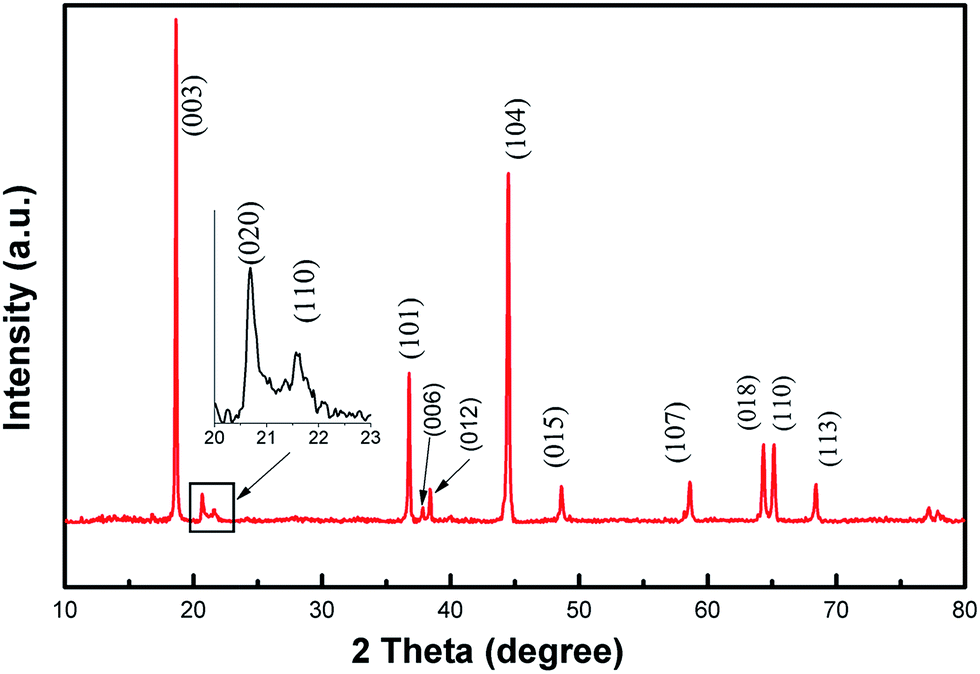 | ||
| Fig. 2 XRD pattern of Li1.2Mn0.6Ni0.2O2 sample. | ||
The morphological structure of the as-prepared Li1.2Mn0.6Ni0.2O2 sample is observed by SEM and TEM. From Fig. 3(a and b), the size of the primary particles is in the range of 50–200 nanometers, which aggregate with each other to form a porous network structure. Furthermore, Fig. 3(c) shows that these nano-particles are in an irregular polyhedral shape with smooth surface and densely stacked in a plane to form a stable network flake structure. The tap density of this sample is determined to be 1.46 g cm−3, which is lower than spherical Li1.2Ni0.2Mn0.6O2 material due to the existence of some tiny cavities between the nanoparticles.42 Such a structure could increase the specific surface area to be 6.9 m2 g−1 compared to the spherical counterpart,42 which contributes significantly to shortening the diffusion path of lithium ions and further raising the high rate capacity. Moreover, there are some tiny cavities observed between these nanoparticles in the densely stacked planes, resulting from the gas release during the decomposition of RF organic carbon-gel and ethanoates. These cavities could accelerate the electrolyte penetration, which is also contributed to the improvement of the cycling performance of the material.
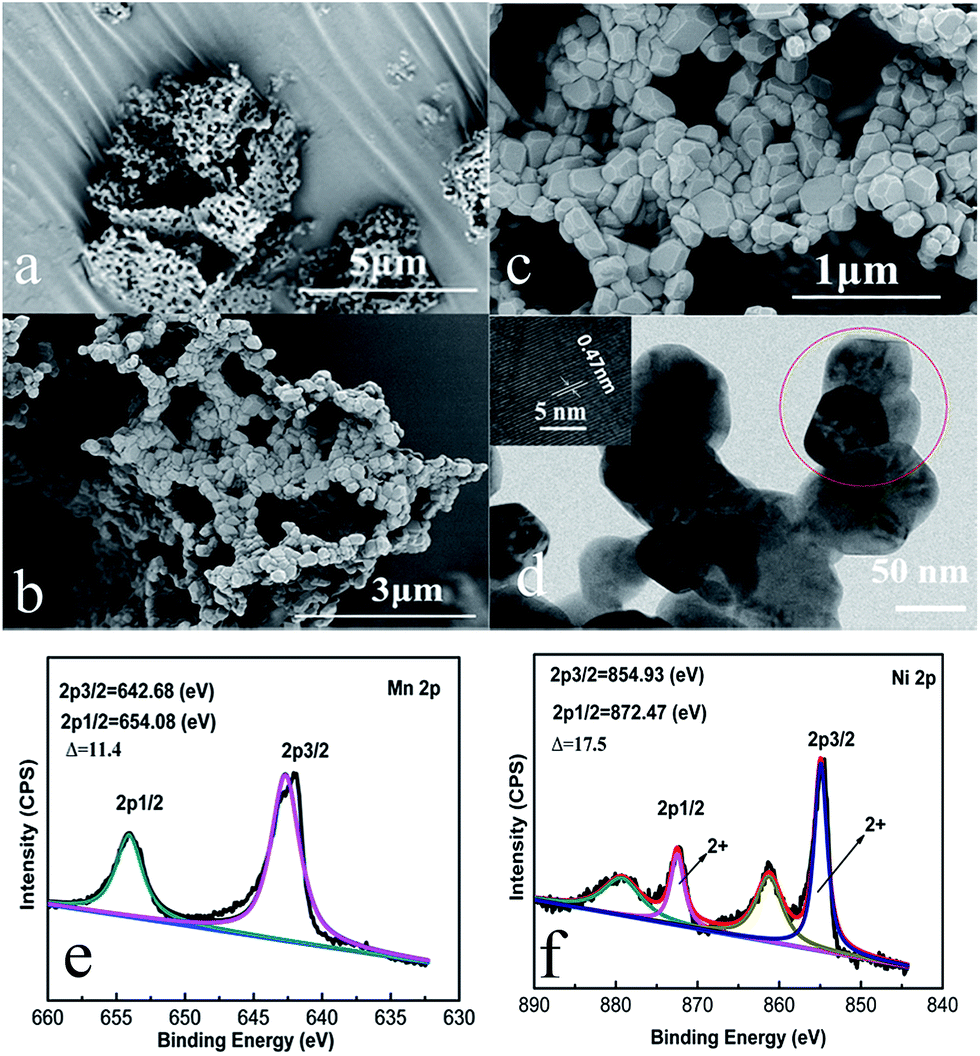 | ||
| Fig. 3 (a–c) SEM and (d) TEM images of Li1.2Mn0.6Ni0.2O2 sample. The inset in (d) shows HRTEM image taken from the red circle. (e and f) XPS spectra of Mn 2p and Ni 2p of Li1.2Mn0.6Ni0.2O2 sample. | ||
The TEM image shown in Fig. 3(d) also indicates that the particles observed are in an irregular polyhedral shape with a particle size of about 50–200 nm. Again, this is due to the gas evolution during the synthesis process, which inhibits the further growth of the Li1.2Mn0.6Ni0.2O2 particles during calcination. The HRTEM image of an individual nanoparticle is displayed in the inset of Fig. 3(d). Distinct lattice fringe is clearly observed and the interlayer spacing is determined to be 0.47 nm, which can be ascribed to the (003) direction of a hexgonal layered structure, indicating the formation of high crystalline oxides.
To obtain more information about the chemical states of the as-prepared samples, X-ray photoelectron spectroscopy (XPS) measurement was employed. XPS spectra for Mn 2p and Ni 2p of samples are shown in Fig. 3(e and f). The Mn 2p spectra exhibits two major peaks at 642.68 and 654.08 eV, and the spin-energy separation is about 11.4 eV. Four signals are observed in the Ni 2p XPS spectra, in which two main sharp peaks of Ni 2p3/2 and Ni 2p1/2 are located at 854.93 and 872.47 eV with a spin-energy separation of about 17.5 eV, strongly suggesting that the oxidation states of Ni and Mn are +2 and +4, respectively.42,43
Fig. 4(a) shows the initial charge–discharge curves of the Li1.2Mn0.6Ni0.2O2 sample at various rates at room temperature. The initial charge and discharge capacity of 357.6 and 273.2 mA h g−1, respectively, with a columbic efficiency of 76.4% at first cycle, better than those in previous reports.44–47 Moreover, there are two distinct regions for the initial charge process: the sloping region below 4.5 V and the flat region at around 4.5 V.
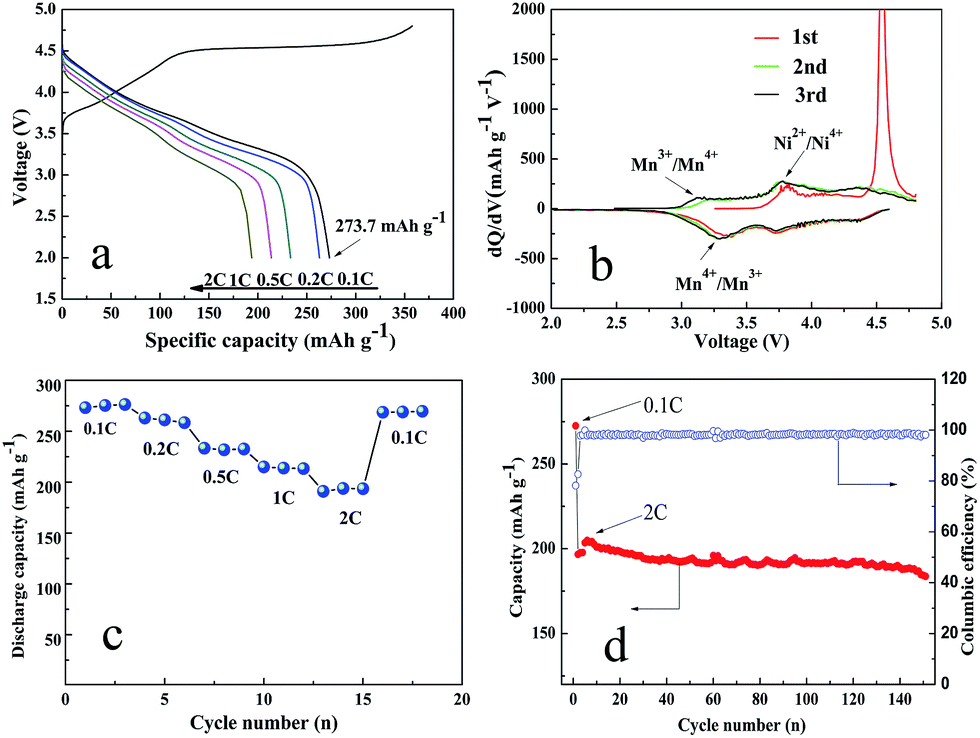 | ||
| Fig. 4 (a) Initial charge–discharge curves of the initial charge–discharge curves at different rates. (b) dQ/dV curves at the first three cycles at 0.1C. (c) Rate capability and (d) cycling performance at 2C of Li1.2Ni0.2Mn0.6O2 sample. Mass of the active material loaded in one electrode: 3.8–4 mg. | ||
The sloping region is attributed to the de-intercalation of Li+ from the structure with space group Rm and the accompanying oxidation of Ni from Ni2+ to Ni4+, which is corresponding to the first oxidation peak at 3.8–3.9 V in Fig. 4(b). The flat region at around 4.5 V could be attributed to the activation of the Li2MnO3 component, accompanying with the oxidation of O2− to facilitate the extraction of Li+ and the following structural rearrangement. This process appears only in the initial cycle, corresponding to the sharp oxidation peak at about 4.6 V in Fig. 4(b). When the discharge process is proceeded, there have two reduction peaks shown in Fig. 4(b). The first reduction peak at about 3.25 V is attributed to the reduction of Mn4+/Mn3+ and the other one is suggested to be the reduction reaction of Ni4+/Ni2+.42
The rate capabilities of the Li1.2Mn0.6Ni0.2O2 sample are shown in Fig. 4(c). The charge–discharge rate is increased successively from 0.1 to 2C, and then decreased back to 0.1C. The discharge capacities of the Li1.2Mn0.6Ni0.2O2 electrodes are 273.2, 263.0, 233.4, 215.0 and 190.9 mA h g−1 at 0.1, 0.2, 0.5, 1 and 2C discharge rates. Furthermore, the high specific capacity could be reversibly recovered 98.7% of the initial discharge capacity (269.6 mA h g−1) after the rate is decreased back to 0.1C, indicating a superior rate performance for Li1.2Mn0.6Ni0.2O2 cathode material. The excellent rate capability can be related to the densely stacked structure. The interior cavities not only accommodates volume change during cycles but also serves as a reservoir of electrolyte and provides more contact area between the electrode and electrolyte. The special structure of the samples comprised of nanoparticles significantly contributes to the remarkable enhancement of electrolyte penetration, charge transfer and ion diffusion, eventually giving rise to the fast lithiation/delithiation reaction kinetics.
In addition, to further evaluate the electrochemical performance of the as-prepared Li1.2Mn0.6Ni0.2O2 sample, the cell is charged/discharged at 0.1C with a reduced polarization for cell activation in the first cycle and then charged/discharged at 2C in the following cycles. Fig. 4(d) exhibits a relatively high initial discharge capacity of 196.7 mA h g−1. The discharge capacity is slightly increased to be 204.9 mA h g−1 in the first three cycles due to the activation of Li1.2Mn0.6Ni0.2O2 cathode material. Surprisingly, a much higher discharge capacity of 183.7 mA h g−1 can be achieved at the completion of 150 cycles with the capacity retention of 93.4%. Table 1 summaries the electrochemical properties of Li1.2Mn0.6Ni0.2O2 sample in comparison with previously reported results. The improved electrochemical properties in our case are superior to or on par with those of the reported Li1.2Mn0.6Ni0.2O2 samples prepared through complex process such as solution combustion synthesis and an ice templating combined coprecipitation method. Again, such an excellent electrochemical performance is mainly due to the fact that the stable network flake structure with the nanosized and densely stacked structure can greatly facilitate the diffusion length for Li+ and electrolyte penetration.
| Preparation method | Initial discharge capacity | Capacity retention | Ref. |
|---|---|---|---|
| Solution method using carbon felt as a carrier | 288 mA h g−1 at 0.1C | 96.4% (246.8 mA h g−1) at 0.1C after 40 cycles | 39 |
| Hydrothermal reaction | 298.5 mA h g−1 at 0.1C | 76.3% at 2C after 150 cycles | 42 |
| 185.8 mA h g−1 at 2C | |||
| Solution combustion synthesis | 252.3 mA h g−1 at 0.1C | 86.7% at 1C after 40 cycles | 48 |
| 152.4 mA h g−1 at 2C | |||
| Sol–gel method | 282.0 mA h g−1 at 0.125C | 138 mA h g−1 at 2.5C after 100 cycles | 49 |
| Ice templating combined coprecipitation strategy | 280.1 mA h g−1 at 0.1C | 91.1% (188.7 mA h g−1) at 2C after 50 cycles | 50 |
| 207.1 mA h g−1 at 2C | |||
| Amorphous complex method | 221.0 mA h g−1 at 0.1C | 81.0% (180.0 mA h g−1) at 0.1C after 50 cycles | 51 |
| Inverse microemulsion route | 248.0 mA h g−1 at 0.125C | 81.0% (210.0 mA h g−1) at 0.125C after 30 cycles | 52 |
| Organic carbon gel assisted-synthesis | 273.3 mA h g−1 at 0.1C | 93.4% (183.7 mA h g−1) at 2C after 150 cycles | This work |
| 196.7 mA h g−1 at 2C |
4. Conclusions
In summary, the Li1.2Ni0.2Mn0.6O2 sample with a high rate performance as cathode material of LIBs was successfully prepared through a facile RF carbon gel-assisted synthesis route in this work. The RF carbon gel is beneficial to the formation of molecular-level dispersion of metal ions, and then results in the well-crystallized Li1.2Ni0.2Mn0.6O2 nanocrystals with high purity. An excellent electrochemical performance of Li1.2Ni0.2Mn0.6O2 is achieved due to the stable network flake structure. The sample can deliver an initial discharge capacity of 273.2 mA h g−1 at a current density of 20 mA g−1 between 2.0 and 4.8 V. Moreover, a high discharge capacity of 196.7 mA h g−1 can be achieved at 2C with a capacity retention of 93.4% after 150 cycles. The Li1.2Ni0.2Mn0.6O2 cathode material prepared by a facile RF organic carbon gel method shows a promising practical potential in high energy density and long cycle life lithium-ion batteries.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (51361006, 51401059, 51461010, 51361005, 51371060, U1501242, and 51461011), the Guangxi Natural Science Foundation (2014GXNSFAA118043 and 2014GXNSFAA118333).References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- A. M. A. Hashem, A. E. Abdel-Ghany, A. E. Eid, J. Trottier, K. Zaghib, A. Mauger and C. M. Julien, J. Power Sources, 2011, 196, 8632–8637 CrossRef CAS.
- Y. D. Zhang, Y. Li, X. Q. Niu, D. H. Wang, D. Zhou, X. L. Wang, C. D. Gu and J. P. Tu, J. Mater. Chem. A, 2015, 3, 14291–14297 CAS.
- X. Miao, Y. Yan, C. Wang, L. Cui, J. Fang and G. Yang, J. Power Sources, 2014, 247, 219–227 CrossRef CAS.
- X. M. He, J. X. Wang, L. Wang and J. J. Li, Materials, 2016, 9, 661 CrossRef.
- F. Fu, Q. Wang, Y. P. Deng, C. H. Shen, X. X. Peng, L. Huang and S. G. Sun, J. Mater. Chem. A, 2015, 3, 5197–5203 CAS.
- C. H. Yang, X. S. Zhang, J. J. Huang, P. Ao and G. Zhang, Electrochim. Acta, 2016, 196, 261–269 CrossRef CAS.
- G. Xu, J. Li, Q. Xue, X. Ren, G. Yan, X. Wang and F. Kang, J. Power Sources, 2014, 248, 894–899 CrossRef CAS.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- Y. Wu and A. Manthiram, Electrochem. Solid-State Lett., 2006, 9, A221–A224 CrossRef CAS.
- J. Liu and A. Manthiram, J. Mater. Chem., 2010, 20, 3961–3967 RSC.
- J. Lu, Q. Peng, W. Y. Wang, C. Y. Nan, L. H. Li and Y. D. Li, J. Am. Chem. Soc., 2013, 135, 1649–1652 CrossRef CAS PubMed.
- J. L. Liu, L. Chen, M. Y. Hou, F. Wang, R. C. Che and Y. Y. Xia, J. Mater. Chem., 2012, 22, 25380–25387 RSC.
- Y. Jiang, Z. Yang, W. Luo, X. Hu and Y. Huang, Phys. Chem. Chem. Phys., 2013, 15, 2954–2960 RSC.
- H. H. Wang, X. W. Li, Q. Zhou, H. Ming, J. Adkins, L. L. Jin, Z. Y. Jia, Y. Fu and J. W. Zheng, J. Alloys Compd., 2014, 604, 217–225 CrossRef CAS.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nature, 3, 31–35 CAS.
- W. C. West, J. Soler and B. V. Ratnakumar, J. Power Sources, 2012, 204, 200–204 CrossRef CAS.
- Z. Zhong, N. Ye, H. Wang and Z. Ma, Chem. Eng. J., 2011, 175, 579–584 CrossRef CAS.
- S. M. Ma, X. H. Hou, Z. R. Lin, Y. L. Huang, Y. M. Gao, S. J. Hu and J. D. Shen, J. Solid State Electrochem., 2016, 20, 95–103 CrossRef CAS.
- S. J. Shi, Z. R. Lou, T. F. Xia, X. L. Wang, C. D. Gu and J. P. Tu, J. Power Sources, 2014, 257, 198–204 CrossRef CAS.
- H. D. Liu, J. Wang, X. F. Zhang, D. Zhou, X. Qi, B. Qiu and J. H. Fang, ACS Appl. Mater. Interfaces, 2016, 8, 4661–4675 CAS.
- R. Kloepsch, G. Schumacher, Z. D. Liu, J. Li, X. H. Hou, Y. L. Huang, S. M. Ma, X. L. Zou, S. J. Hu and Y. P. Wu, Mater. Res. Bull., 2015, 63, 256–264 CrossRef.
- F. Fu, Y. P. Deng, C. H. Shen, G. L. Xu, X. X. Peng, Q. Wang, Y. F. Xu, J. C. Fang, L. Huang and S. G. Sun, Electrochem. Commun., 2014, 44, 54–58 CrossRef CAS.
- X. Wei, S. C. Zhang, Z. J. Du, P. H. Yang, J. Wang and Y. B. Ren, Electrochim. Acta, 2013, 107, 549–554 CrossRef CAS.
- T. Zhang, J. T. Li, J. Liu, Y. P. Deng, Z. G. Wu, Z. W. Yin, J. H. Wu, L. Huang and S. G. Sun, ChemElectroChem, 2016, 3, 98–104 CrossRef CAS.
- F. Li, Y. Y. Sun, Z. H. Yao, J. S. Cao, Y. L. Wang and S. H. Ye, Electrochim. Acta, 2015, 182, 723–732 CrossRef CAS.
- Q. L. Xie, C. H. Zhao, Z. B. Hu, Y. L. Zhou, F. He and K. Y. Liu, J. Sol-Gel Sci. Technol., 2016, 77, 699–707 CrossRef CAS.
- F. Wu, H. Lu, Y. Su, N. Li, L. Bao and S. Chen, J. Appl. Electrochem., 2010, 40, 783–789 CrossRef CAS.
- S. Gao, W. Wei, M. X. Ma, J. J. Qi, J. Yang, S. Q. Chu, J. Zhang and L. Guo, RSC Adv., 2015, 5, 51483–51488 RSC.
- J. Chen, N. Zhao, G. D. Li, F. F. Guo, J. W. Zhao, Y. G. Zhao, T. K. Jia, F. Fu and J. L. Li, Mater. Res. Bull., 2016, 73, 192–196 CrossRef CAS.
- C. H. Shen, L. Huang, Z. Lin, S. Y. Shen, Q. Wang, H. Su, F. Fu and X. M. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 13271–13279 CAS.
- C. H. Lei, J. Bareno, J. G. Wen, I. Petrov, S. H. Kang and D. P. Abraham, J. Power Sources, 2008, 178, 422–433 CrossRef CAS.
- S. J. Shi, J. P. Tu, Y. Y. Tang, Y. X. Yu, Y. Q. Zhang, X. L. Wang and C. D. Gu, J. Power Sources, 2013, 228, 14–23 CrossRef CAS.
- X. H. Hou, X. L. Zou, Y. L. Huang, S. J. Hu, Q. Ru and Y. M. Gao, RSC Adv., 2014, 4, 29534–29541 RSC.
- Z. L. Liu, A. S. Yu and J. Y. Lee, J. Power Sources, 1999, 81, 416–419 CrossRef.
- J. Lin, D. B. Mu, Y. Jin, B. R. Wu, Y. F. Ma and F. Wu, J. Power Sources, 2013, 230, 76–80 CrossRef CAS.
- S. J. Shi, T. Wang, M. Cao, J. W. Wang, M. X Zhao and G. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 11476–11487 Search PubMed.
- S. J. Shi, J. P. Tu, Y. Y. Tang, Y. Q. Zhang, X. L. Wang and C. D. Gu, J. Power Sources, 2013, 240, 140–148 CrossRef CAS.
- Y. Li, Y. Bai, X. X. Bi, J. Qian, L. Ma, J. Tian, C. Wu, F. Wu, J. Lu and K. Amine, ChemSusChem, 2016, 9, 728–735 CrossRef CAS PubMed.
- D. T. Ma, Y. L. Li, P. X. Zhang, A. J. Cooper, A. M. Abdelkader, X. Z. Ren and L. B. Deng, J. Power Sources, 2016, 311, 35–41 CrossRef CAS.
- Y. J. Kang, J. H. Kim, S. W. Lee and Y. K. Sun, Electrochim. Acta, 2005, 50, 4784–4791 CrossRef CAS.
- D. Wang, I. Belharouak, G. Zhou and K. Amine, Adv. Funct. Mater., 2013, 23, 1070–1075 CrossRef CAS.
- X. F. Zhang, X. B. Meng, J. W. Elam and I. Belharouak, Solid State Ionics, 2014, 268, 231–235 CrossRef CAS.
- Y. Zang, C. X. Ding, X. C. Wang, Z. Y. Wen and C. H. Chen, Electrochim. Acta, 2015, 168, 234–239 CrossRef CAS.
- Q. T. Zhang, J. T. Mei, X. L. Xie, X. M. Wang and J. Y. Zhang, Mater. Res. Bull., 2015, 70, 397–402 CrossRef CAS.
- W. W. Yan, Y. N. Liu, S. K. Chong and Y. F. Wu, RSC Adv., 2016, 6, 23677–23685 RSC.
- Y. Li, C. Wu, Y. Bai, L. Liu, H. Wang, F. Wu, N. Zhang and Y. F. Zou, ACS Appl. Mater. Interfaces, 2016, 8, 18832–18840 CAS.
- X. He, J. Wang, L. Wang and J. Li, Materials, 2016, 9, 661 CrossRef.
- S. Duraisamy, T. R. Penki and M. Nookala, New J. Chem., 2016, 40, 1312–1322 RSC.
| This journal is © The Royal Society of Chemistry 2017 |