DOI:
10.1039/C6RA25871A
(Paper)
RSC Adv., 2017,
7, 1348-1356
Tetraphenylethene modified β-ketoiminate boron complexes bearing aggregation-induced emission and mechanofluorochromism†
Received
26th October 2016
, Accepted 5th December 2016
First published on 5th January 2017
Abstract
Two new tetraphenylethene modified β-ketoiminate boron complexes (TTPE-H and TTPE-CN) have been rationally designed and successfully synthesized. The two D–π–A type compounds exhibit typical twisted intramolecular charge-transfer (TICT) emission and evident aggregation-induced emission (AIE) characteristics, remarkable luminescence mechanochromism, and high solid state efficiency (up to 0.785 and 0.672). The mechanofluorochromic (MFC) behaviors of TTPE-H (from bright green to yellow-green emission, λem from 497 to 515 nm) and TTPE-CN (from bright yellow-green to yellow emission, λem from 525 to 565 nm) are reversible upon grinding and fuming treatments. From the X-ray diffraction and differential scanning calorimetry analyses, it was confirmed that the MFC properties of TTPE-H and TTPE-CN should be derived from a phase transition between crystalline and amorphous states. More interestingly, the compound TTPE-CN, which possesses a cyano group in the boron chelating ring, showed more contrast MFC behavior relative to TTPE-H. The reason is that the introduction of the cyano group can enhance the distortion degree of the molecule TTPE-CN because of steric hindrance.
Introduction
As a kind of “smart material”, mechanofluorochromic (MFC) emissive luminogens showing reversible stimuli-responsive emission switching in the solid state have received a great deal of attention from both fundamental research fields of solid-state photochemistry and applications in mechanosensors,1 data storage,2 fluorescent switches,3 security systems,4 and optoelectronic devices.5 Up to now, a good number of MFC molecules have been designed, synthesized and characterized, including the derivatives of pyrene,2c,6 anthracene,7 dibenzofulvene,8 tetraphenylethene,9 silole,10 and cyanostilbene,1c,11 as well as some boron complexes,12,13 these compounds have been shown to be promising MFC materials, and their structure–property relationship has been investigated. High solid-state luminescence efficiency and an obvious color contrast play an important role for the application of mechanochromic fluorescent materials. However, the fluorescence efficiency of many traditional organic luminescent materials often becomes very weak while in the solid state through the aggregation-caused quenching (ACQ) effect caused by both strong intermolecular π–π stacking interactions and non-radiative decay.14 Therefore, there is still much difficulty in the development of MFC compounds based on organic molecules. Several anti-ACQ materials were reported by Tang et al.15 and Park et al.16 in 2001 and 2002, respectively. These substances are called aggregation-induced emission (AIE) or aggregation-induced enhanced emission (AIEE) compounds, which exhibit strong emission when a molecular aggregate occurs in poor solvents or in the solid state. Since then, a large number of AIE luminogens characterized by strongly twisted skeleton bearing rotatable aryl units have been developed.17 Indeed, most of AIE-active molecules have been found to possess MFC nature due to their strong solid-state fluorescence and diverse aggregation morphology. AIE has been considered as a key for opening a treasure chest of MFC materials, and some MFC materials with strong solid-state emission have been developed based on AIE-active molecules. For example, in 2010, Park's group reported the cyano-distyrylbenzene derivative,11a which has mechanofluorochromic aggregation-induced enhanced emission (AIEE) properties. Then a number of new mechanofluorochromic AIE compounds were synthesized by Tang, Chi, Yang and their co-workers.1b,7a–g,8,9a–e,17d Although some novel molecular systems possessing efficient reversible MFC behaviors have been reported, mechanofluorochromic AIE materials are still at the initial state of investigation, and their numbers and the understanding of the phenomenon are limited. Therefore, there is a great demand for the exploitation of new mechanofluorochromic AIE materials and the accumulation of relative structure–property knowledge.
Recently, luminescent organoboron compounds have generated considerable interest because of their large number of advantages over other dyes, such as high fluorescence quantum yield, large absorption coefficient, excellent chemical- and photochemical-stability, long excited-state lifetimes, a large two-photo cross-section, good solubility and narrow emissive band.18 However, many organoboron fluorescent dyes that exhibit strong fluorescence in dilute solutions quench or reduce the fluorescence intensity in the solid state due to ACQ effect. Although some luminescent boron complexes containing chelating N,O- and N,N-chromophores showing AIE effects have been developed,13c,19–21 it is still challenging to rationally design organoboron complexes with excellent AIE properties, especially with both MFC and AIE behaviors. We recently reported a new class of D–π–A conjugated thiazole-based β-ketoiminate boron complexes containing cyano groups functionalized with triphenylamine showing good MFC and AIE properties.22 Here, we present the synthesis and properties of two new D–π–A type β-ketoiminate boron complexes with tetraphenylethene units connected in the β-ketoiminate boron core, namely TTPE-H and TTPE-CN (Scheme 1). The two dyes were designed according to the following considerations: (1) tetraphenylethene (TPE), which is composed of an ethenyl core and multiple rotational phenyl rings, has electron donating characteristics and takes a nonplanar configuration due to the steric repulsion between the adjacent phenyls. The existence of multiple rotational phenyls, nonplanar shape, and weak π–π interaction enable TPE to be a useful building block for the construction of D–π–A conjugated luminogens with ICT, AIE and MFC properties.9 (2) β-Iminoenolate boron unit that contains the nitrogen-based ligand exhibits good electron withdrawing characteristics. β-Ketoiminate boron and its derivatives tend to show strong emission in solutions and solid states.13b,c (3) As a functional unit, the cyano (CN) group has been frequently utilized in the design of advanced optoelectronic materials. The introduction of cyano would increase the electron withdrawing ability of the β-iminoenolate boron unit, favoring ICT emission. At the same time, the internal steric hindrance of the cyano group is advantageous to confer AIE and MFC properties.9c,22 Compared with the organic boron complexes reported at present,12,13,19–22 TTPE-H and TTPE-CN exhibit strong ICT (Δνst = 6364 and 8850 cm−1 in DMSO, respectively) and significant AIE (αAIE = 196 and 224, respectively) characteristics, remarkable luminescence mechanochromism (spectral shift = 18 and 40 nm, respectively), and high solid state efficiency (up to 0.785 and 0.672, respectively). These properties make them potentially promising luminescent materials. In particular, the cyano-containing compound TTPE-CN showed more contrast MFC behavior relative to TTPE-H, it is attributed to the existence of the cyano group can enhance the distortion degree of the molecule TTPE-CN and thus make it have more looser molecular packing in the solid states and better structural transformability.
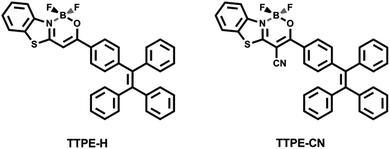 |
| | Scheme 1 The molecular structures of TTPE-H and TTPE-CN. | |
Results and discussion
Synthesis and characterizations
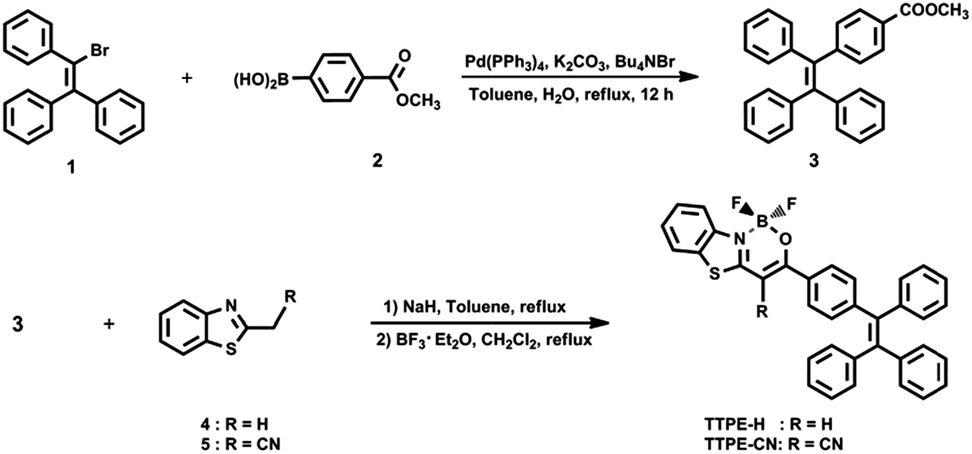
The synthetic routes for boron complexes TTPE-H and TTPE-CN were shown in Scheme 2. Firstly, the tetraphenylethene derivative 3 was synthesized by Suzuki–Miyaura coupling between 1 and boric acid 2 with a high yield of 98%. The target molecules of TTPE-H and TTPE-CN were prepared between methyl ester 3 and the corresponding 2-substituted benzothiazole 4 and 5 in the presence of sodium hydride in anhydrous toluene followed by complexation with boron trifluoride diethyl etherate, to give yields of 62% and 30%, respectively. TTPE-H and TTPE-CN are soluble in CHCl3, CH2Cl2, benzene, toluene, THF and DMSO, but show poor solubility in alcohols (such as methanol and ethanol) and aliphatic hydrocarbon solvents (such as cyclohexane and n-hexane) at room temperature. All the intermediates and target molecules were purified by column chromatography on silica gel, and unambiguously characterized by 1H and 13C NMR, TOF LC/MS spectrum or MALDI-TOF mass spectrometry, and C, H, N elemental analyses.
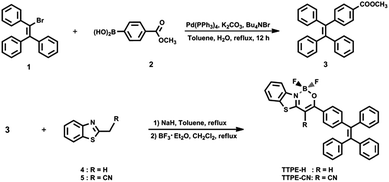 |
| | Scheme 2 Synthetic routes of TTPE-H and TTPE-CN. | |
UV-vis absorption and fluorescent emission spectra in solutions
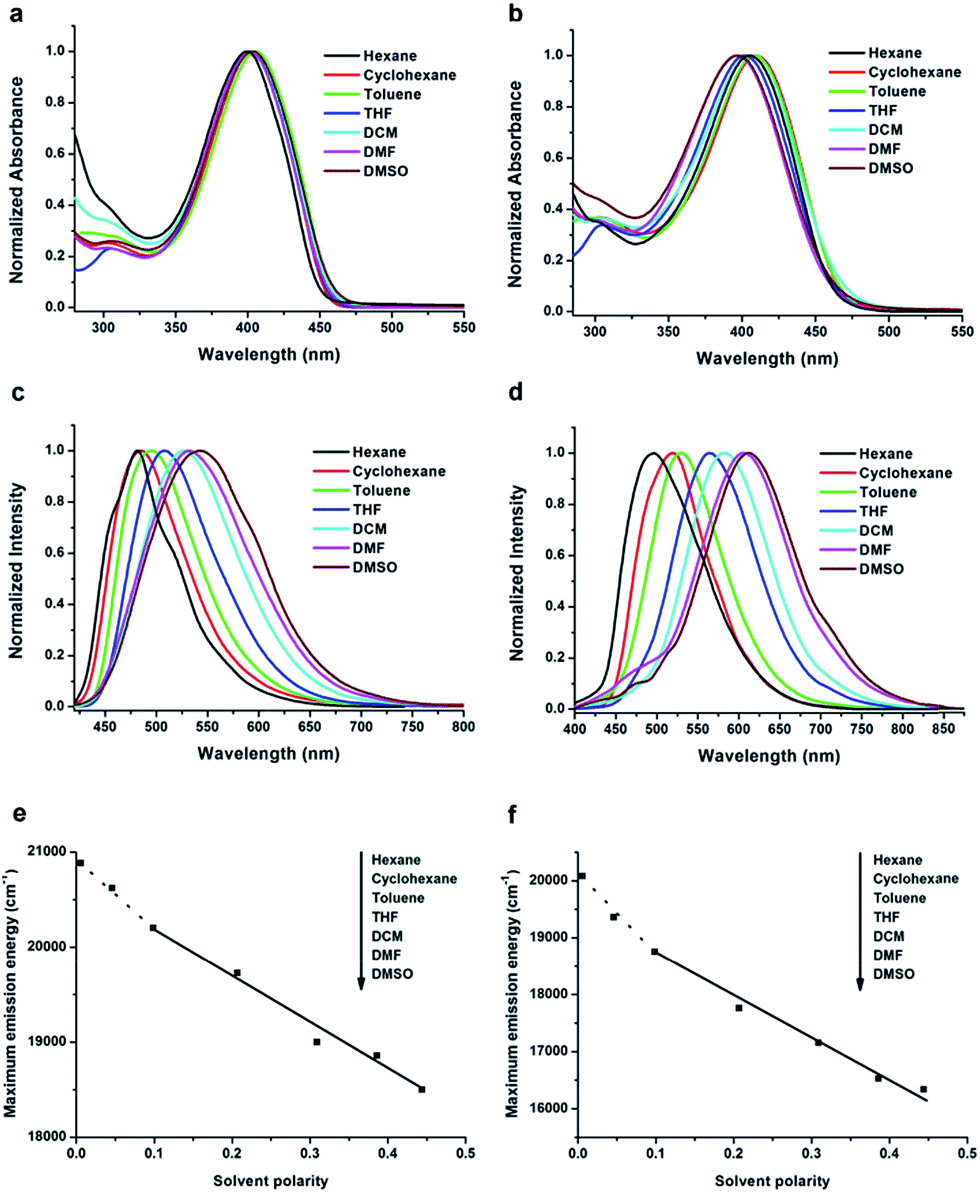
The solvent effect on the absorption and fluorescence properties of TTPE-H and TTPE-CN was examined in various solvents (Fig. 1), and the corresponding photophysical data are collected in Tables S1 and S2 (ESI†). As shown in Fig. 1a, it was obvious that TTPE-H has two strong absorption bands at ca. 300 nm and ca. 400 nm in different solvents (Fig. 1a). The weak absorption bands appearing at ca. 300 nm, which did not shift with increasing polarity of the solvents, was originated from π–π* transitions, and the maximum absorption peaks ca. 400 nm can be attributed to the intramolecular charge transfer (ICT) state between electron donor (tetraphenylethene unit) and electron acceptor (β-iminoenolate boron moiety), in terms of its strong push–pull structure. Fig. 1c showed the solvent-dependent PL spectra of TTPE-H. It was clear that the emission bands became broad and red-shifted significantly with increasing polarity of the solvent. For example, in hexane, TTPE-H gave a strong emission band at 480 nm, but with the increasing solvent polarity, its emission band was red-shifted to 542 nm in DMSO accompanied by emission bands broaden. Combined with the large Stokes shifts (such as 6364 cm−1 in DMSO), the broadening and red-shift of the emission bands, we suggested that ICT transitions of TTPE-H took place in more polar solvents. In addition, it is worth noting that the fluorescence spectra of TTPE-H became structured in non-polar hexane and cyclohexane solvents. We deduced that the emission of TTPE-H in hexane and cyclohexane was from the loc ally excited (LE) state.23 Meanwhile, a plot of the emission maximum energy as a function of the Lippert solvent polarity was given in Fig. 1e to illustrate the conformational change of the excited state surface prior to the emission.24 In such “Lippert–Mataga” plots, the slope can be used to evaluate the variation in dipole moment upon excitation, and the break in the linear relationship indicates the presence of two different excited states. The deviation of the emission maximum energy in hexane and cyclohexane from the linear relationship followed by those in other solvents can further support the notion that the LE state is responsible for the emission in nonpolar solvents25 and the TICT state contributes to the emission in polar solvents.26 Similarly, TTPE-CN also showed the large Stokes shifts, the broadening and red-shift of the emission bands from cyclohexane to DMSO, displaying an TICT character for the excited state (Fig. 1b, d and f).18f,27 Notably, TTPE-CN that contains one cyano on boron complex ring exhibited stronger ICT process in polar solvents compared to TTPE-H. For example, an evident red-shift of the emission bands from 498 to 612 nm accompanied by a remarkable increase in Stokes shifts from 4429 to 8850 cm−1 is observed for TTPE-CN when the solvent changes from a polar (e.g., hexane) to highly polar ones (e.g., DMSO). By contrast, relatively smaller changes of the emission bands and Stokes shifts (from 480 nm and 4104 cm−1 to 542 nm and 6364 cm−1, respectively) are recorded for TTPE-H when the solvent changes from hexane to DMSO. This is because cyano substitution increases the electron affinity of β-iminoenolate boron moiety in TTPE-CN, thus endues them with stronger ICT properties than that of TTPE-H. The fluorescence quantum yields (Φf) of TTPE-H and TTPE-CN were measured using quinine sulfate (Φf = 0.546, 0.5 mol L−1 H2SO4) as the standard. It is found that TTPE-H and TTPE-CN all showed very low Φf (<0.01) in the low-viscosity solvents with different polarity (Tables S1 and S2, ESI†). In order to determine the reason for the low Φf of TTPE-H and TTPE-CN in the tested solvents, time-resolved fluorescence spectroscopy was performed. The fluorescence decays can be fitted with a single exponential function (the lifetime of TTPE-H and TTPE-CN in THF, DCM, DMF, DMSO were too short to measure (<0.1 ns)). Radiative (kf = Φf/τ) and nonradiative (knr = (1 − Φf)/τ) rate constants were calculated (Tables S1 and S2, ESI†). It was clear that the knr of TTPE-H and TTPE-CN was obviously larger than the kf in hexane, cyclohexane, toluene. This suggests that the lower Φf of TTPE-H and TTPE-CN can be ascribed to nonradiative decay.
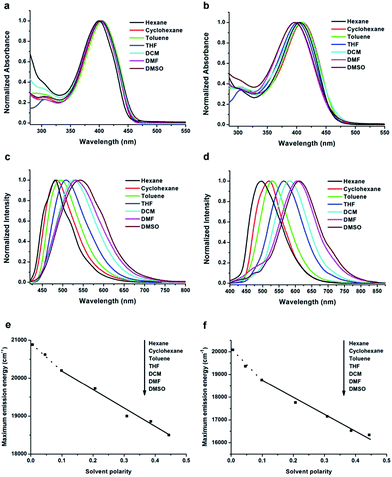 |
| | Fig. 1 Normalized UV-vis absorption spectra of TTPE-H (a) and TTPE-CN (b), and normalized PL spectra of TTPE-H (c) and TTPE-CN (d) excited at 390 nm and 410 nm, respectively, in different solvents (1.0 × 10−5 mol L−1). Lippert–Mataga plot: fluorescence emission maximum energy of TTPE-H (e) and TTPE-CN (f) as a function of solvent polarity. | |
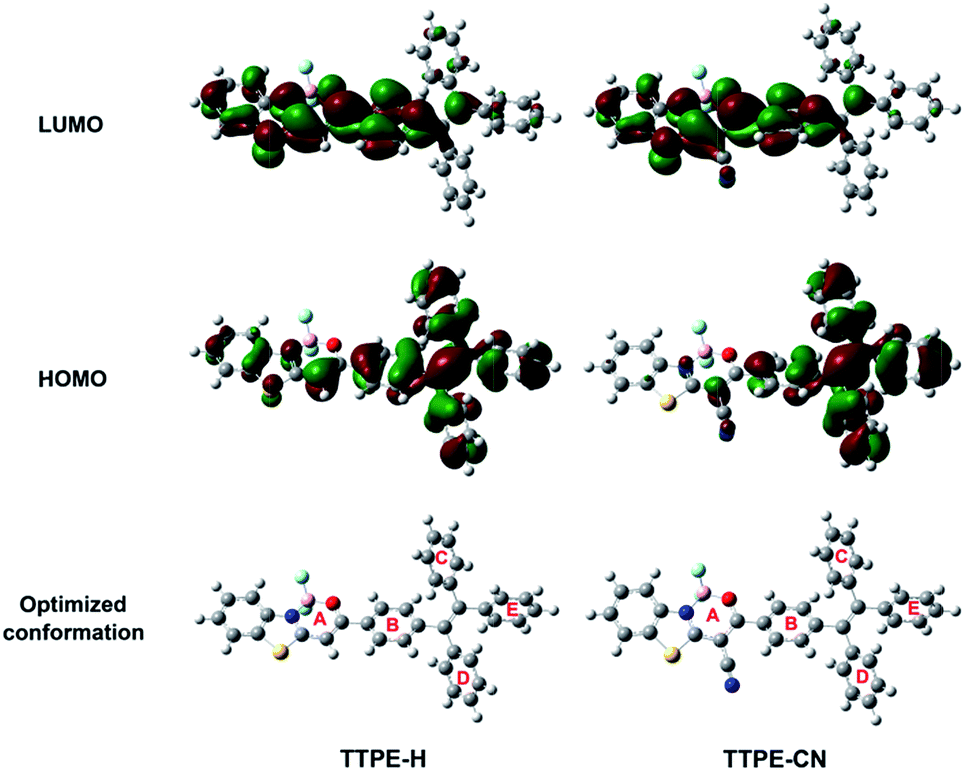
To better understand the observed spectroscopic properties of TTPE-H and TTPE-CN, density functional theory (DFT) calculations were performed on the four complexes at the B3LYP/6-31G(d) level after optimizing their structures to the lowest energy spatial conformations with the Gaussian 09W program. Fig. 2 showed the electron distribution of the HOMOs and LUMOs of TTPE-H and TTPE-CN. It is clear that the LOMO densities of TTPE-H and TTPE-CN are mainly located on the acceptor of the β-iminoenolate boron unit, and the HOMO of TTPE-H was located in the whole molecule. By comparison, the HOMO of TTPE-CN was mainly located in the tetraphenylethene unit. This result suggests that, on one hand, the ICT process of TTPE-H and TTPE-CN was further confirmed to occur from the donor to the acceptor moiety; on the other hand, the introduction of cyano into molecule TTPE-CN significantly enhanced the ICT process, which is consistent with the observed optical property (Fig. 1). Moreover, TTPE-H and TTPE-CN adopt a twisted spatial conformation at their optimized lowest energy states, which disfavors close molecular packing in the solid state and leads to efficient fluorescence. Dihedral angles between boron chelate ring A and phenyl group B in TTPE-H is 12.3°, and it increases to 28.3° after the installation of cyano groups on the boron chelate ring of TTPE-CN. Whereas dihedral angles between phenyl rings in tetraphenylethene units (B–C, B–D, and B–E) remain unchanged. For TTPE-CN, the introduction of cyano group has little effect on the conformations of the tetraphenylethene moiety but can significantly enhance the degree of molecular distortions because of steric hindrance effects.9a,e These increases in molecular distortion can cause the compounds to adopt different molecular packing modes in the solid state and may make them have both AIE characteristics and MFC behaviors.
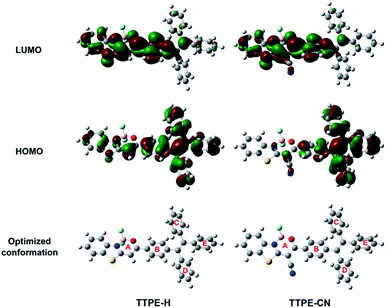 |
| | Fig. 2 Electron density distributions in the HOMO and LUMO states calculated by DFT in Gaussian 09 at the B3LYP/6-31G(d) level, and the optimized conformation structures of TTPE-H and TTPE-CN. | |
Aggregation induced emission (AIE)
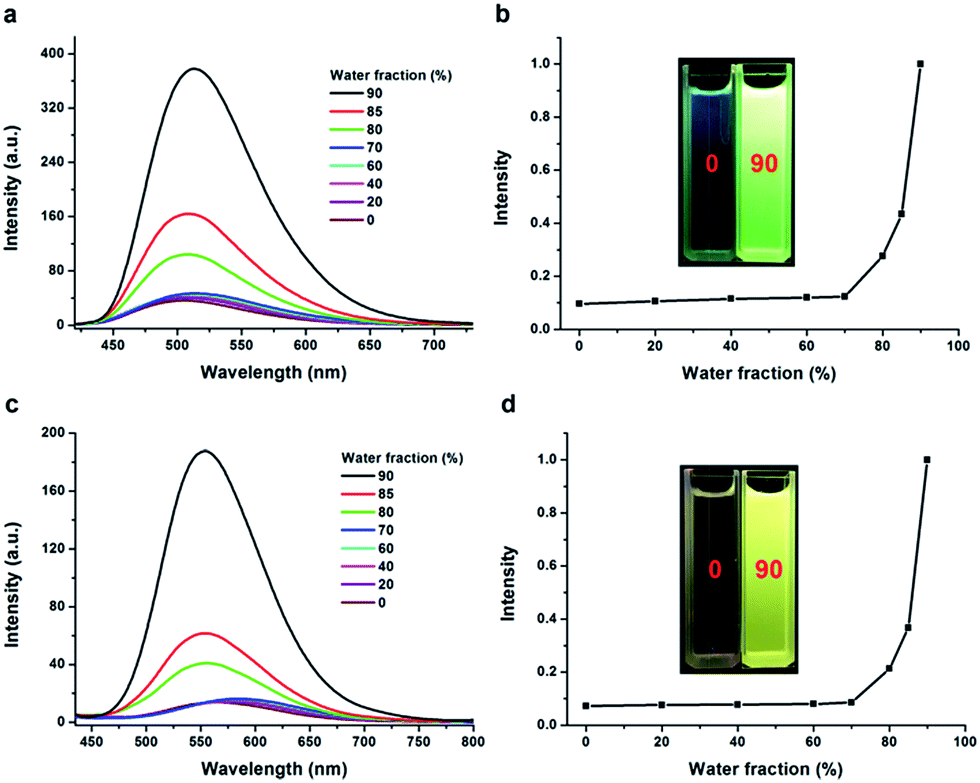
Among the reported AIE systems in the present, the molecules containing tetraphenylethene (TPE) unit have been put into the limelight by researchers for their simple synthetic routes and notable AIE performance.9,28 Moreover, when illuminated under a UV lamp, TTPE-H and TTPE-CN could emit strong green and yellow light in the powder state, respectively, but weak fluorescence was observed from their dilute THF solutions, and thus we speculate that they would possess AIE properties. To determine whether TTPE-H and TTPE-CN have AIE properties, their fluorescent behaviors were studied in tetrahydrofuran (THF)–water mixture with different water fractions (fw, the volume percentage of water in THF/water mixtures). The two compounds were both readily soluble in THF, but have low solubility in water, thus, increasing the water fractions in the mixed solvents would change the existing compound solutions in pure THF into aggregated particles, thereby changing their PL spectra. As shown in Fig. 3, when fw is less than 80%, the PL intensities of TTPE-H and TTPE-CN remained very low. However, but increased swiftly with the water fraction above 80%. As fw is 90%, the THF–water mixture's luminescence of TTPE-H and TTPE-CN significantly increased and the PL intensities are approximately 15 times higher than that in the pure THF, respectively. As a result, the bright yellow green and yellow light emissions (centered at 513 and 553 nm) of TTPE-H and TTPE-CN were observed, respectively. The increase in PL intensities can be attributed to the AIE effect, which can be demonstrated clearly by the fluorescent images of the solvent mixtures at fw = 0% and 90% (Fig. 3b and d), caused by the formation of molecular aggregates when water is added into the THF solution. The aggregated particles obtained could be characterized by using dynamic light scattering (DLS), which show the existence of nano-aggregates as their main constituent in the solvent mixtures with high water contents (fw > 70%, Fig. S1–S6†). To obtain the reason of the AIEE effect, the fluorescence lifetimes (τ) of TTPE-H and TTPE-CN in water–THF mixtures with different water fractions were measured. The results showed that the knr of TTPE-H and TTPE-CN was obviously larger than the kf with the decrease of the proportion of water in mixtures (Table S3, ESI†). It suggests that the lower Φf of TTPE-H and TTPE-CN in the in water–THF mixtures (fw < 80%) are attributed to nonradiative decay. To obtain further insight, the effect of solvent viscosity and temperature on fluorescence was investigated. TTPE-H and TTPE-CN exhibited the extremely weak fluorescence (Φf < 0.01) in low-viscosity solvents (0.31–2.24 cP) (Tables S1 and S2†). By deep contrast, they displayed relatively strong fluorescence in ethylene glycol (Φf = 0.105 (TTPE-H), 0.172 (TTPE-CN)) and glycerol (Φf = 0.165 (TTPE-H), 0.204 (TTPE-CN)) with high viscosity. In addition, Fig. S7 and S8† showed the PL behaviors of TTPE-H and TTPE-CN in THF–ethylene glycol mixture with different ethylene glycol fractions. It was clear that the fluorescence intensity of TTPE-H and TTPE-CN increased gradually with the increase of proportion of ethylene glycol, respectively. The above results indicated that the viscous medium inhibits intramolecular rotation, thereby suppressing the nonradiative process which leads to enhanced fluorescence emission.29 Moreover, with the decreasing of the system temperature, the fluorescence intensity of TTPE-H and TTPE-CN in THF increased obviously, respectively (Fig. S9 and S10†). This suggests that the restriction of intramolecular rotation (RIR) associated with decreasing temperature leads to intensive fluorescence. Therefore, the main reason for AIE of TTPE-H and TTPE-CN is considered to be the RIR associated with the aromatic group. To quantitatively evaluate AIE, the fluorescence quantum yields of TTPE-H and TTPE-CN in solutions (Φf,s) and in the as-prepared solids (Φf,aps) were determined. The Φf,aps value for TTPE-H and TTPE-CN was 0.785 and 0.672, respectively, which was much higher than Φf,s in pure THF (0.004 and 0.003), this can further demonstrate their AIE behaviors. And thus we could get the AIE factors (αAIE = Φf,aps/Φf,s) of TTPE-H and TTPE-CN as 196 and 224, respectively.
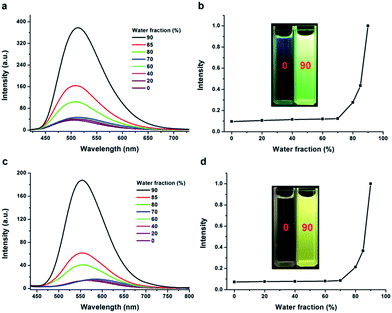 |
| | Fig. 3 PL spectra of TTPE-H (a, λex = 400 nm) and TTPE-CN (c, λex = 420 nm) in water–THF mixtures with different water fractions (0 < fw < 90%). Normalized fluorescent emission intensities of TTPE-H (b) and TTPE-CN (d) in THF–water with different fw. Photographs in (b) and (d) are TTPE-H and TTPE-CN in THF and 10/90 THF–water mixture under UV light (365 nm), respectively. | |
Mechanofluorochromic properties
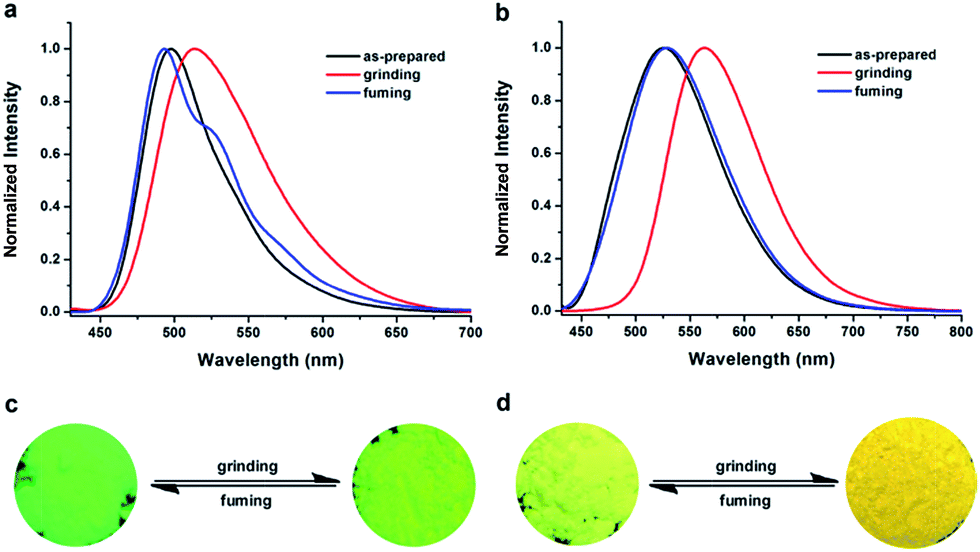
The AIE feature, ICT characteristics, twisted spatial conformation, and high solid-state efficiency render TTPE-H and TTPE-CN promising candidates as stimuli responsive smart materials.9,13a To check whether the two luminogens are mechanochromic, their fluorescence responses toward mechanical and solvent-fuming processes are studied. As shown in Fig. 4, the as-synthesized crystalline powders of TTPE-H could emit strong bright green light under UV irradiation (Fig. S11, ESI†), and the fluorescence color changed into yellow-green after simple grinding them by using a mortar and pestle. Upon further fuming with DCM vapor for 2 min, the original green emission is restored. It means that the emitting color of TTPE-H could be transferred between bright green and yellow-green reversibly through grinding and DCM fuming treatment. The PL spectra were used to monitor such a color transformation under grinding and fuming stimuli. As shown in Fig. 4a, when the as-synthesized sample of TTPE-H was ground, the emission band of original powder is remarkably red-shifted from 497 nm to 515 nm. This indicates that the grinding treatment has induced a spectral red-shift of 18 nm, and through the fuming treatment, its maximum emission wavelength can blue-shifted to the initial wavelength. In addition, it is obvious that the shape of the emission bands of TTPE-H is different between the original powder and the fumed powder, which might be attributed to π–π interactions in the solid states. For TTPE-CN, while the as-synthesized crystalline powders exhibit strong yellow-green emission at 525 nm (Fig. S12, ESI†), they emit yellow light with a maximum at 565 nm upon gently grinding, giving a large spectral shift of 40 nm. The reason for the red-shift in the PL spectra of TTPE-H and TTPE-CN is that the twisted conformation and weaker π–π interactions lead to the relatively loose molecular packing, which are readily destroyed by external forces and thus induce the planarization of molecular conformation or slip deformation. The UV-vis spectra studies show that the absorption bands of TTPE-H and TTPE-CN exhibit a similar change with the emission spectrum between the original samples and the ground samples (Fig. S13 and S14, ESI†). Thus, the mechanism of planarization of molecular conformation is acceptable. The ground powders could also be converted back to emit bright yellow-green light upon fuming with DCM. The MFC behavior of TTPE-H and TTPE-CN can be recycled many times, indicating its reversibility and durability under external stimuli (Fig. S15, ESI†). It is worth noting that TTPE-CN containing a cyano group in the boron chelating ring showed more remarkable MFC behavior relative to TTPE-H. The reason is that the introduction of the cyano group can enhance the distortion degree of the molecule TTPE-CN because of steric hindrance. Therefore, we can infer that the MFC properties of the compounds are related to their distortion degrees; moreover, molecules with higher degrees of distortion display more evident MFC properties under the stimulus of external forces. Thus, introducing a cyano group into a compound can significantly improve its MFC property. The comparative results demonstrate that the cyano group in TTPE-CN plays a crucial role in its mechanofluorochromism, as observed in other stimuli-responsive AIE compounds.
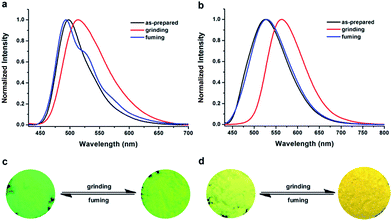 |
| | Fig. 4 Normalized fluorescent spectra of TTPE-H (a) and TTPE-CN (b) in different solid-states: as-prepared, grinding and fuming, λex = 420 nm. Photos of TTPE-H (c) and TTPE-CN (d) color changes under grinding and fuming stimuli. | |
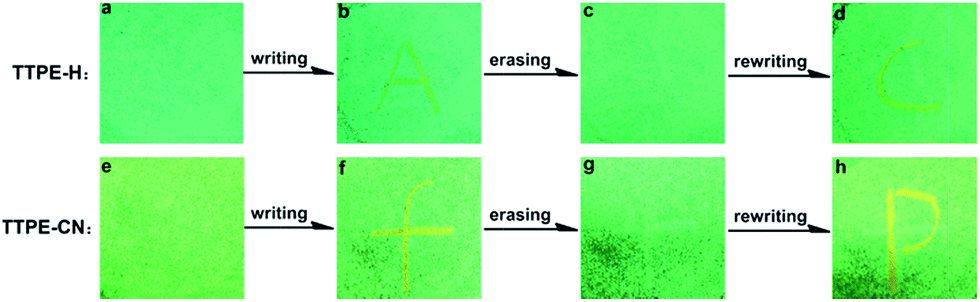
Interestingly, the as-synthesized powder of TTPE-H was spread on a filter paper and letter “A” was written on it with a metal spatula. The change occurred only in the written area. Under the UV lamp, a yellow-green “A” is observed against the green background (Fig. 5b), the letter “A” can be erased after the “paper” was exposed to DCM vapor (Fig. 5c), and the letter “C” can be written again (Fig. 5d). Also, TTPE-CN showed similar fluorescence properties (Fig. 5e–h). These results suggest that TTPE-H and TTPE-CN have the potential for applications as optical recording materials.
 |
| | Fig. 5 Photos of the luminescence writing/erasing process of TTPE-H and TTPE-CN on filter papers under UV light (365 nm): fluorescence emission of as-prepared powder (a and e); mechanochromic fluorescence of the letter of “A” (b) and “f” (f) was written with a spatula; the paper was erased by vapor fuming (the letter “A” (c) and “f” (g) becoming invisible); rewritable mechanochromic fluorescence of the letter of “C” (d) and “p” (h) generated with a spatula. | |
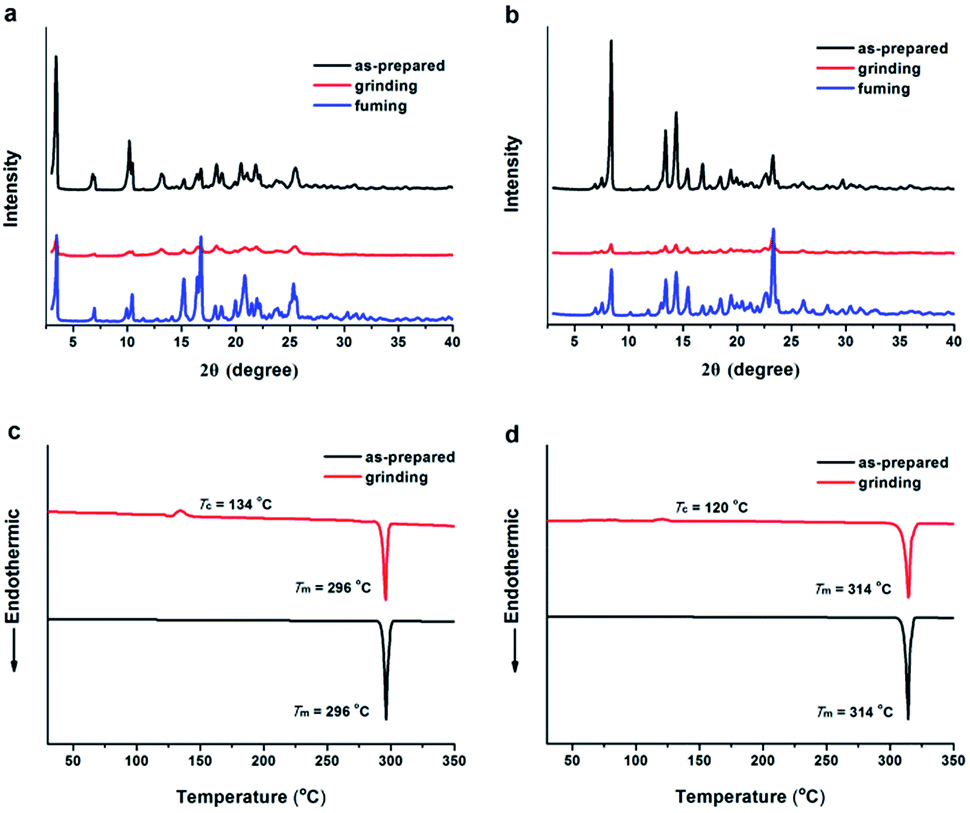
In most cases, the mechanism of the MFC behavior is attributed to the change of molecular packing modes upon external force application.1b To gain insight into the origin of MFC behaviors for TTPE-H and TTPE-CN, powder X-ray diffraction (XRD) and differential scanning calorimetry (DSC) studies were performed. According to the XRD measurements, TTPE-H and TTPE-CN exhibited different molecular aggregation structures before and after grinding treatment. As shown in Fig. 6a and b, it was clearly found that the as-synthesized samples displayed strong and sharp diffraction peaks, indicating the crystalline forms (Fig. S11 and S12, ESI†), although the single crystals of TTPE-H and TTPE-CN suitable for X-ray analysis were not obtained. However, after the as-synthesized samples were ground, their diffraction peaks were very weak, indicating that the well-ordered molecular packing modes were destroyed and disordered molecular packing or amorphous states formed. Upon fuming the ground powders with DCM vapor, the diffraction peaks of the samples came out again, this is because the disordered state changed back to a crystalline state. Furthermore, the XRD patterns of TTPE-H between the pristine and fumed samples were different, which is consistent with that of emission bands. The DSC curves upon heating procedure were shown in Fig. 6c and d, the as-synthesized samples of TTPE-H and TTPE-CN showed strong endothermic peaks at 296 °C and 314 °C, which corresponded to their melt points, respectively. By contrast, when amorphous TTPE-H and TTPE-CN were heated, their DSC curves firstly showed an exothermic recrystallization peak assigned to the crystallization points at relatively lower temperatures (about 134 °C for TTPE-H and 120 °C for TTPE-CN) and then an endothermic melting peak at the melting points of corresponding unground crystals. These results mean that the crystalline and ground samples demonstrably form crystalline and amorphous states, respectively. These data strongly suggest that the MFC behaviors of TTPE-H and TTPE-CN are attributed to the phase transformation between the ordered crystalline and the disordered amorphous states, and this phenomenon is not a chemical change but a microscopic physical change that comes from the arrangement forms of molecules.
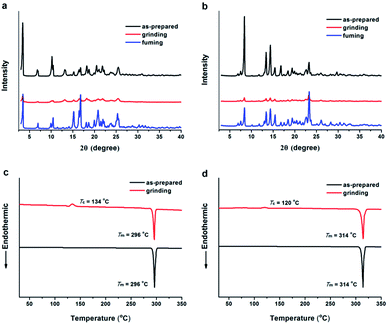 |
| | Fig. 6 XRD patterns of TTPE-H (a) and TTPE-CN (b) in different solid-states: as-prepared, grinding and fuming. DSC curves of TTPE-H (c) and TTPE-CN (d) in the ground and the crystalline states. | |
Conclusions
Two new thiazole-based β-ketoiminate boron complexes containing tetraphenylethene, namely TTPE-H and TTPE-CN, were designed and successfully synthesized. It was found that the two compounds gave TICT properties from the electron donor tetraphenylethene to the electron acceptor β-iminoenolate boron moiety. Furthermore, the two compounds exhibited strong AIE activities and significant MFC properties. The spectroscopic properties and morphological structures were reversibly and repeatedly achieved upon grinding or fuming. It is interesting that the MFC activity of TTPE-CN was considerably enhanced relative to TTPE-H by introduction of cyano groups into the molecular structure. We proposed that the twisted conformation of these compounds is a key factor influencing their MFC activity. The simple molecular structure, easy synthetic procedure, visually defined on/off contrast, and good reproducibility are great advantages for applications in recording, pressure-sensing, and light-emission.
Experimental
Materials and measurements
1H and 13C NMR spectra were recorded with a mercury plus instrument at 400 and 100 MHz by using CDCl3 as the solvents. MS spectra were recorded on an Agilent Technologies 6224 spectrometer and MALDI-TOF MS Performance (Shimadzu, Japan). Elemental analyses were performed with a Perkin-Elmer 240C elemental analyzer by investigation of C, H, and N. UV-visible spectra were collected on a Shimadzu UV-2550 spectrophotometer. Fluorescence measurements were performed on a Cary Eclipse Fluorescence Spectrophotometer. The absolute fluorescence quantum yields for TTPE-H and TTPE-CN were measured on an Edinburgh FLS920 steady state spectrometer using an integrating sphere. The calculations for TTPE-H and TTPE-CN was based on the density functional theory (DFT) and performed at the B3LYP/6-31G(d) level, employing the Gaussian 09W suit of programs. Fluorescence lifetime was measured by time-correlated single photon counting method (Edinburgh FLS920 spectrophotometer). Dynamic light scattering (DLS) measurements were performed on the BI-200SM Laser Light Scattering System (Brookhaven). Differential scanning calorimetry (DSC) curves were was carried out using a DSC/DTA-TG instrument (STA 449F3 Jupiter Netzsch, Germany) at a heating rate of 10 °C min−1 under N2 atmosphere. XRD patterns were obtained on a Bruker D8 Focus Powder X-ray diffraction instrument.
Preparation of the samples for AIE study
The water/THF mixtures with different water fractions were prepared by slowly adding distilled water into solutions of the target molecules in THF under sonication at room temperature, the concentration was maintained at 1.0 × 10−5 M. The fluorescence emission spectral measurement of the mixture was performed immediately.
Preparation of the samples for mechanofluorochromism study
The ground powders were prepared by grinding the as-synthesized powders with a pestle in the mortar. The fumed samples were obtained by fuming the ground powders with CH2Cl2 for 2 min. Ground samples were prepared by grinding using a pestle and mortar.
Synthesis
Toluene was distilled over sodium and benzophenone. CH2Cl2 was dried with sodium hydride. The other chemicals were used as received without further purification. 2-(Benzo[d]thiazol-2-yl)acetonitrile 5 was synthesized according to the literatures.30
Methyl 4-(1,2,2-triphenylvinyl)benzoate (3). A solution of 1 (10.00 g, 2.98 mmol), 2 (6.50 g, 3.61 mmol), Pd(PPh3)4 (100 mg, 0.087 mmol), K2CO3 (7.50 g, 54.27 mmol) in toluene/H2O (150 mL, v/v = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was heated to reflux under nitrogen atmosphere for 24 h. Then the mixture was cooled to room temperature and the organic layer was separated, the aqueous layer extracted with CH2Cl2 (2 × 100 mL), the combined organic layer dried over anhydrous Na2SO4, and evaporated to dryness. The crude product was purified by column chromatography (silica gel; petroleum ether/CH2Cl2, v/v = 1/1) to give a light yellow solid (11.40 g), yield 98%. Mp 144.0–146.0 °C. 1H NMR (400 MHz, CDCl3) δ = 7.81–7.78 (m, 2H), 7.13 (s, 11H), 7.05–7.03 (m, 6H), 3.88 (s, 3H) (Fig. S16†); 13C NMR (100 MHz, CDCl3) δ = 166.99, 148.81, 143.23, 143.14, 143.08, 142.45, 139.95, 131.32, 131.27, 128.99, 127.91, 127.84, 127.71, 126.87, 126.73, 51.97 (Fig. S17†). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C28H23O2 391.1698; found 391.1694 (Fig. S18†). Anal. calcd for C28H22O2: C 86.13, H 5.68; found: C 86.25, H 5.49.
:1) was heated to reflux under nitrogen atmosphere for 24 h. Then the mixture was cooled to room temperature and the organic layer was separated, the aqueous layer extracted with CH2Cl2 (2 × 100 mL), the combined organic layer dried over anhydrous Na2SO4, and evaporated to dryness. The crude product was purified by column chromatography (silica gel; petroleum ether/CH2Cl2, v/v = 1/1) to give a light yellow solid (11.40 g), yield 98%. Mp 144.0–146.0 °C. 1H NMR (400 MHz, CDCl3) δ = 7.81–7.78 (m, 2H), 7.13 (s, 11H), 7.05–7.03 (m, 6H), 3.88 (s, 3H) (Fig. S16†); 13C NMR (100 MHz, CDCl3) δ = 166.99, 148.81, 143.23, 143.14, 143.08, 142.45, 139.95, 131.32, 131.27, 128.99, 127.91, 127.84, 127.71, 126.87, 126.73, 51.97 (Fig. S17†). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C28H23O2 391.1698; found 391.1694 (Fig. S18†). Anal. calcd for C28H22O2: C 86.13, H 5.68; found: C 86.25, H 5.49.
1,1-Difluoro-3-(4-(1,2,2-triphenylvinyl)phenyl)-1H-benzo[4,5]thiazolo[3,2-c][1,3,2]oxazaborinin-10-ium-1-uide (TTPE-H). A mixture of 3 (1.50 g, 3.84 mmol) and 4 (0.86 g, 5.76 mmol) was dissolved in dry toluene (60 mL), and then NaH (60%, 1.15 g, 28.77 mmol) was added. The mixture was refluxed with stirring for 24 h under an atmosphere of nitrogen. After cooling to room temperature, the mixture was acidified with dilute HCl and extracted with CH2Cl2. After solvent removal, the solid residue was dried under vacuum. The obtained solid was dissolved in CH2Cl2 (50 mL), and then DIEA (4.0 mL, 3.13 g, 24.22 mmol) was added to the solution, followed by dropwisely addition of boron trifluoride diethyl etherate (2.90 mL, 3.26 g, 22.97 mmol). The mixture was stirred for 24 h under an atmosphere of nitrogen at room temperature. Then the mixture was poured into the water, and extracted with CH2Cl2. After solvent removal, the crude product was purified by column chromatography (silica gel, CH2Cl2/petroleum ether, v/v = 3/2), affording a yellowish green solid (1.32 g). Yield 62%. Mp 295.0–297.0 °C. 1H NMR (400 MHz, CDCl3) δ = 8.18 (d, J = 8.0 Hz, 1H), 15.53 (t, J = 8.0 Hz, J = 8.4 Hz, 3H), 7.59 (t, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.44 (t, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.15–7.12 (m, 11H), 7.07–7.03 (m, 6H), 6.60 (s, 1H) (Fig. S19†); 13C NMR (100 MHz, CDCl3) δ = 167.92, 166.68, 148.27, 143.27, 143.19, 143.08, 142.59, 139.92, 131.73, 131.35, 131.30, 130.77, 128.26, 128.09, 127.92, 127.85, 127.70, 126.97, 126.76, 126.67, 125.86, 121.77, 118.71, 89.15 (Fig. S20†). HRMS (MALDI-TOF) m/z: [M]+ calcd for C35H24BF2NOS 555.1640; found 555.1637 (Fig. S21†). Anal. calcd for C35H24BF2NOS: C 75.68, H 4.36, N 2.52; found: C 75.52, H 4.22, N 2.65.
4-Cyano-1,1-difluoro-3-(4-(1,2,2-triphenylvinyl)phenyl)-1H-benzo[4,5]thiazolo[3,2-c][1,3,2]oxazaborinin-10-ium-1-uide (TTPE-CN). By following the synthetic procedure for TTPE-H, TTPE-CN was synthesized by using 3 (1.50 g, 3.84 mmol), 5 (1.00 g, 5.76 mmol) and NaH (60%, 1.15 g, 28.80 mmol) as the reagents. The mixture was refluxed with stirring for 48 h under an atmosphere of nitrogen. Then the intermediate was reacted with DIEA (4.0 mL, 3.13 g, 24.22 mmol) and boron trifluoride diethyl etherate (2.90 mL, 3.26 g, 22.97 mmol) in CH2Cl2. The crude product was purified by column chromatography (silica gel, CH2Cl2/petroleum ether, v/v = 2/3), affording a yellow solid (0.67 g). Yield 30%. Mp 313.0–315 °C. 1H NMR (400 MHz, CDCl3) δ = 8.22 (d, J = 8.4 Hz, 1H), 8.02 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.4 Hz, 1H), 7.69 (t, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.58 (t, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 7.18–7.13 (m, 9H), 7.08–7.03 (m, 6H) (Fig. S22†); 13C NMR (100 MHz, CDCl3) δ = 174.26, 150.47, 143.56, 143.03, 142.85, 142.76, 139.52, 131.82, 131.33, 131.27, 131.23, 129.22, 128.94, 128.86, 128.55, 128.05, 127.98, 127.71, 127.25, 127.05, 126.92, 122.15, 119.23, 115.71, 80.05 (Fig. S23†). HRMS (MALDI-TOF) m/z: [M]+ calcd for C36H23BF2N2OS 580.1592; found 580.1588 (Fig. S24†). Anal. calcd for C36H23BF2N2OS: C 74.49, H 3.99, N 4.83; found: C 74.58, H 3.85, N 4.95.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NNSFC, No. 21362027 and 21662028), Young Research Foundation of Qinghai University (2012-QGY-1) and the High level talent project of Qinghai University.
Notes and references
-
(a) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed;
(b) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC;
(c) C. Löwe and C. Weder, Adv. Mater., 2002, 14, 1625–1629 CrossRef;
(d) A. Pucci, F. D. Cuia, F. Signori and G. Ruggeri, J. Mater. Chem., 2007, 17, 783–790 RSC;
(e) D.-H. Park, J. Hong, I. S. Park, C. W. Lee and J.-M. Kim, Adv. Funct. Mater., 2014, 24, 5186–5193 CrossRef CAS;
(f) X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC;
(g) N. Mizoshita, T. Tani and S. Inagaki, Adv. Mater., 2012, 24, 3350–3355 CrossRef CAS PubMed.
-
(a) M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759–760 CrossRef CAS PubMed;
(b) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed;
(c) Y. Sagara, S. Yamane, T. Mutai, K. Araki and T. Kato, Adv. Funct. Mater., 2009, 19, 1869–1875 CrossRef CAS;
(d) C. E. Olson, M. J. R. Previte and J. T. Fourkas, Nat. Mater., 2002, 1, 225–228 CrossRef CAS PubMed;
(e) S. J. Lim, B. K. An, S. D. Jung, M. A. Chung and S. Y. Park, Angew. Chem., Int. Ed., 2004, 43, 6346–6350 CrossRef CAS PubMed;
(f) S. Hirata and T. Watanabe, Adv. Mater., 2006, 18, 2725–2729 CrossRef CAS.
-
(a) D. Yan and D. G. Evans, Mater. Horiz., 2014, 1, 46–57 RSC;
(b) M. S. Kwon, J. Gierschner, J. Seo and S. Y. Park, J. Mater. Chem. C, 2014, 2, 2552–2557 RSC;
(c) X. Cheng, D. Li, Z. Y. Zhang, H. Y. Zhang and Y. Wang, Org. Lett., 2014, 16, 880–883 CrossRef CAS PubMed;
(d) Y. Ooyama and Y. Harima, J. Mater. Chem., 2011, 21, 8372–8380 RSC;
(e) Y. J. Zhang, J. W. Sun, G. L. Zhuang, M. Ouyang, Z. W. Yu, F. Cao, G. X. Pan, P. S. Tang, C. Zhang and Y. G. Ma, J. Mater. Chem. C, 2014, 2, 195–200 RSC;
(f) C. Li, X. Luo, W. Zhao, C. Li, Z. Liu, Z. Bo, Y. Dong, Y. Q. Dong and B. Z. Tang, New J. Chem., 2013, 37, 1696–1699 RSC;
(g) M. Tanioka, S. Kamino, A. Muranaka, Y. Ooyama, H. Ota, Y. Shirasaki, J. Horigome, M. Ueda, M. Uchiyama, D. Sawada and S. Enomoto, J. Am. Chem. Soc., 2015, 137, 6436–6439 CrossRef CAS PubMed;
(h) A. Balch, Angew. Chem., Int. Ed., 2009, 48, 2641–2644 CrossRef CAS PubMed;
(i) M. Teng, X. Jia, S. Yang, X. Chen and Y. Wei, Adv. Mater., 2012, 24, 1255–1261 CrossRef CAS PubMed.
-
(a) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS PubMed;
(b) T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS PubMed;
(c) H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601–3609 Search PubMed;
(d) X. Zhu, R. Liu, Y. Li, H. Huang, Q. Wang, D. Wang, X. Zhu, S. Liu and H. Zhu, Chem. Commun., 2014, 50, 12951–12954 RSC.
-
(a) S. Varughese, J. Mater. Chem. C, 2014, 2, 3499–3516 RSC;
(b) W. Z. Yuan, Y. G. Gong, S. M. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. W. Lu, Z. M. Wang, R. R. Hu, N. Xie, H. S. Kwok, Y. M. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24, 1518–1528 CrossRef CAS;
(c) Y. Y. Gong, Y. R. Zhang, W. Z. Yuan, J. Z. Sun and Y. M. Zhang, J. Phys. Chem. C, 2014, 118, 10998–11005 CrossRef CAS;
(d) Y. Y. Gong, Y. Q. Tan, J. Liu, P. Lu, C. F. Feng, W. Z. Yuan, Y. W. Lu, J. Z. Sun, G. F. He and Y. M. Zhang, Chem. Commun., 2013, 49, 4009–4011 RSC;
(e) J. Gierschner and S. Y. Park, J. Mater. Chem. C, 2013, 1, 5818–5832 RSC.
-
(a) Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed;
(b) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175–5178 CrossRef CAS PubMed.
-
(a) X. Zhang, Z. Chi, J. Zhang, H. Li, B. Xu, X. Li, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. B, 2011, 115, 7606–7611 CrossRef CAS PubMed;
(b) X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, W. Zhou, S. Liu, Y. Zhang and J. Xu, Chem.–Asian J., 2011, 6, 808–811 CrossRef CAS PubMed;
(c) W. Liu, Y. Wang, M. Sun, D. Zhang, M. Zheng and W. Yang, Chem. Commun., 2013, 49, 6042–6044 RSC;
(d) M. Zheng, D. Zhang, M. Sun, Y. Li, T. Liu, S. Xue and W. Yang, J. Mater. Chem. C, 2014, 2, 1913–1920 RSC;
(e) L. Bu, M. Sun, D. Zhang, W. Liu, Y. Wang, M. Zheng, S. Xue and W. Yang, J. Mater. Chem. C, 2013, 1, 2028–2035 RSC;
(f) Y. Wang, W. Liu, L. Bu, J. Li, M. Zheng, D. Zhang, M. Sun, Y. Tao, S. Xue and W. Yang, J. Mater. Chem. C, 2013, 1, 856–862 RSC;
(g) Y. J. Dong, B. Xu, J. B. Zhang, X. Tan, L. J. Wang, J. L. Chen, H. G. Lv, S. P. Wen, B. Li, L. Ye, B. Zou and W. J. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS PubMed;
(h) Y. J. Dong, J. B. Zhang, X. Tan, L. J. Wang, J. L. Chen, B. Li, L. Ye, B. Xu, B. Zou and W. J. Tian, J. Mater. Chem. C, 2013, 1, 7554–7559 RSC.
- X. Luo, J. Li, C. Li, L. Heng, Y. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261–3265 CrossRef CAS PubMed.
-
(a) H. Li, Z. Chi, B. Xu, X. Zhang, X. Li, S. Liu, Y. Zhang and J. Xu, J. Mater. Chem., 2011, 21, 3760–3767 RSC;
(b) C. P. Ma, B. J. Xu, G. Y. Xie, J. J. He, X. Zhou, B. Y. Peng, L. Jiang, B. Xu, W. J. Tian, Z. G. Chi, S. W. Liu, Y. Zhang and J. R. Xu, Chem. Commun., 2014, 50, 7374–7377 RSC;
(c) X. Y. Shen, Y. J. Wang, E. Zhao, W. Z. Yuan, Y. Liu, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2013, 117, 7334–7347 CrossRef CAS;
(d) W. Z. Yuan, Y. Q. Tan, Y. Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. F. Feng, H. H.-Y. Sung, Y. W. Lu, I. D. Williams, J. Z. Sun, Y. M. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed;
(e) Q. Lu, X. Li, J. Li, Z. Yang, B. Xu, Z. Chi, J. Xu and Y. Zhang, J. Mater. Chem. C, 2015, 3, 1225–1234 RSC;
(f) M. P. Aldred, G. F. Zhang, C. Li, G. Chen, T. Chen and M. Q. Zhu, J. Mater. Chem. C, 2013, 1, 6709–6718 RSC.
-
(a) J. Mei, J. Wang, A. Qin, H. Zhao, W. Yuan, Z. Zhao, H. H. Y. Sung, C. Deng, S. Zhang, I. D. Williams, J. Z. Sun and B. Z. Tang, J. Mater. Chem., 2012, 22, 4290–4298 RSC;
(b) J. Shi, N. Chang, C. Li, J. Mei, C. Deng, X. Luo, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675–10677 RSC.
-
(a) S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed;
(b) Y. Sagara, A. Lavrenova, A. Crochet, Y. C. Simon, K. M. Fromm and C. Weder, Chem.–Eur. J., 2016, 22, 4374–4378 CrossRef CAS PubMed;
(c) J. Kunzelman, M. Kinami, B. R. Crenshaw, J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119–122 CrossRef CAS;
(d) C. Dou, D. Chen, J. Iqbal, Y. Yuan, H. Zhang and Y. Wang, Langmuir, 2011, 27, 6323–6329 CrossRef CAS PubMed.
-
(a) G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed;
(b) G. Zhang, J. P. Singer, S. E. Kooi, R. E. Evans, E. L. Thomas and C. L. Fraser, J. Mater. Chem., 2011, 21, 8295–8299 RSC;
(c) P. Galer, R. C. Korošec, M. Vidmar and B. Šket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS PubMed;
(d) T. Liu, A. D. Chien, J. Lu, G. Zhang and C. L. Fraser, J. Mater. Chem., 2011, 21, 8401–8408 RSC;
(e) N. D. Nguyen, G. Zhang, J. Lu, A. E. Sherman and C. L. Fraser, J. Mater. Chem., 2011, 21, 8409–8415 RSC.
-
(a) R. Yoshii, K. Suenaga, K. Tanaka and Y. Chujo, Chem.–Eur. J., 2015, 21, 7231–7237 CrossRef CAS PubMed;
(b) Z. Zhang, P. Xue, P. Gong, G. Zhang, J. Peng and R. Lu, J. Mater. Chem. C, 2014, 2, 9543–9551 RSC;
(c) X. Wang, Q. Liu, H. Yan, Z. Liu, M. Yao, Q. Zhang, S. Gong and W. He, Chem. Commun., 2015, 51, 7497–7500 RSC.
-
(a) J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 CrossRef CAS PubMed;
(b) Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC;
(c) A. Hepp, G. Ulrich, R. Schmechel, H. Von Seggern and R. Ziessel, Synth. Met., 2004, 146, 11–15 CrossRef CAS;
(d) D. Zhang, Y. Wen, Y. Xiao, G. Yu, Y. Liu and X. Qian, Chem. Commun., 2008, 4777–4779 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
-
(a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed;
(b) Y. Dong, J. W. Y. Lam, A. Qin, Z. Li, J. Sun, H. H.-Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40–42 RSC;
(c) J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS PubMed;
(d) J. Huang, R. Tang, T. Zhang, Q. Li, G. Yu, S. Xie, Y. Liu, S. Ye, J. Qin and Z. Li, Chem.–Eur. J., 2014, 20, 5317–5326 CrossRef CAS PubMed.
-
(a) G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS PubMed;
(b) K. Umezawa, Y. Nakamura, H. Makino, D. Citterio and K. Suzuki, J. Am. Chem. Soc., 2008, 130, 1550–1551 CrossRef CAS PubMed;
(c) W. Zhao and E. M. Carreira, Angew. Chem., Int. Ed., 2005, 44, 1677–1679 CrossRef CAS PubMed;
(d) B. Domercq, C. Grasso, J.-L. Maldonado, M. Halik, S. Barlow, S. R. Marder and B. Kippelen, J. Phys. Chem. B, 2004, 108, 8647–8651 CrossRef CAS;
(e) G. M. Fischer, E. Daltrozzo and A. Zumbusch, Angew. Chem., Int. Ed., 2011, 50, 1406–1409 CrossRef CAS PubMed;
(f) X. Liu, X. Zhang, R. Lu, P. Xue, D. Xu and H. Zhou, J. Mater. Chem., 2011, 21, 8756–8765 RSC.
-
(a) R. Yoshii, A. Nagai, K. Tanaka and Y. Chujo, Chem.–Eur. J., 2013, 19, 4506–4512 CrossRef CAS PubMed;
(b) R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, Chem.–Eur. J., 2014, 20, 8320–8324 CrossRef CAS PubMed;
(c) R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131–18139 CrossRef CAS PubMed;
(d) R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CrossRef CAS.
-
(a) Y. Wu, Z. Li, Q. Liu, X. Wang, H. Yan, S. Gong, Z. Liu and W. He, Org. Biomol. Chem., 2015, 13, 5775–5782 RSC;
(b) S. Gong, Q. Liu, X. Wang, B. Xia, Z. Liu and W. He, Dalton Trans., 2015, 44, 14063–14070 RSC;
(c) Q. Liu, X. Wang, H. Yan, Y. Wu, Z. Li, S. Gong, P. Liu and Z. Liu, J. Mater. Chem. C, 2015, 3, 2953–2959 RSC.
-
(a) L. Quan, Y. Chen, X.-J. Lv and W.-F. Fu, Chem.–Eur. J., 2012, 18, 14599–14604 CrossRef CAS PubMed;
(b) Y. Kubota, S. Tanaka, K. Funabiki and M. Matsui, Org. Lett., 2012, 14, 4682–4685 CrossRef CAS PubMed;
(c) Y. Yang, X. Su, C. N. Carroll and I. Aprahamian, Chem. Sci., 2012, 3, 610–613 RSC.
- L. Zhou, D. Xu, H. Gao, A. Han, Y. Yang, C. Zhang, X. Liu and F. Zhao, RSC Adv., 2016, 6, 69560–69568 RSC.
- G. Jones II, W. R. Jackson, C. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294–300 CrossRef.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- S. Nad and H. Pal, J. Phys. Chem. A, 2001, 105, 1097–1106 CrossRef CAS.
- F. Loiseau, S. Campagna, A. Hameurlaine and W. Dehaen, J. Am. Chem. Soc., 2005, 127, 11352–11363 CrossRef CAS PubMed.
- J. S. Yang, K. L. Liau, C. M. Wang and C. Y. Hwang, J. Am. Chem. Soc., 2004, 126, 12325–12335 CrossRef CAS PubMed.
-
(a) A. Rananaware, D. D. La and S. V. Bhosale, RSC Adv., 2015, 5, 56270–56273 RSC;
(b) A. Rananaware, D. D. La, S. M. Jackson and S. V. Bhosale, RSC Adv., 2016, 6, 16250–16255 RSC;
(c) M. H. Chua, Y. Ni, M. Garai, B. Zheng, K.-W. Huang, Q.-H. Xu, J. Xu and J. Wu, Chem.–Asian J., 2015, 10, 1631–1634 CrossRef CAS PubMed.
- P. F. Barbara, S. D. Rand and P. M. Rentzepis, J. Am. Chem. Soc., 1981, 103, 2156–2162 CrossRef CAS.
- S. Koji, K. Satoshi and N. Yukihiro, Synth. Commun., 1983, 210–212 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Photophysical data, DLS data, PL spectra, reversibility of MFC processes, 1H and 13C NMR, TOF LC/MS, or MALDI-TOF MS, fluorescence microscopy images of target molecules. See DOI: 10.1039/c6ra25871a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Aixia Hana,
Lin Zhoua,
Chao Zhanga,
Yan Yanga and
Wenlin Lia
*a,
Aixia Hana,
Lin Zhoua,
Chao Zhanga,
Yan Yanga and
Wenlin Lia