
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An environmentally benign approach to achieving vectorial alignment and high microporosity in bacterial cellulose/chitosan scaffolds†
Guohui Lia,
Avinav G. Nandgaonkarab,
Youssef Habibic,
Wendy E. Kraused,
Qufu Wei*a and
Lucian A. Lucia *abd
*abd
aKey Laboratory of Eco-Textiles, Ministry of Education, Jiangnan University, 1800 Lihu Avenue, Jiangsu Province, Wuxi 214122, P. R. China. E-mail: lalucia@ncsu.edu; qfwei@jiangnan.edu.cn
bKey Laboratory of Pulp and Paper Science & Technology, College of Light Chemical & Environmental Engineering, Qilu University of Technology, Jinan, 250353, P. R. China
cDepartment of Materials Research and Technology (MRT), Luxembourg Institute of Science and Technology (LIST), 5 Avenue des Hauts-Fourneaux, L-4362 Esch-sur-Alzette, Luxembourg
dFiber and Polymer Science Program, North Carolina State University, 2401 Research Dr, Campus Box 8301, Raleigh, NC 27695-8301, USA
First published on 28th February 2017
Abstract
Bacterial cellulose (BC) nanofibers secreted by Komagataeibacter xylinus 10245 were applied alone or in combination with chitosan to prepare highly aligned and porous scaffolds through a combined liquid nitrogen-initiated ice “templating” and freeze-drying process. Their morphology and physical properties were controlled by adjusting the concentration of chitosan over a range of 1, 1.5, and 2% (wt%) and analyzed by Scanning Electron Microscopy (SEM), Brunauer–Emmett–Teller (BET), and X-ray diffraction methods. The SEM images confirmed a distribution of fibrils vectorially aligned in the freezing axis direction, while chitosan contributed to the development of a dense network, superior mechanical properties, and biomedical relevance of the final scaffolds. It was found that as the chitosan concentration increased, the crystallinity index decreased from 89% to 79% likely because of strong intermolecular bonding. However, the scaffolds containing chitosan demonstrated excellent shape recovery and structural stability after compressive tests and may act as excellent scaffolds for potential cartilage tissue engineering applications.
Introduction
Although cellulosic materials are ubiquitous in nature, in general, isolation of pure cellulose from terrestrial or aquatic biospheres requires expenditure of significant energy to remove contaminants such as lignin, heteropolysaccharides, and related biomolecules. Bacterial cellulose (BC), however, is a cellulosic material with the same chemical structure as plant- or animal-based cellulose, but it does not contain lignin in contrast to plant cellulose and hence is much purer. It is known that various genera of bacteria such as Komagataeibacter within the appropriate environment can produce highly porous and fibrous BC networks. The biomaterial is characterized by high water sorption, gel-forming ability, biocompatibility, high modulus of elasticity, and extremely high purity. It has found applications in pulp & paper products, audio components (e.g., speaker diaphragms), conductive materials, and soft tissue engineering.1–3 It possesses unique morphological and mechanical properties as evidenced by nanoscale fibrillar structure and high tensile strength, respectively; most importantly, it displays biocompatibility from analogous chemical motifs to the extracellular matrix (ECM) component of naturally occurring tissues, i.e., collagen, that make it a desirable building block for soft tissue engineering.3–8Chitosan, like cellulose, is an abundant and ubiquitous polysaccharide widely known for biomedical applications. It is a linear polysaccharide composed of glucosamine and N-acetyl glucosamine residues linked in a β-(1–4) regular structural pattern. It can acquire a cationic nature because of the –NH2 groups on the chitosan backbone to modulate electrostatic interactions with anionic glycosaminoglycans (GAGs), proteoglycans, and sundry negatively charged molecules. The electrostatic property is of great importance because most growth factors are attached to GAG to help chitosan-based scaffolds to maintain the growth factors secreted by cells.9 Chitosan has secured significant interest for as a tissue engineered scaffold because in addition to its charge interactions with GAGs, it aids in wound healing, drug delivery, and displays antibacterial activity.
Engineered polymer-based tissue engineered scaffolds require several properties for successful application. Among them, the scaffold material must provide a platform for cell seeding, growth, and proliferation. In fact, human cells require a complex selection of pores, ridges, and fibrous materials within the extracellular matrix. Therefore, factors that enhance cell attachment, migration, and nutrient delivery to such cells include fiber size, porosity (pore size & structure), the void volume, and the architecture of the system.10 Scaffold porosity is among the most important parameters for cell migration and ultimately for vascularization. A porous scaffold will enable facile diffusion of nutrients, oxygen, and waste within implanted tissue. Finally, a high specific surface area will facilitate strong cell attachment.10,11 The phenotypic cell expression, however, can be altered if the fiber diameter does not coincide with the pre-requisite size of the cells within the ECM.12 Furthermore, uniform fiber directionality is an essential parameter in engineering scaffolds because it influences not only cell phenotypic expression, attachment, and proliferation, but final scaffold mechanics.13–15
Different techniques have been adopted to mimic natural tissue properties such as pore size and fiber directionality. Among them are gas foaming,16 solvent casting/particulate leaching,17 phase separation,18 fiber mesh generation,19,20 and electrospinning.21 In the gas-foaming technique, the scaffolds have closed cellular structures, whereas in solvent casting/particulate leaching, the morphology of the resulting scaffold cannot be controlled because chemical modifications occur during dissolution and removal of the solvent that alter final material properties.11 Although electrospinning and fiber mesh techniques facilitate fiber orientation, the final scaffolds typically lack both acceptable structural stability and mechanical strength.22
The freeze-drying method has been used to produce porous scaffolds because of its simplicity, low cost, and ease of use. It requires sublimation under specific temperature and pressure profiles over a defined time, but does not require chemical reactions. Thus, properties such as pore structure, size, and networking can be modulated and fine-tuned.11 In addition, the method is considered highly functional because it allows production of porous materials with vectorially-guided morphologies. In fact, aligned scaffold porosity is critical to successful tissue engineering to ensure directional growth of cells.23 Various functional polymer-based scaffolds possessing tailored microstructures and physical properties have been prepared by freeze-drying.24
Freeze-drying is an approach for removing water from BC,8,25–27 but until this report, no work has shown vectorially-guided final morphologies by using liquid nitrogen. In the past, homogenous and well-structured foams of cellulose have been prepared by dissolving cellulose in solvents to develop gels, while vectorial alignment in cellulose nanofibers was obtained by dispersing fibers in water under high-pressure homogenization. However, the latter techniques tend to result in a loss of desirable physical and chemical qualities.28,29
The current study has developed a porous BC scaffold by using a green approach that consists of an environmentally benign liquid nitrogen-initiated ice-templated freezing methodology coupled with freeze-drying. Chitosan was added to the system to impart relevant biological properties; the systems were evaluated in terms of their morphological, chemical, and physical characteristics.
Experimental section
Materials
Komagataeibacter xylinus (ATCC 10245) was purchased from American Type Culture Collection. Yeast extract, bacto-peptone (becton, Dickinson and Company), di-sodium phosphate, and citric acid were used for preparing the appropriate culture media. All the reagents were purchased from Sigma unless otherwise stated. Chitosan was obtained from Vanson Company. Methanol (99%) was used to wash the chitosan samples. Hydrochloric acid solution, 0.1 N (N/10) (Certified) and sodium hydroxide solution 0.1 N (N/10) (Certified), both obtained from Fisher Chemical, were used in acid–base titrations to determine the degree of deacetylation (% DD) found to be 90%. Sodium acetate anhydrous (Fused Crystals/Certified ACS) was obtained from Fisher Chemical. All reagents and polymers were used as received unless stated otherwise.Methods
The BC film after the cultivation period was sterilized with washing with a 1 N NaOH solution at 80 °C for 90 min to dissolve remaining bacteria. The samples were rinsed thoroughly with deionized water. The purified BC was dried to constant weight at 80 °C and then weighed to ascertain the production of BC. Finally, the purified wet cellulose film was stored in distilled water at 4 °C for future experiments.
The degree of deacetylation was measured by acid–base titration. A detailed explanation is provided elsewhere.31,32 Briefly, a fixed amount of chitosan was placed in a 250 ml beaker and dissolved by adding 10 ml of 0.1 N hydrochloric acid. Next, 90 ml of distilled water was added to the beaker in addition to a few drops of phenolphthalein as an indicator. The conductivity meter (Orion conductivity cell, model 013030) was immersed into the solution while the solution was kept at constant stirring. The solution was then titrated with 0.1 N NaOH with by a 10 ml burette. The readings were noted after adding the NaOH solution and plotted as volume of NaOH solution versus conductivity.
The molecular weight of chitosan was determined by viscosity measurements using a Ubbelohde viscometer. A series of dilute solutions with increasing chitosan concentrations were prepared by dissolving chitosan in a solution of 0.2 M CH3COOH/0.1 M CH3COONa. A solution of 10 ml was then placed into a Cannon–Ubbelohde semi-micro viscometer (size 75 J-134) and the instrument was immersed into a constant water bath at 30 °C. It was kept for 20 min to equilibrate before the readings were recorded. The viscosity average molecular weight (Mv) was calculated from the measured intrinsic viscosity [η] and found to be 8.2 × 105 by use of the Mark–Houwink–Sakurada equation, [η] = kMa where k = 1.64 × 10−30 × (% DD) and a = −1.02 × 10−2 × (% DD) + 1.82.32
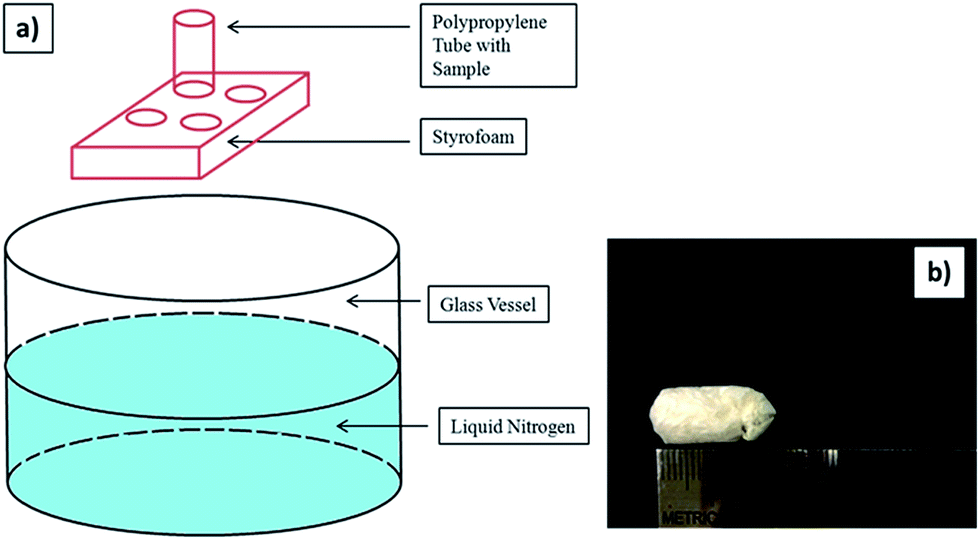 | ||
| Fig. 1 Schematic procedure of preparing porous sample: (a) the sample is placed into a polypropylene tube which can contact with the liquid nitrogen in the glass vessel to induce a bottom-to-top freezing event instantly. (b) A digital photograph of the final ice-templated/freeze-dried sample. | ||
To fabricate a composite scaffold of chitosan, a wet BC film was placed between two filter papers to remove excess water. Then the BC film was immersed in 1% chitosan solution (previously dissolved in a 1% aqueous acetic acid solution) at 50 °C for 24 hours. Afterwards, the BC film was withdrawn from the chitosan solution beaker and excess chitosan solution was removed by dabbing with filter paper. Afterwards, the BC film that was immersed in the chitosan solution was put in polypropylene tube and vectorially aligned frozen in a similar manner to above (vide infra). Thus, BC–Ch-x was prepared where BC is bacterial cellulose, Ch is chitosan, and x represents the wt% of chitosan solution. The BC and BC-Ch-x scaffolds after freeze-drying were removed and stored in silica gel desiccators for further characterization.
Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times original sizes. Those images were used to evaluate fiber diameter and consistency. Revolution software and ImageJ software were used to process the SEM images to determine the surface morphology and the cross-sectional image of BC and composite scaffolds. Fifty readings were measured from two different SEM images and the readings were averaged to determine the mean fiber diameter and fiber distribution. The porosity of the scaffolds was analyzed based on a literature method using ImageJ software. The SEM images were first converted into gray scale images and then the threshold value was adjusted to observe the first layer of the porous structure and measure the number of filled versus unfilled pixels.
000 times original sizes. Those images were used to evaluate fiber diameter and consistency. Revolution software and ImageJ software were used to process the SEM images to determine the surface morphology and the cross-sectional image of BC and composite scaffolds. Fifty readings were measured from two different SEM images and the readings were averaged to determine the mean fiber diameter and fiber distribution. The porosity of the scaffolds was analyzed based on a literature method using ImageJ software. The SEM images were first converted into gray scale images and then the threshold value was adjusted to observe the first layer of the porous structure and measure the number of filled versus unfilled pixels.The percent adsorption was calculated using following equation:
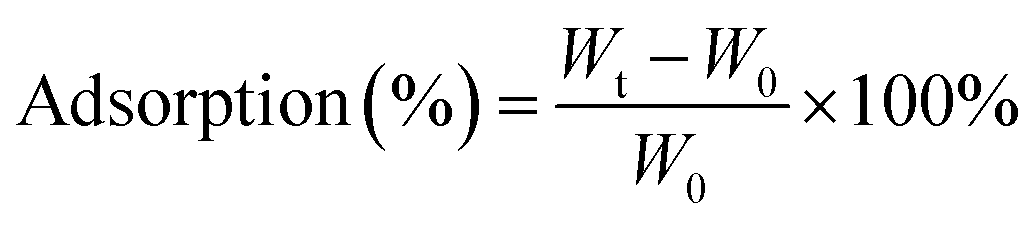 | (1) |
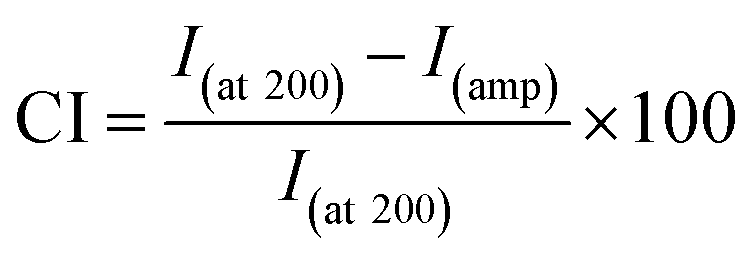 | (2) |
Results and discussion
Preparation and characterization of bacterial cellulose nanofibers
The original random morphology of BC film was aligned by a vectorially aligned freeze-drying method to assert a level of biomimetic control of scaffold architecture for tissue engineering. Because BC hydrogels upon mechanical loading cannot recover their original shape, chitosan was added as a reinforcing polymer within the BC network in addition to providing useful biological properties. SEM was used to observe the BC film after purification and drying. Fig. 2 shows the SEM image of the macroscopic sponge-like material that at the microscopic level displays an interconnected nonwoven nanofibrous network. The average diameter of these nanofibers is 64 ± 15 nm. | ||
| Fig. 2 SEM image of BC produced by Komagataeibacter xylinus. | ||
The freeze-drying process can freeze the water in the cellulose network and dry it (remove solvent) below the glass transition or melting temperature of water (sublimation). The removed frozen solvent consequently leaves behind void spaces that characterize the resultant porous structure.34 Even when various amounts of chitosan were added and freeze-dried, all the morphological analyses by SEM revealed a highly porous structure (Fig. 3).
 | ||
| Fig. 3 SEM photographs of freeze-dried pure BC and BC–Ch composites possessing different levels of chitosan (a, b: pure BC; c, d: BC–Ch-1%; e, f: BC–Ch-1.5%; g, h: BC–Ch-2%). Top row is the cross section and bottom row is the inner wall. (all images are at 5000× magnification.) | ||
Fig. 3a–d provide cross sections of pure BC and BC–chitosan scaffolds. Pure BC (Fig. 3a) did not possess pores that were only circular; rather they were circular and ovaloid within an interconnected network. Pores were created in the cross-sectional area perpendicular to the freezing direction. The channel-like porosity was created in the sample in the cross section parallel to the freezing direction, a phenomenon shown in Fig. 3b. The channel pores are connected with short nanofibrils in which the red arrow indicates the direction of (aligned) porosity in the sample. It should be noted that this alignment was not present in the original bacterial cellulose nanofibrous network (Fig. 2). The freeze-drying method requires that the bottom surface first contacts the liquid nitrogen to induce nanofibers to stretch and display random morphology because the ice crystals nucleate and propagate from underneath. Thin film formation was observed on the outer scaffold surface that was in contact with the inner wall of the polystyrene/glass tube. The film shows nonwoven nanofibrous morphology; however, it does not display any pore formation, a result that has been documented in other reports.9,35
The SEM analyses were done to determine the effect of chitosan concentration on the morphological changes inside the scaffold. Fig. 3c shows the interconnected pores within a three-dimensional network in which the structure of the pure BC nanofibrils was lost due to the incorporation of chitosan. The inner section of BC–Ch-1% demonstrates an alignment of the porosity. The channel like formation can be attributed to a vectorially aligned freezing of the sample as the porosity developed from the bottom to the top of the sample. BC with 1 wt% chitosan (Fig. 3a) was even more porous than the other two samples that had chitosan concentrations of 1.5 and 2 wt% (Fig. 3e and g). They displayed an interconnected porous network within a three-dimensional network similar to the pure BC scaffold. The distance between two channels was largely due to chitosan penetration inside the BC pores to form a film on the individual surfaces. It was found, not unlike what has been previously reported, that the surfaces became more compact when chitosan concentration increased.27 Mean pore diameter and porosity of BC and composite scaffolds are listed in Fig. 4a and b.
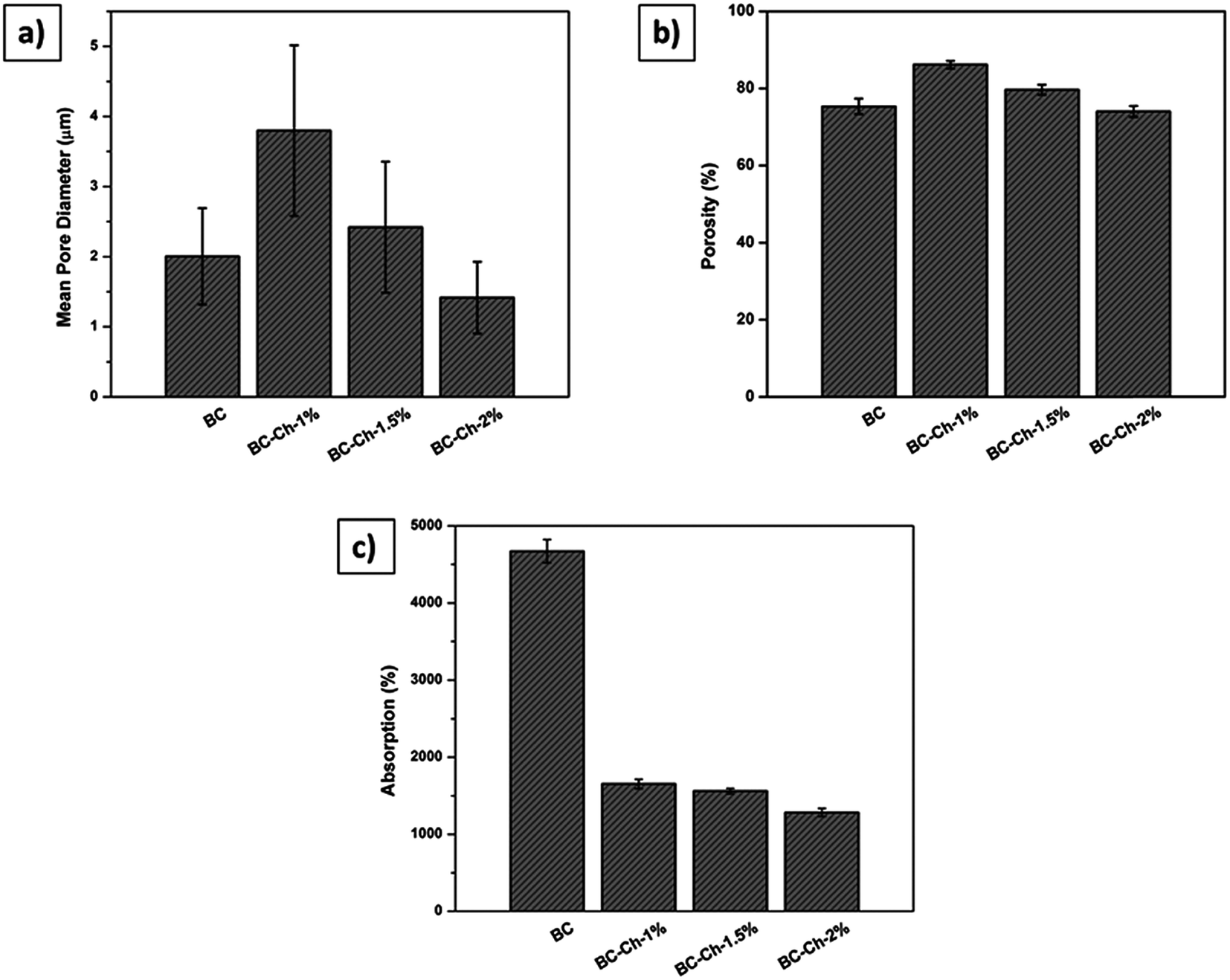 | ||
| Fig. 4 (a) Mean pore diameter, (b) porosity, and (c) absorption properties of pure BC and composite scaffolds possessing different weight percentages of chitosan (1%, 1.5%, and 2%). | ||
As the concentration of chitosan increased to 1.5%, the distance between two channels became shorter because of chitosan aggregation inside the channels (Fig. 3f). The channels displayed a specific degree of alignment due to increased penetration of chitosan clearly witnessed at the 2 wt% loading mark (Fig. 3h) that induced layer formation on the BC film. However, the channels were still interconnected by cellulose nanofibrils.
From the cross-sectional SEM images, mean pore diameters (Fig. 4a), and porosities (Fig. 4b), it appeared that the pores at 1 wt% chitosan were larger compared to pure BC. The mean pore diameter and porosity in the pure scaffold was 2 ± 1 μm and 75 ± 2%, respectively, whereas, for BC–Ch-1%, it was 4 ± 2 μm and 87 ± 1%. The increase in pore diameter and porosity was due to increased concentration as well as the viscosity of the overall sample that facilitated the BC film to rearrange themselves into highly oriented pores and channels. Water acted as a solvent during BC scaffold preparation. Many small size ice crystals tended to form during the freezing process to induce small pores and reduce the porosity in the final sample. The overall porous structure was reduced as the chitosan concentration increased from 1 to 2 wt% as shown in Fig. 3c, e, g and 4a, b. The pore diameter and the porosity of BC–Ch-1.5% was 2.5 ± 1 μm and 80 ± 1%, while for BC–Ch-2%, it was 1.4 ± 1 μm and 74 ± 2%, respectively. This may be because increasing chitosan concentration interfered with ice crystal nucleation during the freezing step. Water is sequestered, while the “spectator” chitosan macromolecules prevented the water molecules from rearranging and forming larger ice crystals during a rapid freezing process. Another reason for the random and reduced pore distribution might be due to the viscosity of the chitosan solution. As the concentration increased, the viscosity of the solution also increased. Thus, during the freezing step, a higher viscosity sample prevented water molecules from rearranging and thus hindered ice crystal nucleation and gave rise to random crystal shapes.35 Therefore, as the concentration of chitosan increased, the distance between two juxtaposing channels decreased as did the pore size. The 2 wt% chitosan sample had a random distribution of channels with relatively small pore size. However, all the scaffolds retained a porous structure. This was observed in a previous effort.35 The current result indicates that adjusting chitosan concentration may control the nano-structure of the BC/chitosan scaffold. The reduced pore size may also be beneficial to retain synthesized matrix molecules.36 The porous structure and the interconnection of fibrils are clearly also critical for cellular attachment and proliferation, thus making it an attractive scaffold for tissue engineering.
Absorption properties
The absorption properties of pure BC and associated composite scaffolds were tested by immersion in PBS at 37 °C for 24 hours. The results showed that pure BC has the highest absorption percentage because of the abundance of hydrophilic functionalities. Additionally, because the scaffold dimensions are fixed before adsorption, the scaffold tended to swell, accommodating as much as water inside the porous network as possible and thereby increasing its volume. During absorption, the hydroxyl groups attracted water because of hydrogen bonding forces. When chitosan (1%, 1.5%, and 2%) was introduced into the scaffold, water sorption reduced as shown in Fig. 4c. The water penetration was limited due to bond formation between chitosan –NH2 groups and the hydroxyl groups of BC; thus, the original porous structure was retained because chitosan limited the scaffold swelling given that it has a tendency to retain its morphology and hence the original porous structures/dimensions. The results were similar to former reports that demonstrated that intermolecular hydrogen bonds affected the interaction between water and polymer chains directly.37,38 Though the absorption properties decreased with inclusion of chitosan, this may have been offset by the favorable mechanical and biological activities of chitosan. Very fortuitously, chitosan may act as a structural stabilizer for optimizing tissue engineering applications.Mechanical properties
Compressive properties were tested in both the dry and wet states. The wet studies were specifically executed as close to in vivo conditions as possible. Table 1 shows the compressive moduli of pure BC and composite scaffolds.| Sample name | Surface area (m2 g−1) | Crystallinity index (%) | Dry compressive modulus (MPa) | Wet compressive modulus (MPa) |
|---|---|---|---|---|
| BC | 201 | 89.45 | 9.51 ± 0.23 | 37.32 ± 1.07 |
| BC–Ch-1% | 29.5 | 83.57 | 8.04 ± 0.47 | 0.58 ± 0.17 |
| BC–Ch-1.5% | 22.06 | 83.94 | 9.13 ± 0.40 | 0.82 ± 0.15 |
| BC–Ch-2% | 19.85 | 79.46 | 4.74 ± 1.00 | 9.64 ± 0.46 |
The mechanical strength of pure BC was highest, 9.51 ± 0.23 MPa, among all the scaffolds in the dry state. This is due to the reduced pore size and porosity of the scaffold. During the initial loading, the fibers had better chances of aligning themselves between the channels of the nanofibrils. This rearrangement might be the reason for the higher modulus of pure BC. The addition of chitosan tends to decrease the compressive modulus, but the values were approximately similar for BC containing 1% and 1.5% chitosan. The addition of chitosan increased the structural integrity and formed networks within the BC nanofibrils, except for the scaffold containing 2% chitosan. This reduction in dry state might be due to the coating of individual BC nanofibrils that upon drying become brittle and thus under normal loading display lower compressive strengths.
The compressive properties of the scaffold have mixed results in the wet state. For pure BC, the modulus increased 3.6× compared to its dry state. This is because the BC has a strong affinity for water that tends to fill the empty pores thereby swelling and increasing the volumetric dimensions of the scaffold and directly affecting the mechanical properties. In addition, the scaffold undergoes many physical and chemical changes during water absorption that are highest for pure BC. The water content helps the nanofibers slide over each other such that upon mechanical loading can rearrange. Though the pure BC possesses high mechanical strength, the scaffold can recover its original shape even after 2 hours at room temperature.39 The compressive moduli for BC–Ch-1% (0.58 ± 0.17) and BC–Ch-1.5% (0.82 ± 0.15) were found to be much lower than in their dry state. Because the BC–Ch-composite display reduced absorption and chitosan shows a tendency to retain its original shape, the scaffold was lubricated between the chitosan and BC nanofibers to reduce internal friction, ultimately reducing the compressive strength.40 The wet compressive modulus of the BC–Ch-2% was found to be higher (9.64 ± 0.46) than in its dry state (4.74 ± 1.00), despite its lower absorption properties. This might be due to the chitosan having a high degree of deacetylation (90%), which means it has more amino groups. Moreover, the chitosan had an entanglement concentration of 1.85 wt% (it tends to form a charged complex in acidic solution) that surpassing causes an increase in intermolecular hydrogen bonding and restriction of individual polymer chains.
In addition, the lower pore size facilitates a viscous chitosan charged complex inside the porous network upon uniform compressive loading, ultimately increasing its compressive strength.31 All the wet scaffolds showed higher compressive moduli at the same order of magnitude as native human cartilage (0.677 ± 0.198).41 All the scaffolds containing chitosan showed 100% recovery after two minutes of removing the load (Fig. 5). This type of scaffold is useful for waste removal during compression and diffusion and exchange of nutrients during the recovery of scaffolds given that natural articular cartilage lacks the necessary blood vessels for such functions. Additionally, previous studies confirmed these potential applications: the porous scaffolds and three-dimensional scaffolds having high compressive strength could be used for meniscus regeneration, culturing primary human fibroblasts, periosteal cells, osteoblasts, and other cellular tissues.42–45
 | ||
| Fig. 5 Photographs of the BC–Ch-1% scaffold (original dimensions: 8 mm diameter × 6 mm height) that show a compressed form (at 0 s) that undergoes shape recovery after a two-minute interval to virtually its original dimensions. | ||
Surface area measurements
From inspection of the SEM images, pure BC scaffold has a more open structure than the BC–chitosan scaffold. The BET technique was used to obtain the surface area to give a total surface area for pure BC of 201 m2 g−1 (Table 1). However, the total surface area drastically reduced with incorporation of chitosan. The total surface areas were 29.5 m2 g−1 for BC–Ch-1%, 22.06 m2 g−1 for BC–Ch-1.5%, and 19.85 m2 g−1 for BC–Ch-2%. Solution facilitates chitosan penetration inside the BC interconnected fibrils to block empty pores. Moreover, a higher content of chitosan decreased pore size as clearly seen from SEM images in Fig. 3. The fibrils having a close arrangement became dense and compact ultimately leading to a lower surface area. Another reason for the reduction in surface area was due to a strong interaction of chitosan molecules with the BC chains to form a dense network of fibrils and ultimately close the empty spaces among the BC nanofibrils.46XRD analysis of prepared scaffolds
Fig. 6b shows the recorded X-ray diffractogram. The XRD of BC films were recorded in parallel with the X-ray beam. Three main peaks were observed in both spectra. The 14.80° was assigned to (1 −1 0), the 16.6° was assigned to (1 1 0), and the 22.93° was assigned to (2 0 0) crystallographic planes. These results confirm the presence of cellulose I in the BC films.26,27,33 The crystallinity index of BC was 89.45%. Thus, the data gleaned from the specific chemical peak position and the crystal structure from the combined results of FTIR and XRD confirm the presence of cellulose I production by Komagataeibacter xylinus.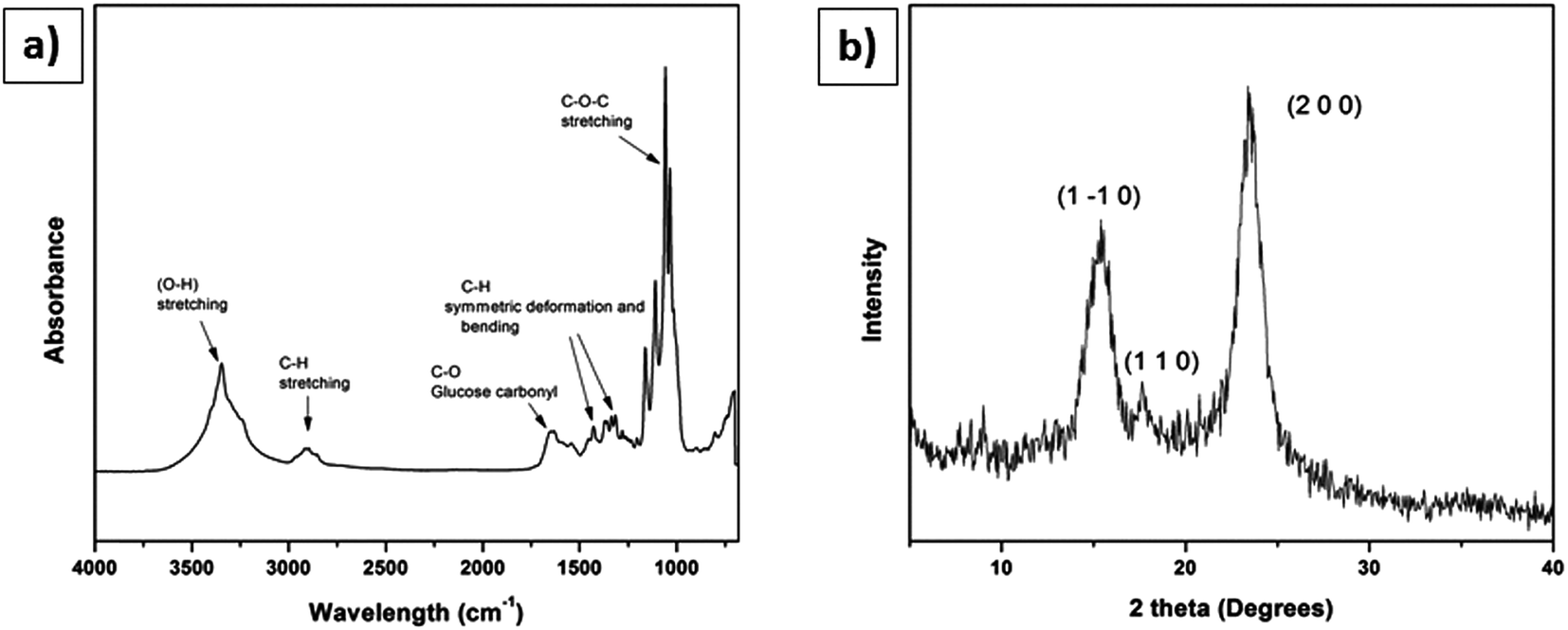 | ||
| Fig. 6 Characterization of BC membrane used for making scaffolds: (a) FT-IR spectra, (b) XRD diffractogram. | ||
From Fig. 6b, the presence of cellulose I in the BC cultured from mannitol as a carbon source was confirmed. One method employed to measure the crystallinity of the cellulose I was peak height from X-ray diffraction. This technique measures the difference in intensity of the highest peak (2 0 0) and the lowest peak that is attributed to the amorphous region. Fig. 7 shows the XRD pattern and Table 1 shows the crystallinity indices of pure BC and composite scaffolds, respectively.
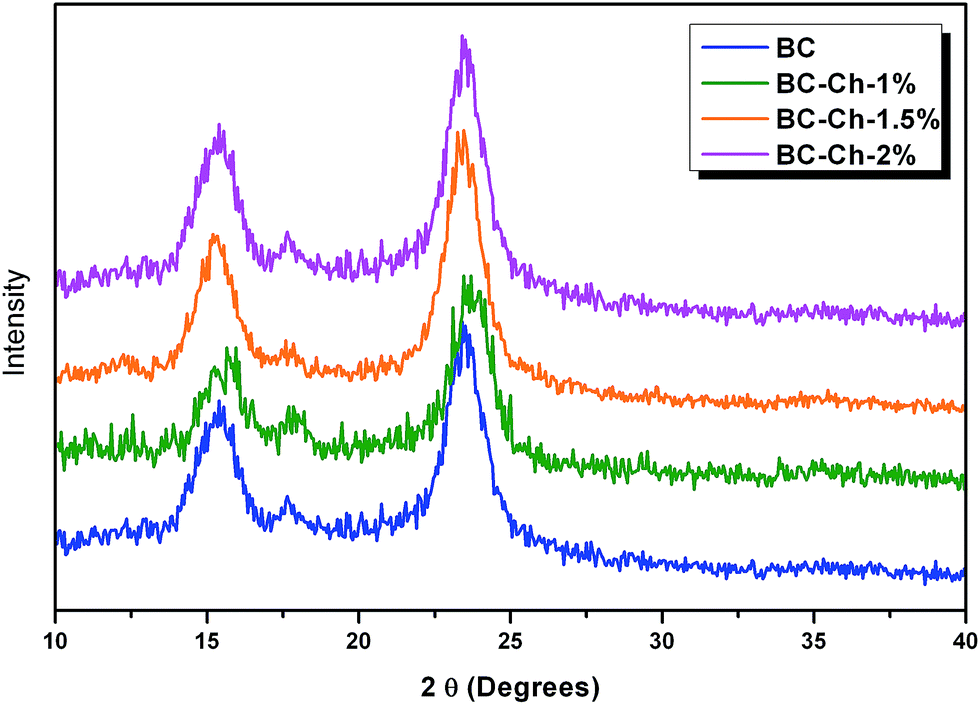 | ||
| Fig. 7 XRD patterns of pure BC and at different weight-percentages of chitosan. | ||
For the pure BC and composite scaffolds, three major peaks were found at 14.80° that are assignable to (1 −1 0), 16.6° to (1 1 0), and 22.93° to (2 0 0) crystallographic planes. These findings confirm the presence of cellulose I in the BC film. The crystallinity of the composite scaffolds decreased with an increase in chitosan concentration. The crystallinity index of pure BC was 89.45% that reduced to 83.57% for BC–Ch-1%, 83.94% for BC–Ch-1.5%, and then further reduced to 79.46% for BC–Ch-2%. This indicates that the high crystallinity of the pure BC can be reduced by addition of chitosan. Similar results have been found in which the crystallinity index of cellulose I reduced with the addition of additives.26,27,33 This decrease was due to penetration of chitosan inside in the BC inter-fibrillar network. The chitosan molecules interact with the BC network through intermolecular hydrogen bonding to significantly alter the BC network by creating packing disruptions that lead to a reduction in the crystallinity.
FTIR analysis of prepared scaffolds
The FTIR results (Fig. 6a) confirmed the presence of stretching vibrations for O–H and C–H at 3347 cm−1 and 2897 cm−1, respectively. A sharp and intense band appears at 1651 cm−1 represents the glucose carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of cellulose. Two important bands appeared at 1427 cm−1 and 1364 cm−1 characteristic of the symmetric deformation and bending vibration of C–H, respectively. The sharp band at 1033 cm−1 represents C–O–C stretching.27
O) of cellulose. Two important bands appeared at 1427 cm−1 and 1364 cm−1 characteristic of the symmetric deformation and bending vibration of C–H, respectively. The sharp band at 1033 cm−1 represents C–O–C stretching.27
Fig. 8 shows the IR spectra of pure BC and BC–chitosan composite scaffolds with different weight percentages of chitosan.
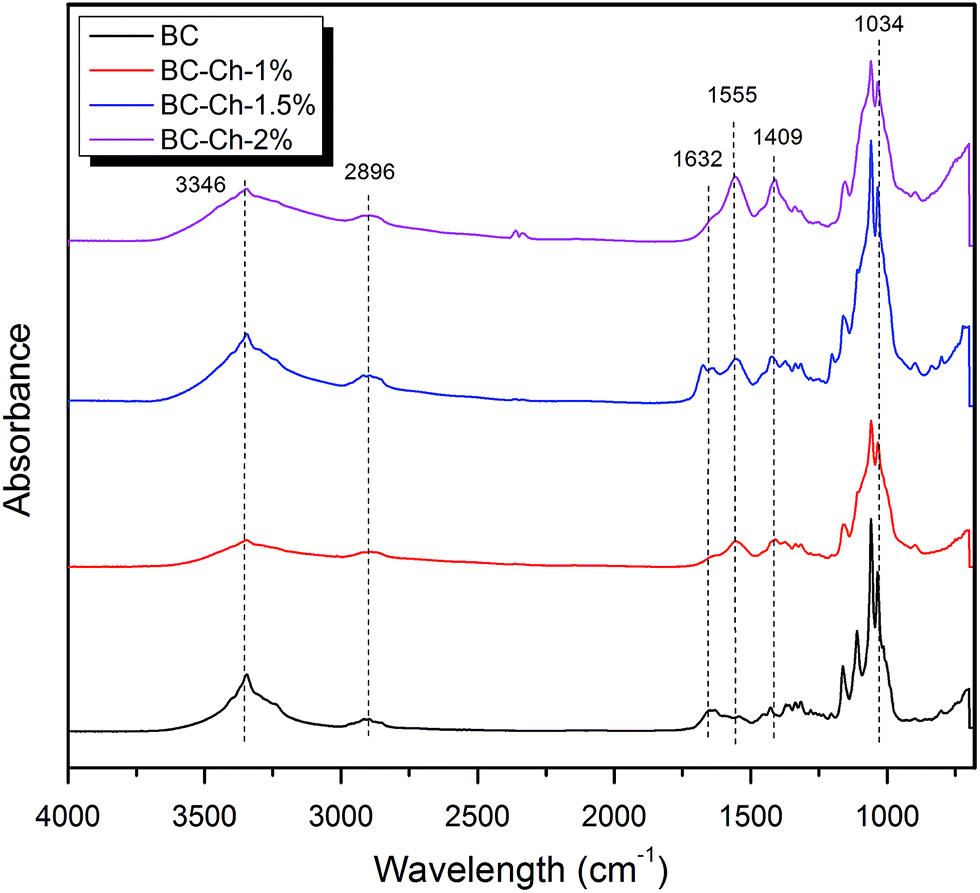 | ||
| Fig. 8 FTIR spectra of pure BC and at different weight-percentages of chitosan. | ||
The BC scaffold results confirmed the presence of the stretching vibrations of O–H and C–H at 3346 cm−1 and 2896 cm−1, respectively. An intense band appears at 1632 cm−1 represents the glucose carbonyl stretch of cellulose. Two more bands at 1427 cm−1 and 1370 cm−1 showed the symmetric deformation and bending vibration of CH, respectively. The band over 1035 to 1033 cm−1 represents the C–O–C stretching (Table 2).
| Pure BC sample wave number (cm−1) | Functional group | BC–Ch sample wave number (cm−1) |
|---|---|---|
| 3346 | O–H/N–H | 3345 |
| 2896 | C–H | 2894 |
| 1632 | C–O | — |
| — | NH2 | 1555 |
| 1427 | C–H deformation | — |
| 1370 | C–H bending | 1373 |
| — | C–N stretching (primary amide) | 1409 |
| 1035 | C–O–C | 1034 |
The spectral analysis of BC–Ch shows all the characteristic bands of BC. For the BC–chitosan scaffold, a broad band appears at 3245 cm−1 (combination of O–H stretching and N–H stretching). C–O-stretching of BC–Ch decreased due to increased hydrogen bonding in the composite and more interactions between BC and Ch. The –NH2 deformation vibration at 1555 cm−1 indicates the presence of chitosan molecules inside BC. The C–N stretching peak at 1409 cm−1 was due to the presence of primary amides. All the data indicate that the chitosan molecules interact with the BC through intermolecular hydrogen bonding networks and penetrate well within the BC fibrils.27
Mechanism of vectorially aligned freezing
The freezing step is the most critical step for creating a porous scaffold. Because BC contains water (solvent), these solvent molecules are converted in form to ice crystals during freezing. The randomly distributed web of BC nanofibers (Fig. 2) is vectorially aligned (in the direction of the freezing – from low to high temperature gradient) by freezing and subsequent freeze-drying. The BC nanofibrillar network becomes disentangled during liquid nitrogen-induced freezing process by the ice-templating effect; in other words, the nanofibrils follow vectorial alignment (growth) of the ice nuclei by hydrogen bonding to the water as it freezes. The liquid nitrogen induced rapid formation of ice nuclei which consequently propagated in the direction of freezing.34 The ice-templating effect thus had a profound impact on the nanofibrillar alignment. The samples were vectorially aligned by the liquid nitrogen freezing of the water solvent. More specifically, the water-ice crystals grew parallel to the direction of freezing (i.e., from the low temperature to high temperature gradient). After drying, the scaffold displayed a highly vectorially aligned channel-like character that possessed a huge number of small voids (hence, its highly porous character). In sum, rapid cooling by liquid nitrogen of the water within the BC nanofibrils caused the formation of many small sized ice nuclei to result in a scaffold of dimensionally small pore size. Fig. 9 provides an illustration of the process that allows for ready visualization of how the wet BC film was frozen in one direction.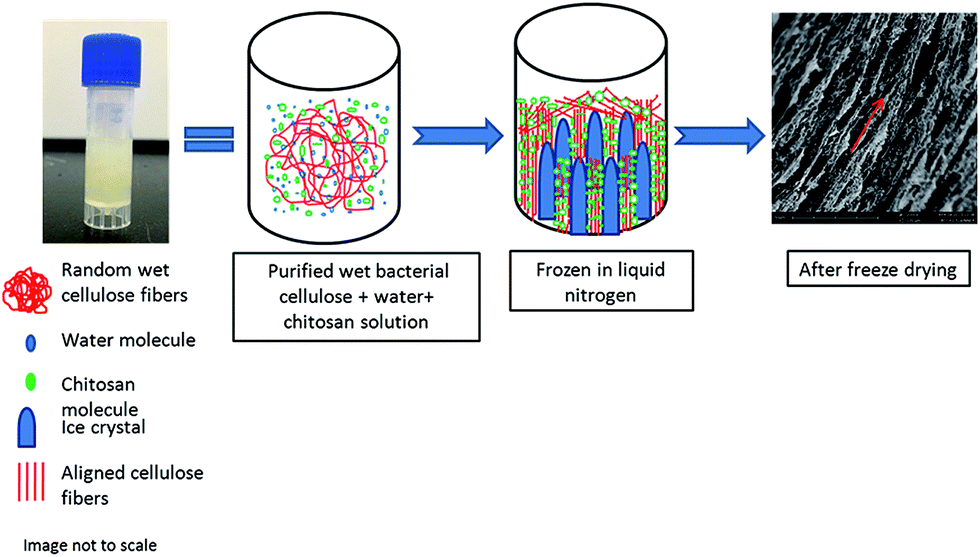 | ||
| Fig. 9 Mechanism of orienting randomly distributed bacterial cellulose/composite scaffold by the vectorially aligned freeze-drying method. During freezing, ice crystals grow in freezing direction (vertically) that exclude the bacterial cellulose fibrils and chitosan molecules and aligns the nanofibrils in the spaces between the ice crystals (parallel to direction of freezing). | ||
The ice crystals started to grow from the bottom to the top of the sample due to differences in the temperature gradient. The random nonwoven BC film fibrils were stretched and pulled by hydrogen bonding forces toward the ice crystals and consequently, they both aligned themselves parallel to the freezing direction. Interestingly, the chitosan macromolecules tended to separate from the frozen solvent likely due to their milder hydrophobicity relative to the cellulose (see representation using green dots in Fig. 9). These chitosan macromolecules then aggregated and aligned themselves around the ice crystals and into the pores of the BC fibrils.
In general, the method is extremely simple, useful, and powerful for obtaining aligned porosity from a randomly nonwoven cellulose nanofibrillar network. The preparation of aligned porous materials with micrometre-sized pores is of particular importance for applications such as tissue engineering that require subsequent modification with entities such as biological cells, which can attach along the length of the channels of aligned porous scaffolds. Also, the synthetically aligned porous structures may also be used to mimic natural aligned materials such as bone.23
Conclusions
Pure BC and BC–chitosan scaffolds were prepared by vectorially aligned freeze-drying. The incorporation of chitosan inside the BC network increased the interaction between BC nanofibrils and chitosan macromolecules that after freeze-drying displayed a vectorial alignment of fibrils along the freezing direction. Morphology considerations such as pore size and distance between channels could be altered by adjusting chitosan concentration; however, the crystallinity index tended to decrease with increasing chitosan concentration likely because of its well known strong intermolecular interactions with the cellulose backbone. However, chitosan penetration inside BC fibrils facilitated the formation of a dense network that allowed the scaffolds to maintain their structural integrity, display excellent shape recovery, and provide superior overall mechanical properties relative to pure BC after load removal, a finding that was very comparable to native human cartilage. These unique properties of the current prepared scaffolds may specifically be tailored for cartilage tissue regeneration.Acknowledgements
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. These authors contributed equally. The authors thank Judy Elson for help in running the FE-SEM. We also thank the United Soybean Board (USB) for providing partial funding for this work and thank Jiangnan University for the Foreign Talent Professorship that allowed LAL to complete parts of this work.References
- R. M. Brown, Microbial Cellulose: A New Resource for Wood, Paper, Textiles, Food and Specialty Products, May, 2013, http://www.botany.utexas.edu/facstaff/facpages/mbrown/position1.html Search PubMed.
- A. Nandgaonkar, Q. Wang, K. Fu, W. E. Krause, Q. Wei, R. Gorga and L. A. Lucia, One-Pot Biosynthesis of Reduced Graphene Oxide (RGO)/Bacterial Cellulose (BC) Nanocomposites, Green Chem., 2014, 16(6), 3195–3201 RSC.
- N. Petersen and P. Gatenholm, Bacterial Cellulose-Based Materials and Medical Devices: Current State and Perspectives, Appl. Microbiol. Biotechnol., 2011, 91(5), 1277–1286 CrossRef CAS PubMed.
- H. Backdahl, G. Helenius, A. Bodin, U. Nannmark, B. R. Johansson, B. Risberg and P. Gatenholm, Mechanical Properties of Bacterial Cellulose and Interactions with Smooth Muscle Cells, Biomaterials, 2006, 27(9), 2141–2149 CrossRef PubMed.
- J. Kim, Z. Cai and Y. Chen, Biocompatible Bacterial Cellulose Composites for Biomedical Application, J. Nanotechnol. Eng. Med., 2010, 1(1), 11006–11011 CrossRef.
- F. A. Muller, L. Muller, I. Hofmann, P. Greil, M. M. Wenzel and R. Staudenmaier, Cellulose-Based Scaffold Materials for Cartilage Tissue Engineering, Biomaterials, 2006, 27(21), 3955–3963 CrossRef PubMed.
- H. Pulkkinen, V. Tiitu, E. Lammentausta, E. R. Hamalainen, I. Kiviranta and M. J. Lammi, Cellulose Sponge as a Scaffold for Cartilage Tissue Engineering, Bio-Med. Mater. Eng., 2006, 16, S29–S35 CAS.
- A. Svensson, E. Nicklasson, T. Harrah, B. Panilaitis, D. L. Kaplan, M. Brittberg and P. Gatenholm, Bacterial Cellulose as a Potential Scaffold for Tissue Engineering of Cartilage, Biomaterials, 2005, 26(4), 419–431 CrossRef CAS PubMed.
- S. V. Madihally and H. W. Matthew, Porous Chitosan Scaffolds for Tissue Engineering, Biomaterials, 1999, 20(12), 1133–1142 CrossRef CAS PubMed.
- P. K. Dutta, K. Rinki and J. Dutta, Chitosan: a Promising Biomaterial for Tissue Engineering Scaffolds, in Chitosan for Biomaterials II, Springer, Berlin, Heidelberg, 2011, pp. 45–79 Search PubMed.
- E. Carletti, A. Motta and C. Migliaresi, Scaffolds for Tissue Engineering and 3D Cell Culture, in 3D Cell Culture: Methods and Protocols, Humana Press, 2011, pp. 17–39 Search PubMed.
- E. Boland, G. Wnek, D. Simpson, K. Pawlowski and G. Bowlin, Tailoring Tissue Engineering Scaffolds Using Electrostatic Processing Techniques: a Study of Poly(Glycolic Acid) Electrospinning, J. Macromol. Sci., Part A: Pure Appl.Chem., 2001, 38(12), 1231–1243 CrossRef.
- A. Frenot and I. S. Chronakis, Polymer Nanofibers Assembled by Electrospinning, Curr. Opin. Colloid Interface Sci., 2003, 8(1), 64–75 CrossRef CAS.
- Z. W. Ma, M. Kotaki, R. Inai and S. Ramakrishna, Potential of Nanofiber Matrix as Tissue-Engineering Scaffolds, Tissue Eng., 2005, 11(1–2), 101–109 CrossRef CAS PubMed.
- A. Subramanian, D. Vu, G. F. Larsen and H. Y. Lin, Preparation and Evaluation of the Electrospun Chitosan/PEO Fibers for Potential Applications in Cartilage Tissue Engineering, J. Biomater. Sci., Polym. Ed., 2005, 16(7), 861–873 CrossRef CAS PubMed.
- L. D. Harris, B. S. Kim and D. J. Mooney, Open Pore Biodegradable Matrices Formed with Gas Foaming, J. Biomed. Mater. Res., 1998, 396–402 CrossRef CAS.
- Z. Xiang, R. Liao, M. S. Kelly and M. Spector, Collagen–GAG Scaffolds Grafted onto Myocardial Infarcts in a Rat Model: a Delivery Vehicle for Mesenchymal Stem Cells, Tissue Eng., 2006, 12(9), 2467–2478 CrossRef CAS PubMed.
- L. A. Smith, J. A. Beck and P. X. Ma, Nanofibrous Scaffolds and their Biological Effects, in Nanotechnologies for the Life Sciences, Wiley-VCH Verlag GmbH & Co. KGaA, 2007 Search PubMed.
- A. N. Brown, B. S. Kim, E. Alsberg and D. J. Mooney, Combining Chondrocytes and Smooth Muscle Cells to Engineer Hybrid Soft Tissue Constructs, Tissue Eng., 2000, 6(4), 297–305 CrossRef CAS PubMed.
- G. Chen, T. Ushida and T. Tateishi, Development of Biodegradable Porous Scaffolds for Tissue Engineering, Mater. Sci. Eng., C, 2001, 17(1), 63–69 CrossRef.
- W. Y. J. Chen and G. Abatangelo, Functions of Hyaluronan in Wound Repair, Wound Repair and Regeneration, 1999, 7(2), 79–89 CrossRef CAS PubMed.
- B. Subia, J. Kundu and S. C. Kundu, Biomaterial Scaffold Fabrication Techniques for Potential Tissue Engineering Applications, in Tissue Engineering, ed. D. Eberli, InTech, 2010, http://www.intechopen.com/books/tissue-engineering/biomaterial-scaffold-fabricationtechniques-for-potential-tissue-engineering-applications Search PubMed.
- H. F. Zhang, I. Hussain, M. Brust, M. F. Butler, S. P. Rannard and A. I. Cooper, Aligned Two- and Three-Dimensional Structures by Directional Freezing of Polymers and Nanoparticles, Nat. Mater., 2005, 4(10), 787–793 CrossRef CAS PubMed.
- H. Ko, C. Sfeir and P. N. Kumta, Novel Synthesis Strategies for Natural Polymer and Composite Biomaterials as Potential Scaffolds for Tissue Engineering, Philos. Trans. R. Soc., A, 2010, 368(1917), 1981–1997 CrossRef CAS PubMed.
- J. Andersson, H. Stenhamre, H. Backdahl and P. Gatenholm, Behavior of Human Chondrocytes in Engineered Porous Bacterial Cellulose Scaffolds, J. Biomed. Mater. Res., Part A, 2010, 94(4), 1124–1132 Search PubMed.
- Z. J. Cai, C. W. Hou and G. Yang, Preparation and Characterization of a Bacterial Cellulose/Chitosan Composite for Potential Biomedical Application, J. Appl. Polym. Sci., 2012, 126(6), 2078 CrossRef CAS.
- M. Ul-Islam, N. Shah, J. H. Ha and J. K. Park, Effect of Chitosan Penetration on Physico-Chemical and Mechanical Properties of Bacterial Cellulose, Korean J. Chem. Eng., 2011, 28(8), 1736–1743 CrossRef CAS.
- J. Han, C. Zhou, Y. Wu, F. Liu and Q. Wu, Self-assembling Behavior of Cellulose Nanoparticles During Freeze-Drying: Effect of Suspension Concentration, Particle Size, Crystal Structure, and Surface Charge, Biomacromolecules, 2013, 14(5), 1529–1540 CrossRef CAS PubMed.
- N. Yin, S. Chen, Z. Li, Y. Ouyang, W. Hu, L. Tang, W. Zhang, B. Zhou, J. Yang and Q. Xu, Porous Bacterial Cellulose Prepared by a Facile Surfactant-Assisted Foaming Method in Azodicarbonamide–NaOH Aqueous Solution, Mater. Lett., 2012, 81, 131–134 CrossRef CAS.
- H. Son, M. Heo, Y. Kim and S. Lee, Optimization of Fermentation Conditions for the Production of Bacterial Cellulose by a Newly Isolated Acetobacter, Biotechnol. Appl. Biochem., 2001, 33(1), 1–5 CrossRef CAS PubMed.
- A. Nandgaonkar and W. Krause, Electrospinning of Chitosan and its Correlation with Degree of Deacetylation and Rheological Property, M.Sc. thesis, NC State University, 2011.
- K. El-Tahlawy and S. M. Hudson, Chitosan: Aspects of Fiber Spinnability, J. Appl. Polym. Sci., 2006, 100(2), 1162–1168 CrossRef CAS.
- C. Castro, R. Zuluaga, C. Álvarez, J. Putaux, G. Caro, O. J. Rojas and P. Gañán, Bacterial Cellulose Produced by a New Acid-Resistant Strain of Gluconacetobacter Genus, Carbohydr. Polym., 2012, 89(4), 1033–1037 CrossRef CAS PubMed.
- L. Qian and H. Zhang, Controlled Freezing and Freeze Drying: a Versatile Route for Porous and Micro-/Nano-Structured Materials, J. Chem. Technol. Biotechnol., 2011, 86(2), 172–184 CrossRef CAS.
- X. Wu, Y. Liu, X. Li, P. Wen, Y. Zhang, Y. Long, X. Wang, Y. Guo, F. Xing and J. Gao, Preparation of Aligned Porous Gelatin Scaffolds by Unidirectional Freeze-Drying Method, Acta Biomater., 2010, 6(3), 1167–1177 CrossRef CAS PubMed.
- S. Grad, K. Gorna, S. Gogolewski and M. Alini, Scaffolds for Cartilage Tissue Engineering: Effect of Pore Size, Eur. Cells Mater., 2004, 7(2), 3 Search PubMed.
- L. Chen, Y. Du and X. Zeng, Relationships between the molecular structure and moisture-absorption and moisture-retention abilities of carboxymethyl chitosan: II. Effect of degree of deacetylation and carboxymethylation, Carbohydr. Res., 2003, 338(4), 333–340 CrossRef CAS PubMed.
- W. Thein-Han and R. Misra, Biomimetic chitosan–nanohydroxyapatite composite scaffolds for bone tissue engineering, Acta Biomater., 2009, 5(4), 1182–1197 CrossRef CAS PubMed.
- T. G. Chiciudean, A. Stoica, T. Dobre and M. Van Tooren, Synthesis and Characterization of Poly(Vinyl Alcohol)–Bacterial Cellulose Nanocomposite, UPB Buletin Stiintific, Series B: Chemistry and Materials Science, 2011, 73(2), 17–30 CAS.
- S. Molladavoodi, M. Gorbet, J. Medley and K. H. Ju, Investigation of Microstructure, Mechanical Properties and Cellular Viability of Poly(L-Lactic Acid) Tissue Engineering Scaffolds Prepared by Different Thermally Induced Phase Separation Protocols, J. Mech. Behav. Biomed. Mater., 2013, 17, 186–197 CrossRef CAS PubMed.
- J. Jurvelin, M. Buschmann and E. Hunziker, Optical and Mechanical Determination of Poisson's Ratio of Adult Bovine Humeral Articular Cartilage, J. Biomech., 1997, 30(3), 235–241 CrossRef CAS PubMed.
- R. Heijkants, R. Van Calck and J. De Groot, Design, synthesis and properties of a degradable polyurethane scaffold for meniscus regeneration, J. Mater. Sci.: Mater. Med., 2004, 15(4), 423–427 CrossRef CAS PubMed.
- D. Hutmacher, T. Schantz and I. Zein, Mechanical properties and cell cultural response of polycaprolactone scaffolds designed and fabricated via fused deposition modeling, J. Biomed. Mater. Res., Part A, 2001, 55(2), 203–216 CrossRef CAS.
- J. Williams, A. Adewunmi and R. Schek, Bone tissue engineering using polycaprolactone scaffolds fabricated via selective laser sintering, Biomaterials, 2005, 26(23), 4817–4827 CrossRef CAS PubMed.
- N. Naseri, J. Poirier and L. Girandon, 3-Dimensional porous nanocomposite scaffolds based on cellulose nanofibers for cartilage tissue engineering: tailoring of porosity and mechanical performance, RSC Adv., 2016, 6(8), 5999–6007 RSC.
- M. Ul-Islam, T. Khan and J. K. Park, Water Holding and Release Properties of Bacterial Cellulose Obtained by In Situ and Ex Situ Modification, Carbohydr. Polym., 2012, 88(2), 596–603 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26049g |
| This journal is © The Royal Society of Chemistry 2017 |