
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermally robust: triarylsulfonium ionic liquids stable in air for 90 days at 300 °C†
Benjamin Siua,
Cody G. Cassityb,
Adela Bencheab,
Taylor Hamby b,
Jeffrey Hendrichb,
Katie J. Stricklandb,
Andrzej Wierzbickia,
Richard E. Sykorab,
E. Alan Saltera,
Richard A. O'Brienb,
Kevin N. West*a and
James H. Davis Jr.*b
b,
Jeffrey Hendrichb,
Katie J. Stricklandb,
Andrzej Wierzbickia,
Richard E. Sykorab,
E. Alan Saltera,
Richard A. O'Brienb,
Kevin N. West*a and
James H. Davis Jr.*b
aDepartment of Chemical & Biomolecular Engineering, University of South Alabama, Mobile, Alabama 36688, USA. E-mail: kevinwest@southalabama.edu
bDepartment of Chemistry, University of South Alabama, Mobile, Alabama 36688, USA. E-mail: jdavis@southalabama.edu
First published on 23rd January 2017
Abstract
Select triarylsulfonium salts constitute ionic liquids with outstanding long-term, high-temperature aerobic stability (no mass loss in 90 days at 300 °C in air), making them among the most thermally stable organic materials known. A detailed analysis of their thermophysical properties reveals that lowering melting points in these salts by increasing ion size or lowering ion symmetry cannot be assumed, but remains an iterative process.
Introduction
The utilization of ionic liquids (ILs) in a range of energy applications has recently been reviewed by MacFarlane et al.1 Among these are uses – thermal energy storage, for example – in which long-term, high-temperature stability on the part of the ionic liquid is absolutely essential. Likewise, their use as high-performance lubricants or lubricant additives2 is similarly dependent on their remaining stable towards oxidation and high temperatures for extended periods, as will be their utility as additives (plasticizers, anti-static agents) to high-performance polymers,3 and their suitability as solvents for high-temperature materials synthesis.4 Yet, while ionic liquids have been widely touted as thermally stable, the unfortunate fact is that most are commonly unstable at high temperatures for extended periods of time.5 Although short-term stability (minutes to perhaps hours) in the 200–300+ °C range can be achieved, ILs which remain stable at such temperatures for days, weeks, or longer have proven elusive.In 2013, forearmed with knowledge of the mechanisms by which typical IL-ions thermally decompose,5 we set out to design/identify ions which would out-perform species currently in widespread use. We opted to first focus our efforts on cations, since it seemed to us on the basis of prior work that they were (especially in comparison to anions such as Tf2N−, BETI−, and PF6−) the thermal Achilles' heel of most ILs.5 Those initial efforts culminated in the discovery that ionic liquids pairing the Tf2N− anion with select tetraarylphosphonium (TAP) cations manifest thermal stabilities far superior to salts of typical imidazolium-, quaternary ammonium, or tetralkylphosphonium cations paired with Tf2N− or other anions.6 Indeed, insofar as we can determine, several of those perarylated phosphonium salts are the most thermally stable organic-ion ionic liquids/molten salts heretofore described, and even more recent work buttresses that conclusion.7
It was our postulate that we could achieve the foregoing favourable outcome by using only aryl moieties as the structural points-of-contact between the organic components of the cation and its heteroatom locus of positive charge. We reasoned that by doing so we would circumvent the two dominant mechanisms by which organic cations most commonly thermally decompose: the SN2 or E2 removal of an organic group from the heteroatom centre of the cation.5 Note that neither SN2 nor E2 reactions are generally observed with aryl substrates. Additionally, detailed studies carried out in the early 1960s pertaining to the thermal stability of organic compounds pointed to aromatic moieties as being particularly stable.8
Wishing to determine if achieving increased thermal stability through cation per-arylation could be extended to species with other heteroatoms at their core, we turned our attention to salts based upon triarylsulfonium cations.9 We can now report that Tf2N− and BETI− salts (Fig. 1) of carefully chosen representatives of the genre can be added to the list of ILs of superior thermal stability. Better yet, some of the new salts have Tm values <100 °C, one being only slightly above normal room temperature.
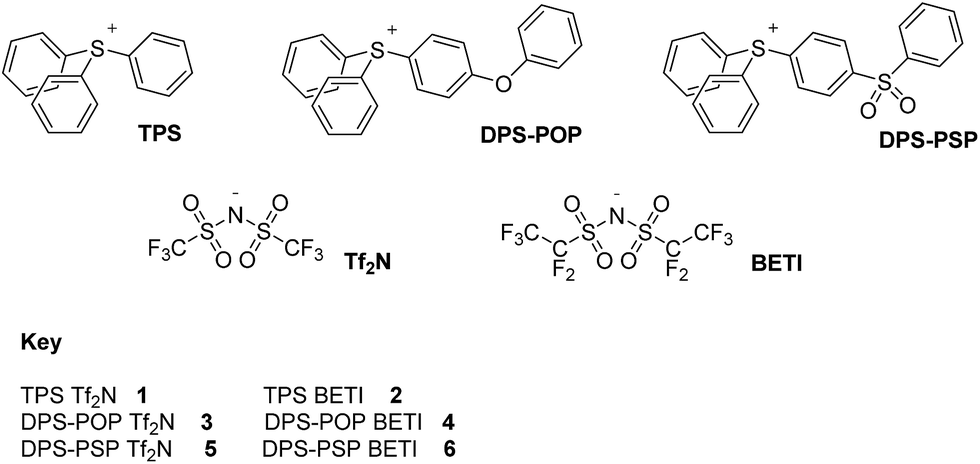 | ||
| Fig. 1 Cations and anions of new ILs 1–6. Acronyms: TPS – triphenylsulfonium; DPS-POP – diphenyl(phenoxyphenyl)sulfonium; DPS-PSP – diphenyl(phenylsulfonylphenyl)sulfonium; Tf2N− – bis(trifluoromethanesulfonyl)amide; BETI – bis(pentafluoroethanesulfonyl)amide. | ||
Experimental
Background
While a number of synthetic routes to the parent triphenylsulfonium cation and its various congeners have been developed,9 one which is quite convenient, and which was used in the present work, is that shown in Scheme 1.10 As is apparent from the scheme, triarylsulfonium cations prepared in this fashion are (by default) isolated as TfO− salts. Those of the triphenylsulfonium and diphenyl(phenoxyphenyl)sulfonium cations are known materials, while we believe DPS-PSP to be original to this report. Its synthesis (Scheme 2) involves the preparation of a (previously known) diphenylsulfonium(diphenylthioether) triflate precursor, followed by oxidation of the thioether sulfur using 30% aqueous H2O2 in a water–CH2Cl2 biphasic system. Note that this oxidation step can be performed prior or subsequent to the exchange of the TfO− anion for Tf2N− or BETI− (vide infra).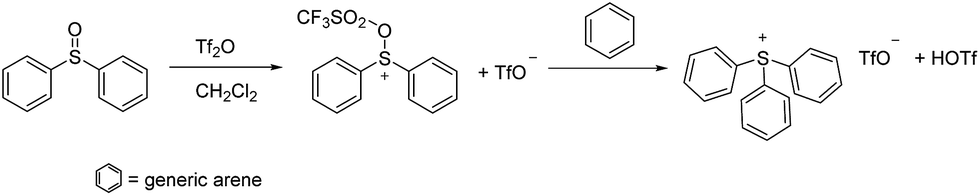 | ||
| Scheme 1 Common synthesis of triarylsulfonium triflate salts. | ||
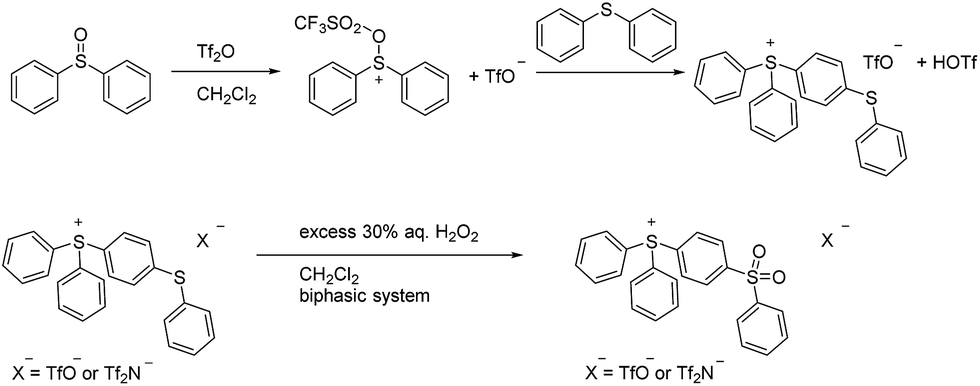 | ||
| Scheme 2 Synthesis of IL 5. | ||
In this study we elected to evaluate perarylsulfonium salts of the Tf2N− and BETI− anions in lieu of the initially isolated TfO− salts. Our decision was grounded in our earlier work with perarylphosphonium cations.6,7 It was done using Tf2N− salts, that anion having been chosen at the time because it is generally regarded (after PF6−) as the most thermally stable anion apt to form ion pairs with low (or relatively low) melting points.5 Hence, its use in the present case would (it was our expectation) allow us to make salts with melting points as low as might be achievable, and it would allow us to directly compare thermal stability data on the sulfonium salts to that of their phosphonium counterparts. In turn, the BETI− series of salts was prepared because we had ready access to a stock of this relatively hard-to-acquire material, thus it seemed to be both an easy and useful study to undertake. Finally, we chose to defer (for the moment) a thermal stability study on the triflate salts of the sulfonium cations because the literature seems less-than-clear on the relative thermal stability characteristics of this anion,5 so it seemed appropriate to undertake a separate, systematic, more broadly-based study of the thermal stability of its salts with a variety of cations at a later date.
Naturally, in order to study the Tf2N− and BETI− salts of the TPS, DPS-POP, and DPS-PSP cations, they first had to be prepared. And, since the cations were initially isolated as TfO− salts, anion metathesis was required. In candour, it did not seem likely to us that the replacement of one fluorous anion by another would be a trivial exercise. Even so, we were rewarded for trying a direct approach – it proved to be remarkably straightforward. The process was essentially identical to that commonly used in IL synthesis when replacing a halide with a fluorous anion. Specifically, the TfO− salt of the sulfonium cation was suspended in water and heated to near boiling. Methanol was then added to the rapidly stirred mixture until the salt dissolved or was nearly so. Then, to this was added a pre-made solution of KTf2N or LiBETI in water. Addition of the latter to the former solution results in the immediate formation of a milky suspension which, with continued stirring and heating, splits into two clearly separate liquid phases, the denser of which is the desired IL. That these seemingly unlikely metatheses were cleanly achieved is validated by satisfactory elemental analysis of each new salt (see ESI†), the 19F-NMR spectra (especially of the BETI− salts, see Fig. 2), and by a single crystal X-ray structure of IL 1 (Fig. 3).
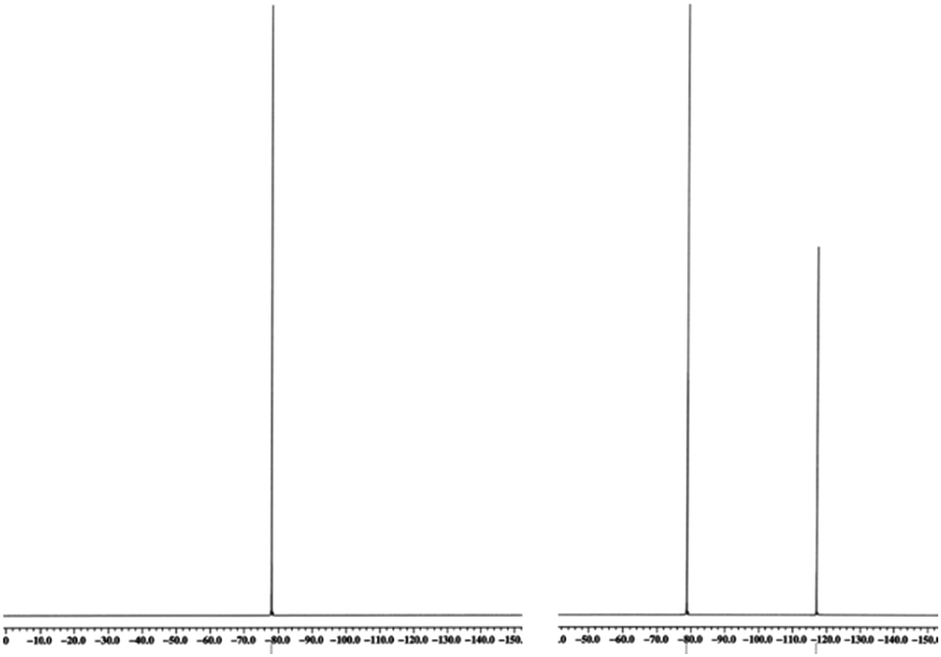 | ||
| Fig. 2 19F-NMR of Ph3S+ TfO− (left) with its single resonance from CF3, and after anion exchange to form the corresponding BETI salt with both CF2 and CF3 resonances. | ||
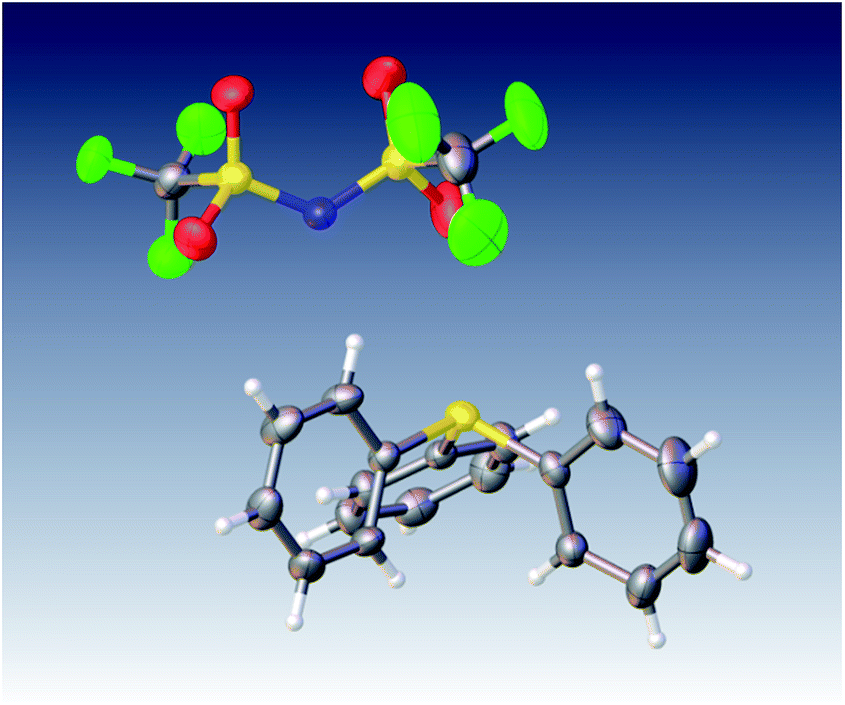 | ||
| Fig. 3 ORTEP of new IL 1. | ||
After isolation of the dense IL phase following each metathesis, the materials were dissolved in CHCl3, dried over anhydrous MgSO4, and isolated after filtration and removal of the solvent in vacuo. Compounds 1, 2, 5 and 6 were recovered as solids and compounds 3 and 4 were obtained as room temperature liquids. Compound 3 crystallized from its melt at room temperature after one month, but attempts to directly crystallize 3 and 4 from solution were unsuccessful. All of the new ILs were isolated in analytically pure form (C, H, N values all within ±0.4 percent of theoretical – see ESI†).
Materials and representative synthesis
Commercial reagents were obtained from Aldrich Chemical and used without further purification. 1H, 19F, and 13C NMR were recorded on a 500 MHz JEOL spectrometer using CDCl3 as a solvent at room temperature. All chemical shifts for 1H and 13C NMR were reported downfield using tetramethylsilane (TMS, at δ = 0.00 ppm). Elemental analyses were obtained as a commercial technical service from Atlantic Microlabs, Inc., Norcross, Georgia, USA.Synthesis of DPS-PSP·Tf2N, IL 5
A 500 mL, one-neck, round-bottomed flask was charged with a magnetic stir bar and 10.11 g (0.05 mol) diphenylsulfoxide, 10.24 g (0.055 mol) diphenylsulfide, and 150 mL of dichloromethane. The flask was then fitted with a pressure equalizing dropping funnel, into which was introduced 50 mL of a 1 M solution (0.05 mol) of triflic anhydride in dichloromethane, the funnel then being sealed with a glass stopper. Stirring was commenced, and the triflic anhydride solution added dropwise to the stirred, colourless solution of diphenylsulfide and diphenylsulfone. Upon addition of the triflic anhydride, the solution of the combined reagents gradually became a dark golden colour, and a slight warming of the solution was noted.After stirring for 1 h at room temperature, 100 mL of deionized water was added, and the biphasic mixture stirred vigorously for 30 min. Stirring was stopped, and the aqueous and organic layers then separated. The dense organic layer was dried over MgSO4, which was then removed by filtration and the solvent removed in vacuo to give a golden oil which slowly crystallizes upon standing into an ivory coloured mass (18.5 g, 71.2%). The identity of the material as [Ph2S-(p-C6H4)-S-(C6H5)]OTf was confirmed by comparison of its 1H- and 13C-NMR spectra to those from a commercial sample of the same material.
In a 1 L beaker charged with a magnetic stir bar, 15.0 g of [Ph2S-(p-C6H4)-S-(C6H5)]OTf [diphenyl(diphenylthioether) trifluoromethanesulfonate] was suspended in about 150 mL of water. The suspension was heated to near boiling, during which time the salt melted and partly dissolved. Methanol was added to the stirred solution in portions of approximately 5 mL until the IL was almost completely dissolved. Then, to this rapidly stirred solution was added, in one portion, a solution of 11.0 g of KTf2N [potassium bis(trifluoromethanesulfonyl)amide] dissolved in 100 mL of water. On mixing, the combined solution instantly became milky in appearance. Heating was discontinued but stirring maintained for 4 h, during which time the milky suspension coalesced into a clear (upper) aqueous phase and a likewise clear, ivory-colored, denser ionic liquid phase. The aqueous layer was decanted, and the IL dissolved in CHCl3. After drying over anhydrous MgSO4, filtration, and removal of the solvent in vacuo, a pale yellow glass was obtained. This was used without further isolation in the next step (100% yield of anion-exchanged product was assumed).
The round-bottomed flask containing the starting salt (above) was charged with a magnetic stir bar and 200 mL of chloroform. While vigorously stirring, 50 mL of aqueous H2O2 (25 mL of 30% H2O2 diluted to 50 mL) was added in one portion. Stirring was continued overnight after which time the layers were separated, the CHCl3 phase washed with water, then dried over anhydrous MgSO4. Filtration, followed by removal of the solvent in vacuo, left the product as a pale ivory solid, 18.54 g (94%). Tm: 104.6 °C. Elemental analysis (theory; observed): C (45.68; 45.48), H (2.80; 2.82), N (2.05; 2.11). NMR: 1H (CDCl3, 500 MHz): δ 8.14–8.18 (dd, 2H), 7.88–7.92 (dd, 2H), 7.74–7.80 (m, 4H), 7.64–7.72 (m, 8H), 7.58–7.63 (m, 1H), and 7.50–7.55 (m, 2H) ppm; 13C (CDCl3, 125 MHz): δ 147.51, 139.21, 135.37, 134.42, 132.02, 131.75, 131.35, 130.25, 129.89, 129.80, 128.14, 123.20, 122.84, 121.03, 118.47, and 115.92 ppm; 19F (CDCl3, 470 MHz): δ −78.64 ppm.
Differential scanning calorimetry
Melting points, glass transitions and enthalpies of fusion were measured using Differential Scanning Calorimetry (DSC) on a TA Instruments Q2000. Entropies of fusion were calculated as the enthalpy of fusion divided by the absolute temperature. In this work, the melting points are reported as the transition from a solid state (crystalline or amorphous) to the isotropic liquid state. 5–20 mg samples were loaded into open DSC pans and heated to 110 °C for 10 minutes to remove any water absorbed from the environment or volatile contaminants. The samples were then cooled to −80 °C and heated at a ramp rate of 10 °C per minute to 300 °C. Their melting points are reported as the melting onset temperature as calculated by the analysis software. All measurements were carried out under a nitrogen atmosphere. The reported values are the average of at least 5 measurements and the standard uncertainty is calculated as the standard deviation of the mean.The material examined in this crystallographic study diffracted extremely well and was non-problematic in regards to data collection and structure analysis. The program suite SHELX was used for structure solution (XS) and least-squares refinement (XL).12 The initial structure solution was carried out using direct methods and the remaining heavy-atom atomic positions were located in difference maps. The final refinement included anisotropic displacement parameters for all non-hydrogen atoms and isotropic refinements for all H positions. Refinement was performed against F2 by weighted full-matrix least squares and semi-empirical absorption corrections were applied.
Computational methods
All calculations, including the generation of electrostatic potential energy maps, were performed with Spartan'08 (Wavefunction, Inc., Irvine, CA).13 The isolated cations and anions were optimized in the gas phase using the B3LYP density functional method REF and the 6-31G* basis set. All structures were confirmed as stable by computing analytic vibrational frequencies.13Results and discussion
Differential scanning calorimetry
Fig. 4 shows the DSC melting curves for compounds 1–3, 5 and 6, while Fig. 5 shows the DSC glass transition curves for compounds 3 and 4. The scales of these plots are significantly different. Compounds 1–3, 5 and 6 exhibit typical melting behavior. In addition to the melting event, 1 exhibits a weak glass transition at approximately −30 °C and a crystallization event at approximately −24 °C, the position of which varies slightly depending on the thermal history of the sample. Compounds 3 and 4, recovered from synthesis as room temperature liquids, were not observed to have a crystallization or melting thermal event in the DSC, only the glass transition observed for both at approximately −15 °C. The DSC scan melting point for compound 3 was obtained from the sample that crystallized at room temperature over an extended time.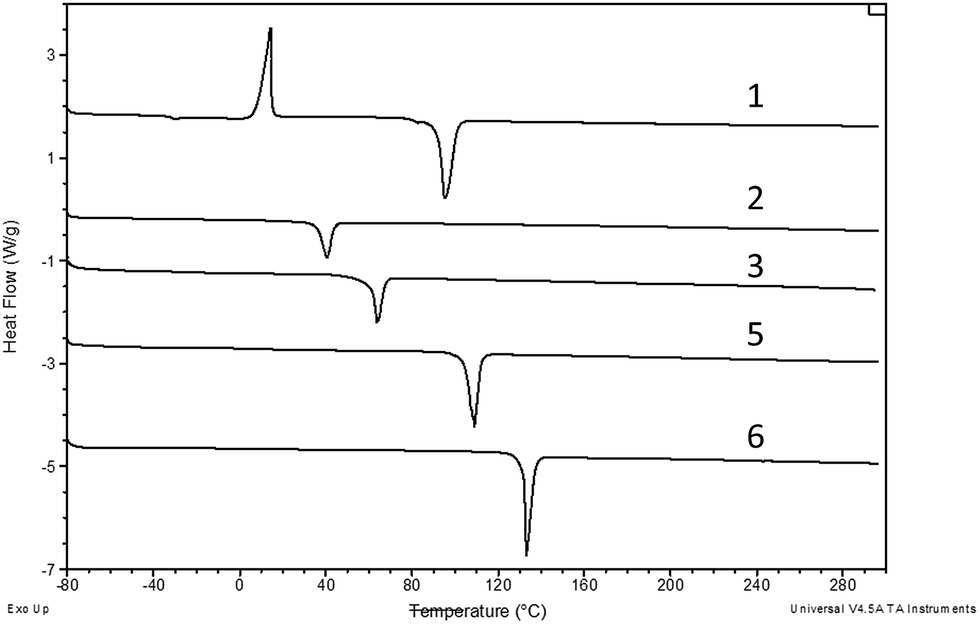 | ||
| Fig. 4 DSC curves compounds 1–3, 5, 6. Measurements were taken in open pans under nitrogen at ramp rate of 10 °C min−1. The curves have been offset but not rescaled. | ||
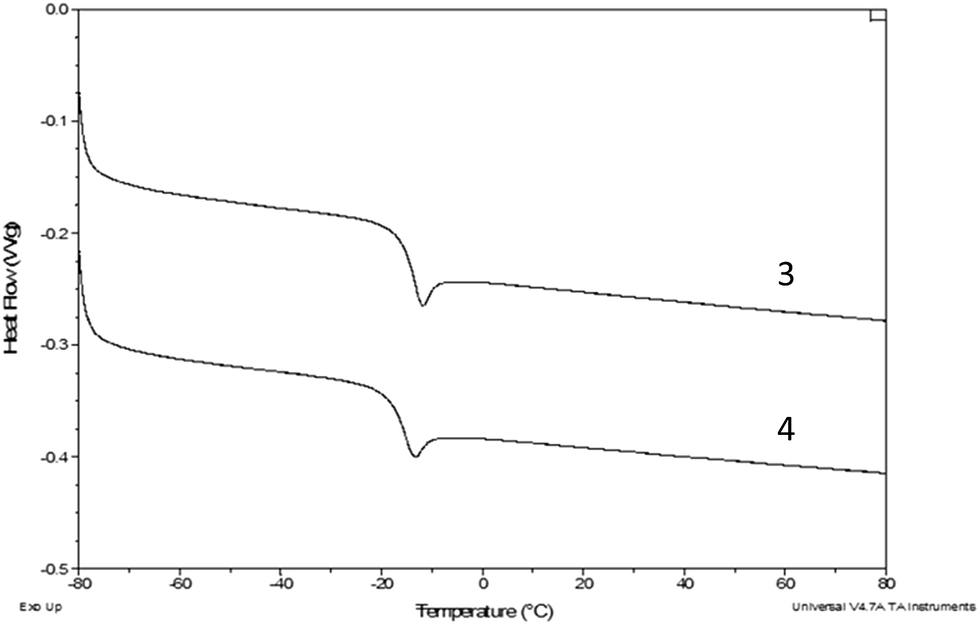 | ||
| Fig. 5 DSC curves for compounds 3 and 4. The curves have been offset, but not rescaled. | ||
Long-term thermal stability evaluation
Once in hand, samples (approximately 0.25 g) of each new IL were charged into porcelain crucibles which were then placed in a muffle furnace at 300 °C. The samples were subjected to this heating for 90 days, being removed periodically for a few minutes for the sample weight to be checked, after which time they were returned to the oven. Remarkably, in each case there was no mass loss of IL during the 90 day thermal stressing period – and little to no degree of discoloration (generally colourless to a light tea colour when observed). Multinuclear NMR spectra of the post-heat samples betrayed no discernible changes from their pre-heating appearances, leaving little doubt about the exceptional degree of thermal robustness of these new materials.While the impressive stability of these compounds is manifestly self-apparent, it is also instructive to view them in juxtaposition to data from another truly long-term IL thermal stability study, conducted by Fox and co-workers.14 In it, the stability of seven ‘classic’ ILs – [C4mim]Tf2N, [N4111]Tf2N, [C4mmim]Tf2N, [C4mpyrr]Tf2N, [C4N11pyr]Tf2N, [N1114CO2]Tf2N, and [C4mim]BETI (all ion acronyms per the nomenclature in ref. 14) – was evaluated over a 15 week period, in static air in a muffle furnace at 200 °C. While no mass loss data was reported, under these conditions only two ILs survived unchanged (per appearance, time-dependent TGA, and FTIR): [C4mim]Tf2N and [C4mim]BETI. While we note that the duration of the study by Fox et al. was 25% longer than that in the present, the temperature to which the ILs were exposed for that period of time was 100 °C lower. Significantly, the differences do not appear to be offsetting. Specifically, we found that a sample of [C4mim]Tf2N heated to 300 °C for only 96 hours was completely reduced to ash. In short, as we observed in our initial work6 with tetraarylphosphonium salts and comparisons of their thermal stability to ‘classic’ ILs under high-temperature, long-duration conditions, the stability of the present triarylsulfonium ILs is likewise clearly superior.
Analysis of computational, enthalpic and entropic data
Seeing in these results encouragement to continue future development of this line of ionic liquids, we decided to undertake a detailed analysis of their thermodynamic properties in light of their structures and insights into their electronic characteristics provided by quantum calculations. Specifically, we examined how the enthalpy and entropies of fusion of the different ion pairs affect and drive the melting point behavior, and how these may be influenced by the charge distributions within the various ions.Rather than exhibiting relatively simple trends relating to overall polarity of the ions or their symmetry (as observed with some imidazolium or alkylammonium salts) a more complex pattern emerges that is highly dependent on the interplay between the ions. Thus, insight into the effects of the cation and anion on the solid/liquid phase transitions can be obtained by inspecting the relative enthalpies and entropies of fusion, extracted from the DSC evaluations of each compound (Fig. 5) and compiled in Table 1. Note that increases in ΔHfus will serve to increase melting points and increases in ΔSfus will serve to decrease melting points (and vice versa). Five comparisons can be made: two are obtained by holding the cation constant while changing the anion (2/1 and 6/5) and three more are obtained by holding the anion constant and varying the cation (3/1, 5/1–5 and 6/2). To facilitate this analysis, the comparisons are made by examining the ratios of the melting points, enthalpies of fusion and entropies of fusion for each of the pairs. This analysis is shown in Table 2, and we note that for these ratios, a number closer to 100% represents a smaller difference.
| Cation | Anion | Tm (°C) | Tm (K) | ΔHfus (kJ mol−1) | ΔSfus (J mol−1 K−1) | |
|---|---|---|---|---|---|---|
| 1 | TPS | Tf2N− | 92.4 | 365.7 ± 0.1 | 34.3 ± 0.1 | 93.5 ± 0.4 |
| 2 | TPS | BETI | 36.3 | 309.5 ± 0.5 | 14.1 ± 0.2 | 45.6 ± 0.5 |
| 3 | DPS-POP | TfN2− | 62.7 | 335.9 ± 0.5 | 18.1 ± 0.2 | 53.8 ± 0.6 |
| 4 | DPS-POP | BETI | — | — | — | — |
| 5 | DPS-PSP | Tf2N− | 104.6 | 377.8 ± 0.1 | 30.9 ± 0.3 | 81.8 ± 0.6 |
| 6 | DPS-PSP | BETI | 132.4 | 405.6 ± 0.1 | 38.0 ± 0.2 | 93.7 ± 0.5 |
| (a) Effect of the anion: NTf2 → BETI | |||||
|---|---|---|---|---|---|
| Cation |
|
ΔHBETI/ΔHTf2N | ΔSBETI/ΔSTf2N | Effect and dominant driving force | |
| TPS | 2/1 | 85% | 41% | 49% | Tm decrease, enthalpically driven |
| DPS-PSP | 6/5 | 107% | 123% | 115% | Tm increase, enthalpically driven |
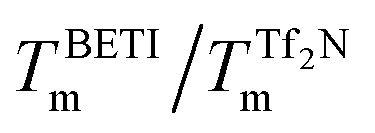
| (b) Effect of the cation: TPS → DPS-POP | |||||
|---|---|---|---|---|---|
| Anion | TDPS-POPm/TTPSm | ΔHDPS-POP/ΔHTPS | ΔSDPS-POP/ΔSTPS | Effect and dominant driving force | |
| Tf2N | 3/1 | 92% | 53% | 58% | Tm decrease, enthalpically driven |
| BETI | 4/2 | — | — | — | Tg only, no Tm observed |
| (c) Effect of cation: TPS → DPS-PSP | |||||
|---|---|---|---|---|---|
| Anion | TDPS-PSPm/TTPSm | ΔHDPS-PSP/ΔHTPS | ΔSDPS-PSP/ΔSTPS | Effect and dominant driving force | |
| Tf2N | 5/1 | 103% | 90% | 87% | Slight Tm increase, entropically driven |
| BETI | 6/2 | 131% | 270% | 205% | Significant Tm increase, enthalpically driven |
For four of these comparisons, the differences in the enthalpy of fusion is found to be the driving force for melting point difference (2/1, 6/5, 3/1 and 6/2), while for one of the comparisons (5/1), the entropy of fusion is the driving force. Each of these is discussed below. However, to aid in understanding the observed trends, we first give a brief description of the differences between the cations and between the anions, informed by the quantum chemical calculations (Fig. 6).
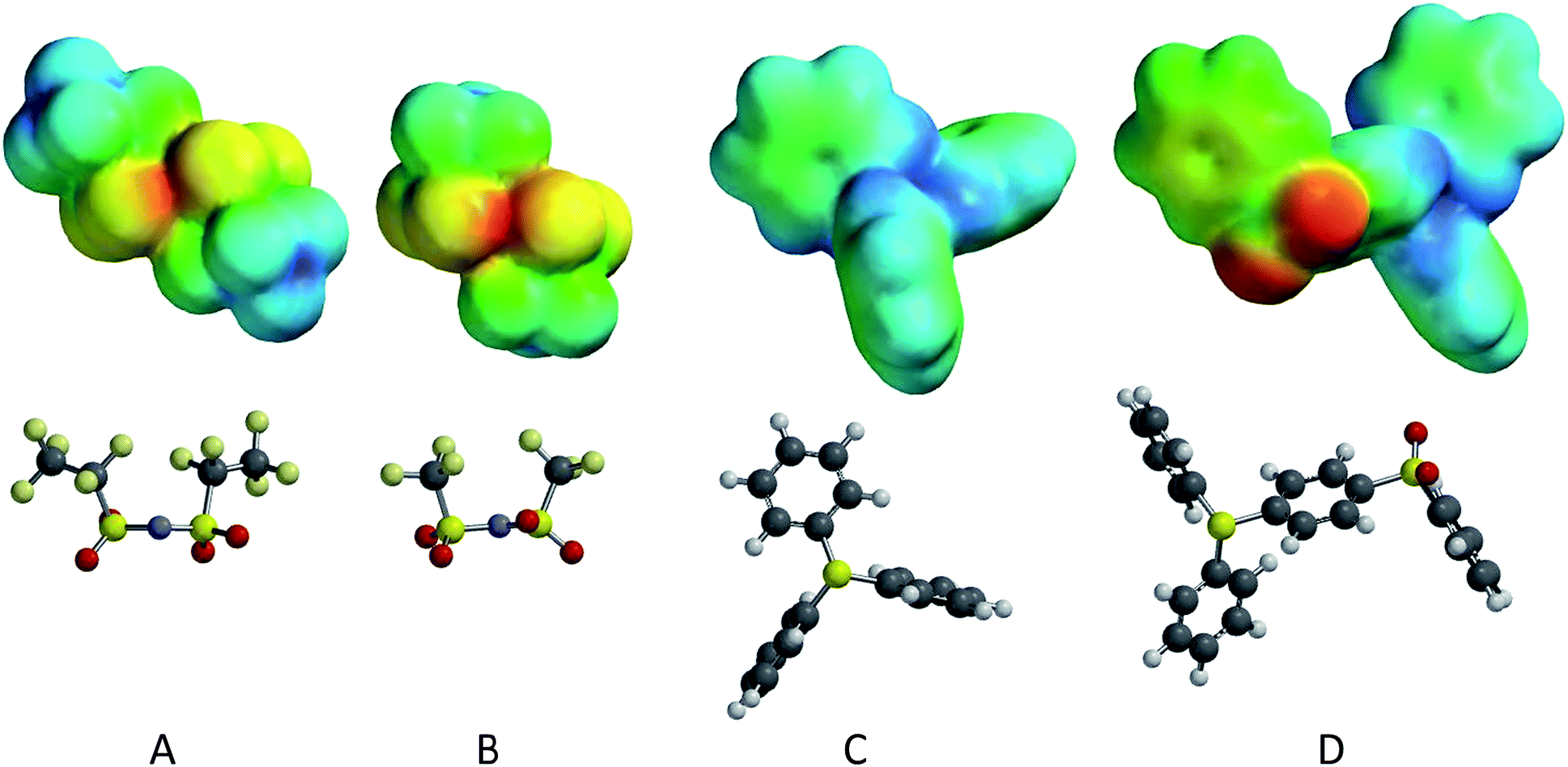 | ||
| Fig. 6 Optimized geometries and electrostatic potential energy maps for four isolated ions: (A) – [BETI] anion; (B) – [Tf2N] anion; (C) – TPS cation; (D) – DPS-PSP cation. | ||
In comparing the cations, TPS is relatively small and symmetric (the lowest energy conformation has C3 symmetry) with a significant amount of charge delocalization, as the positive charge is delocalized through the three phenyl groups with their collective electron delocalization. The electrostatic energy values range from +236.7 to +416.3 kJ mol−1, with the most positive locations near the central sulfur (typically +400 to +415 kJ mol−1), but not on the sulfur itself. A shift in these energy maxima may be caused by the lone pair of electrons on the sulfur atoms; note that there is a local energy minimum of ∼+310 kJ mol−1 in the vicinity of sulfur lone pair. The least positive regions (∼+236 kJ mol−1) broadly cover the π surfaces of the phenyl groups.
The DPS-POP cation is similar to the TPS cation; however the –O–Ph group on the third phenyl ring introduces asymmetry to the system and may inhibit packing in the solid structure. For the DPS-POP, the electrostatic energy values range from +120.0 to +397.3 kJ mol−1, also with the most positive sites near the central sulfur (typically +385 to +396 kJ mol−1) and a local minimum on the sulfur lone pair (+296 kJ mol−1). The π faces exhibit the least positive charges on the cation, with the π face of the phenoxy moiety syn to the oxygen lone pairs having values in the range of +120 to +140 kJ mol−1 (comparable to the oxygen lone pair region) and the π face anti to the oxygen lone pair having values in the range of +150 to +165 kJ mol−1. The authors note that the DPS-POP has generally lower electrostatic charge relative to the TPS, due to higher surface area and more charge delocalization.
The DPS-PSP cation also has significant charge delocalization; however, the third group on the central sulfur (–PhSO2Ph) is significantly larger, making it more polarizable, and has a much more polar moiety: the –SO2– group. The electrostatic energy values range from +8.0 to +429.9 kJ mol−1 and, similar to the other cations, the most positive locations are near the central sulfur (typically +410 to +425 kJ mol−1), but not upon the sulfur itself. Here again the possibility exists that these maxima are shifted over by the sulfur lone pair. In the vicinity of the central sulfur's lone pair, there is a local energy minimum of ∼+319 kJ mol−1. The least positive domain associated with the sulfone oxygens (ranging from +8 to +120 kJ mol−1) is far less positive than anywhere on [Ph2SPhSO2Ph], as is most of the terminal phenyl group. The π region charges range from +135 to +150 kJ mol−1 on the syn face and from +170 to +180 kJ mol−1 on the anti face (anti/syn relative to the sulfone oxygens). Furthermore, the charge distribution of the three other phenyl groups resembles those of the analogous phenyl groups on TPS.
The anions share a common structure, save that the BETI− anion has bis-pentafluoroethyl groups, while the Tf2N− anion has bis-trifluoromethyl group. The additional fluorine content on the BETI− ion serves to inductively delocalize the negative charge more so than the smaller fluorous groups on the Tf2N−. This lower charge density on the BETI− could potentially lead to weaker interactions. Additionally, the larger fluorous groups on the BETI− may allow them to align better in the solid or liquid phases, possibly interdigitating to create fluorous domains. Further insight into the differences in the anions can be obtained by analyzing the computational results for the isolated ions.
For the Tf2N− anion, the trans structure (C2 symmetry) was found to be more stable than cis by 0.82 kcal mol−1 (B3LYP/6-31G*), with a(S–N–S) = 123.9° (trans) versus 124.9° (cis). The electrostatic energy values (for trans) range from −534.2 to −215.4 kJ mol−1. The most negative sites are between the proximal oxygens (∼−534 kJ mol−1), followed by the backside of the central nitrogen atom (site of formal charge) at (∼−496 kJ mol−1). The least negative regions are found at the backsides of the –CF3 groups (∼−215 kJ mol−1).
For the BETI− anion, as above for the Tf2N− anion, the trans structure (C2 symmetry) was found to be more stable than cis by 1.59 kcal mol−1 (B3LYP/6-31G*), with a(S–N–S) = 123.9° (trans) versus 125.1° (cis). The electrostatic energy values (for trans) range from −517.9 to −184.2 kJ mol−1. Also, as above, the most negative sites are between the most proximal oxygens (∼−517 kJ mol−1), followed by the backside of the central nitrogen atom (site of formal charge) at (∼−483 kJ mol−1). The least negative regions are at the backsides of the terminal –CF3 groups (∼−184 kJ mol−1). In comparison with Tf2N−, BETI− has greater delocalization of the anionic charge with moderated (i.e., less negative) electrostatic energy potentials everywhere, which agrees with the reasoning presented above.
Having examined the electrostatic natures of the ions, these insights can now be used to understand how these natures affect the phase behavior of the various ion pairings.
Effect of the anion
For the first comparison, we examine the effect of the changing the anion from Tf2N− to BETI− while holding the cation constant (2/1 and 6/5 in Table 2(a)). Beginning with the TPS cation, we observe that as the anion is changed from Tf2N− to BETI− (1 → 2), the melting point decreases by 56 °C due to ΔHfus and ΔSfus values for 2 which are 41% and 49%, respectively, that of 1. This indicates that the change in the enthalpy of fusion is dominant driving force in the melting point. This change can be explained by the increased charge delocalization in the BETI− anion as well as its increased fluorous content, which will decrease van der Waals interactions.Surprisingly, the opposite effect on melting point is observed when cation is DPS-PSP and the anion is changed (5 → 6). As the anion is changed from Tf2N− to BETI− for this cation, the melting point increases 28 °C with ΔHfus and ΔSfus for 6 that are 123% and 115%, respectively, compared to that of 5. Therefore, this demonstrates that the increase in the enthalpy of fusion is the dominant factor in the melting point increase. This is in contrast to the observation made above for the TPS cation where the enthalpy of fusion also dominated, but leads to a melting point decrease rather than increase. The size of the DPS-PSP cation may play a role in the increase in enthalpy of fusion of the BETI− salt relative to that of the Tf2N− salt. The larger cation/larger anion combination may provide more room in the crystal lattice each ion to interact, serving to maximize the interaction and increase the energy required to melt the solid.
Effect of cation
Now we examine the effect of the cation while holding the anion constant, focusing first on changing the cation from TPS to DPS-POP for Tf2−N (1 → 3). Here we note that the Tf2N− salt of DPS-POP was eventually obtained as a solid after several months at room temperature as an oil, while the BETI− the salt was never observed to solidify. The melting point of 3 is 30 °C lower than that of 1, with ΔHfus and ΔSfus for 3 that are 53% and 58%, respectively, that of 1, demonstrating that the change in melting point is driven by the enthalpy differences. Interestingly, it would be logical to assume that the DPS-POP salt would have a lower melting point than the TPS salt based on entropic arguments, as the DPS-POP cation introduces asymmetry into the salt. However, the measurements indicate that both the enthalpy and entropy of fusion decrease, with the enthalpy change driving the melting point reduction, while the entropy change partially counteracting the enthalpic effect.Next we consider the effect of changing the cation from TPS to DPS-PSP while holding the anion constant as Tf2N− (1 → 5) and then as BETI− (2 → 6) (Table 2(c)). In comparing 1 and 5, the melting point is observed to increase by 12 °C with ΔHfus and ΔSfus values for 5 that are 90% and 87%, respectively, those of 1. Therefore, it can be determined that the driving force for this temperature increase is the decrease in the entropy of fusion, either decreasing the entropy of the liquid or increasing the entropy of the solid, as ΔSfus = Sl − Ss. Potentially, both effects could be happening. The addition of the –SO2– group in the cation could create a polar site which interacts with the anion better than the TPS cation, thereby decreasing the entropy of the liquid phase. Additionally, the decreased symmetry of the DPS-PSP cation could decrease packing efficiency in the solid phase. This decrease in packing efficiency would increase the solid entropy and also potentially decreasing the enthalpy of fusion as well, since decreased packing efficiency can prevent potential energetic interactions from being otherwise stronger. However, we note that these effects are likely small as the overall change in properties is small for this comparison.
A very different effect is observed as the cation is changed for the BETI− anion (2 → 6). While the melting point also increases, this time by 96 °C, the ΔHfus and ΔSfus values for 6 are 270% and 205%, respectively, those of 2. This indicates that the increase in the enthalpy of fusion is the driving force for the melting point increase. It is worth noting that this large increase in ΔHfus observed for the BETI− when comparing the different cations may simply be due to the low ΔHfus of 2, as the ΔHfus of 1, 5 and 6 vary by less than 25%, while the ΔHfus of 2 is significantly less than these and more similar to that of 3. The small value of ΔHfus for 2 is likely due to the pairing of the TPS cation with BETI−, each of which have significant charge delocalization (the BETI− anion more so than the Tf2N− anion, with additional fluorous content). With the introduction of the more polar and larger –PhSO2Ph group in 6, the enthalpy of fusion increases, resulting in a higher melting point.
Conclusions
Although ionic liquids are often regarded as having high thermal and oxidative stability, many of the most commonly employed cations and anions survive for only short periods of time at elevated (>250 °C) temperatures. However, previous work has established that select tetraarylphosphonium Tf2N− salts are considerably more stable, at higher temperatures, and for longer periods of time (96+ hours). In the present work we have shown that carefully conceived triarylsulfonium salts of the Tf2N− and BETI− ions are likewise thermally stable at high temperature and in air; moreover, in the work presented here, we have established that these salts remain stable for at least 90 days under these thermally demanding conditions. Also, juxtaposed with prior work by Fox and co-workers, it becomes clear that ILs 1–6 can be added to any list of the most thermally stable ionic liquids known. Better still, several of these salts can be obtained as (apparently) highly persistent supercooled liquids, or melt at temperatures only slightly above ambient, extending the practical long-duration liquidus range for ionic liquids.It is also important to note that the thermochemical analysis of the present salts provides extremely useful insights into structure–property (i.e., Tm) relationships that reflect on ion design. Specifically, it has been the accepted wisdom for some time in IL design that making ions more charge diffuse and/or less symmetric will tend to induce lower Tm values in the resulting salts. The present ΔHfus and ΔS analyses paint a much more complex picture. Specifically, that achieving lower melting points with perarylated cation salts is not as simple a matter as making them less symmetric or more charge-diffuse. Rather, the structural modifications one makes to do so may bring with them offsetting changes in enthalpic factors that can negate or even exceed in a countervailing fashion the salutary effects that these modifications are intended to elicit. In short, there is (at the moment) no way to conclude a priori that, at least for perarylated sulfonium salts, particular ion modifications or combinations will yield predictable (even if just qualitatively so) changes in the Tm of a salt. Hence, until computational approaches evolve to the point where structure–property relationships such as those seen here can be predicted, the empirical [conceive structure → synthesize → evaluate] paradigm will continue to prevail in the development of these and related materials.
Acknowledgements
This work was supported by the National Science Foundation under grant number CHE-1464740. JHD also thanks the donors of the Petroleum Research Fund, administered by the American Chemical Society, for support of this research.Notes and references
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis Jr., M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232 CAS.
- J. C. J. Bart, E. Gucciardi and S. Cavallaro, Advanced Lubricant Fluids, Series in Energy 46, Woodhead Publishing, 2013, ch. 2 and 3 Search PubMed.
- A. H. Tullo, Chem. Eng. News, 2016, 94(9), 23 Search PubMed.
- D. Sugioka, T. Kameyama, T. Yamamoto, S. Kuwabata and T. Torimoto, Chem. Commun., 2016, 52, 12241 RSC.
- C. Maton, N. De Vos and C. V. Stevens, Chem. Soc. Rev., 2013, 42, 5963 RSC.
- C. G. Cassity, A. Mirjafari, N. Mobarrez, K. J. Strickland, R. A. O'Brien and J. H. Davis Jr, Chem. Commun., 2013, 49, 7590 RSC.
- M. Scheuermeyer, M. Kusche, F. Agel, P. Schreiber, F. Maier, H.-P. Steinrück, J. H. Davis Jr, F. Heym, A. Jess and P. Wasserscheid, New J. Chem., 2016, 40, 7157 RSC.
- I. B. Johns, E. A. McElhill and J. O. Smith, Ind. Eng. Chem. Prod. Res. Dev., 1962, 1, 2 CrossRef CAS; I. B. Johns, E. A. McElhill and J. O. Smith, J. Chem. Eng. Data, 1962, 7, 277 CrossRef; E. S. Blake, W. C. Hammann, J. W. Edwards, T. E. Reichard and M. R. Ort, J. Chem. Eng. Data, 1961, 6, 87 CrossRef; I. C. Lewis and T. Edstrom, J. Org. Chem., 1963, 28, 2050 CrossRef; M. A. Fabuss, A. S. Borsanyi, B. M. Fabuss, J. O. Smith, J. Chem. Eng. Data, 1963, 8, 64 CrossRef; K. L. McHugh and L. R. Stark, ASLE Trans., 1966, 9, 13 CrossRef.
- For example, see: S. R. Akhtar, J. V. Crivello and J. L. Lee, J. Org. Chem., 1990, 55, 4222 CrossRef CAS; J. V. Crivello and J. H. W. Lam, J. Org. Chem., 1978, 43, 3055 CrossRef; J. V. Crivello and J. H. W. Lam, Journal of Polymer Science, Symposium Series, 1976, 56, 383 Search PubMed; G. H. Wiegand, Ph.D. thesis, University of Massachusetts, 1965.
- J.-H. Park, D.-C. Seo, S.-J. Park and S.-J. Kim, European patent EP 972761, 2000, CAS, 2000:50072.
- CrysAlisPro, v1.171.37.35, Rigaku Oxford Diffraction, 2015, Rigaku Corporation, Oxford, UK Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef; S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef; P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef.
- E. B. Fox, L. T. Smith, T. K. Williamson and S. E. Kendrick, Energy Fuels, 2013, 27, 6355 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Elemental analyses and multinuclear NMR data on all new compounds. CCDC 1506146. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra25774g |
| This journal is © The Royal Society of Chemistry 2017 |