
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring complex transitions between polymorphs on a small scale by coupling AFM, FTIR and DSC: the case of Irganox 1076® antioxidant
Johanna Saunier *a,
Jean-Marie Herryb,
Najet Yagoubia and
Christian Marlièrec
*a,
Jean-Marie Herryb,
Najet Yagoubia and
Christian Marlièrec
aIFR 141, EA 401, UFR de Pharmacie, Univ. Paris Sud, Université Paris-Saclay, 92290 Chatenay Malabry, France. E-mail: johanna.saunier@u-psud.fr
bMicalis Institute, INRA, AgroParisTech, Université Paris-Saclay, 78350 Jouy-en-Josas, France
cISMO, UMR CNRS 8214, Univ. Paris Sud, Université Paris-Saclay, 91405 Orsay cedex, France
First published on 13th January 2017
Abstract
This study illustrates the significant interest of using atomic force microscopy (AFM) in force curve imaging mode for discovering and studying not easily detectable solid/solid transitions between polymorphs: we show that AFM in this imaging mode is a powerful means for studying in situ these transitions as they can be (i) detected in a very early step because of the high spatial resolution (at nanometer scale) of AFM and (ii) be distinguished from melting/recrystallization processes that can occur in the same temperature range. This was illustrated with the case of Irganox 1076®. This compound is a phenolic antioxidant currently used in standard polymers; it can bloom on the surface of polymer-based medical devices and its polymorphism might affect the device surface state and thus the biocompatibility. In a previous paper, the polymorphism of this compound was studied: four forms were characterized at a macroscopic level and one of them (form III) was identified on the surface of a polyurethane catheter. However, it was difficult to characterize the transitions between the different forms with only classical tools (DSC, FTIR and SAXS). In the present study, to evidence these transitions, we use AFM measurements coupled with a heating stage and we correlate them to ATR-FTIR measurements and to DSC analysis. This new study put into evidence a solid–solid transition between form III and II.
Introduction
Polymorphism is a subject of great importance, in particular in the pharmaceutical industry.1–5 Depending on the type of polymorph, properties of the compound, such as solubility, friability, interaction ability can drastically change. It can modify also dissolution rate, stability, hardness, compatibility, handling, flow, and blending, properties which are important for drugs because they can have important medical and process implications.6,7 For example, because of differences in solubility, one form may be more active therapeutically than another.1,8In our case we were interested in the polymorphism of polymer additives, which is a much less studied field,9–11 but which might have an impact on
(i) The surface properties of the polymer.
(ii) The leaching of the additive in liquid media in contact with the polymer surface.
Many additives such as antioxidants (AO) can indeed bloom12–15 that is to say they can migrate through the polymer bulk to the surface, exudate and crystallize on it. Depending on the polymorph that crystallizes, the impact on surface properties and leaching might be different. We considered more specifically in this paper the case of additives contained in a polymer used as a medical device. It is indeed well known that surface properties will condition the device biocompatibility and the bioadhesion on it.16,17 Furthermore, leaching of low molecular weight compounds into patient's body fluids should be avoided. In the medical field and, despite its potential importance, the phenomenon of blooming was much less studied than the migration of additives in the body fluids. However, blooming was shown to have an impact on the blood compatibility18 (platelet adhesion), protein adsorption,19,20 bacterial adhesion21 and resistance to enzymatic degradation.22
Polymorphism is not always easy to understand and to predict and its comprehension needs the use and association of different analytical techniques.23,24 Two kinds of polymorphism exist: either a form is metastable relative to another one (monotropic system) or there is a transition temperature between two stable forms, a low temperature and a high temperature ones (enantiotropic system). However, the determination of such relations might be very difficult, because some metastable forms are very stable-like at ambient temperature for kinetic reasons and because these transitions can thus be very slow and difficult to put into evidence by differential scanning calorimetry (DSC).25
Irganox 1076®, one AO, was chosen for this study because its blooming was observed on the surface of polyurethane catheters after storage in a cold environment.12,13 Irganox 1076® is a sterically hindered phenol having a long alkyl chain (C18) with an ester group and its polymorphism was shown to impact the catheter surface properties.12,26 It can at least crystallize in four different polymorphs (named I, II, III, IV, the lowest number corresponding to the form melting at the highest temperature) and the bloomed AO on the catheter was identified as mainly being form III.13 The form IV was a low temperature form obtained by rapid cooling after melting; it melted at 5 °C and was shown to be a metastable form. But, in this former study, it was difficult to assess the transitions between the three other forms which melt between 45 and 51 °C. However, these transitions are important because they might be involved in the accelerated aging of the surface after a plasma treatment when it was stored at 45 °C.26
To elucidate these transitions, we realized spincoated polyurethane films with blooming of Irganox 1076® and we followed the surface evolution with temperature by using AFM spectroscopy force imaging. Complementary, FTIR-ATR in heating mode and DSC experiments were realized on Irganox 1076® powder. AFM is proved to be a powerful method for evidencing the slow speed phase transitions. When only using classical tools like DSC or FTIR, it is indeed difficult to very early detect changes in structures as they appear at a nano-level on the crystal surface. AFM is thus a convenient tool for this research.27 It has been already used as a performing tool that allows to visualize the early stages of crystal growth, and to understand crystal nucleation and growth kinetic and mechanism,28–33 but not yet to follow polymorphic transformations of bloomed species on the surface of polymers.
Results and discussion
DSC
Firstly, the impact of the heating rate on the DSC thermograms was studied. The thermograms of the form I and of the form II were quite similar when increasing the heating rate, but the shape of the thermogram of the form III changed in a significant way (Fig. 1a–e). For 2 °C min−1, only one endothermic peak occurred with an onset at 47.7 °C (Fig. 1a). For 1 °C min−1, the same one was observed, but moreover, two small and very close endothermic peaks appeared (the melting began at 50.5 °C and the enthalpy of fusion was of 2.5 J g−1). For these both experiments, the enthalpy of the main endothermic peak was around 150 J g−1. For 0.5, 0.1 and 0.05 °C min−1, very different thermograms were obtained (Fig. 1c–e): there was a first endothermic peak (mean onset at 47.5 ± 0.4 °C) with a concomitant exothermic one (the onset cannot be calculated but the maximum was at 48 °C for sample between 1 and 2 mg) and then a new endothermic one. The exothermic peak increased in intensity when decreasing the heating speed. Indeed, at 0.5 °C min−1, two main endothermic peaks were clearly observed, but the exothermic peak was almost totally overlapped by the first endothermic peak. It was thus difficult to calculate enthalpies of the two first peaks for the lowest heating rates due to the fact that the endo- and exothermic events happen almost at the same time.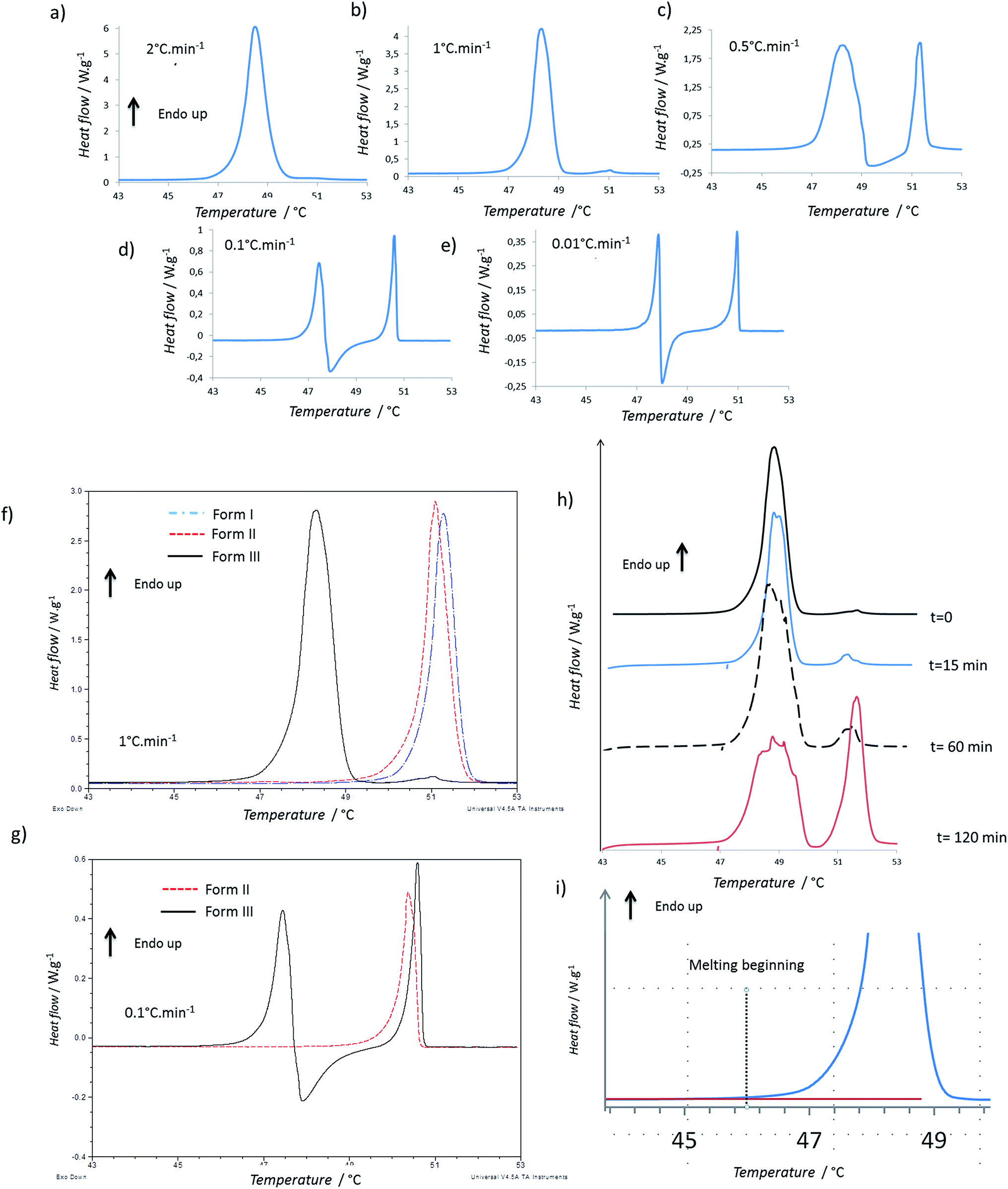 | ||
| Fig. 1 DSC analysis: (a–e) thermograms of form III powder for different heating rates; (f and g) comparison of the thermograms of the different forms at 1 °C min−1 and 0.1 °C min−1; (h) thermogram of form III powder after different annealing times at 47 °C (heating rate 1 °C min−1); (i) thermogram of form III powder: zoom on the beginning of melting. | ||
When we compare the thermograms of the forms III, II, I at 1 °C min−1, we observe mainly an endothermic peak for each form that should correspond to the melting of each form (Fig. 1f). The presence of the small endotherms for form III can thus be associated with the melting of form II and I. At 0.1 °C min−1, the melting of form III was followed by a recrystallization, and then a peak that matched with the melting peak of the form I was observed (Fig. 1g). However it was difficult to exclude the previous melting of the form II because it was shown that, at this heating rate, forms I and II have very close melting temperature and enthalpies of fusion.13 When the form III was annealed at 47 °C for several times (Fig. 1h), a growth of the melting peaks around 50 °C were observed and the area of the form III melting peak decreased.
To compare the enthalpies of fusion, three samples of each form were analyzed at 2 °C min−1, a speed rate for which no apparent recrystallization of form III and no melting peaks characteristic of form I or form II were observed. The enthalpies of fusion were calculated for form I, II and III and were respectively equal to 119.5 ± 3.0 J g−1, 122.5 ± 4.0 J g−1, and 141.5 ± 8.5 J g−1.
The enthalpy of fusion of the form III which melt at lower temperature than forms I and II was greater than those of the two other forms (Table 1). This suggests the form II and form III are enantiotropes and the form III is the stable form at low temperature (Burger rule34,35). The greater variation observed in the enthalpy of fusion for form III could be related to the difficulty of avoiding totally the transitions previously described when melting the form III. The relationship between form I and form II, is more difficult to establish because their enthalpies of fusion were very close.
| I | II | III | IV15 | |
|---|---|---|---|---|
| Tfus [°C] | 51.0 ± 0.2 | 50.9 ± 0.2 | 47.5 ± 0.4 | 5.3 ± 0.3 |
| ΔfusH [J g−1] | 119.5 ± 3.0 | 122.5 ± 4.0 | 141.5 ± 8.5 | 60.5 ± 3.0 |
We estimated the transition temperature using the following relationship, which was used quite successfully by other authors:36,37
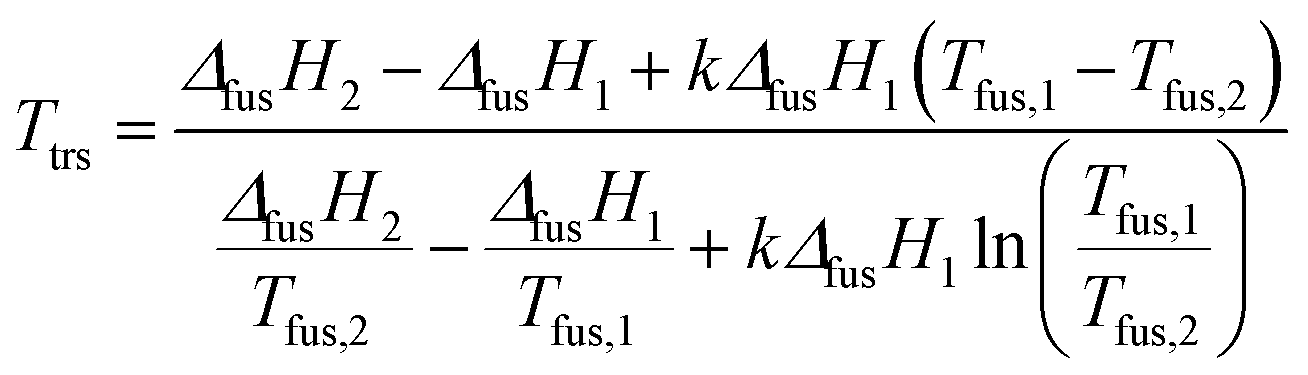 | (1) |
By using eqn (1), transition temperatures were found at around 29 °C for III → II and 47 °C for II → I. However these temperatures are only a rough estimation as our enthalpy data may have a quite high standard deviation. For example, if we use the extreme ranges of enthalpies of fusion of form II and III, the transition temperature is found between 26 °C and 36 °C.
It should be noted that the onset of the melting temperature for form III was found at 47.5 °C, but the melting began at least below 46 °C, as shown on Fig. 1i. Same phenomenon was observed for the melting of the other forms which began at around 48.5 °C. This wider melting range might result from a premelting process which occurs because of the presence of impurities in the compound (using the rate of melting of the sample, the purity analysis by DSC indicated a purity of 99.7% for the Irganox 1076® powder).
AFM
The first important point is that, by using Force Spectroscopy Mode (FSM), it was possible to distinguish the liquid melt AO from the PU surface and the PU surface from the AO crystals (Fig. 2) by the two following properties: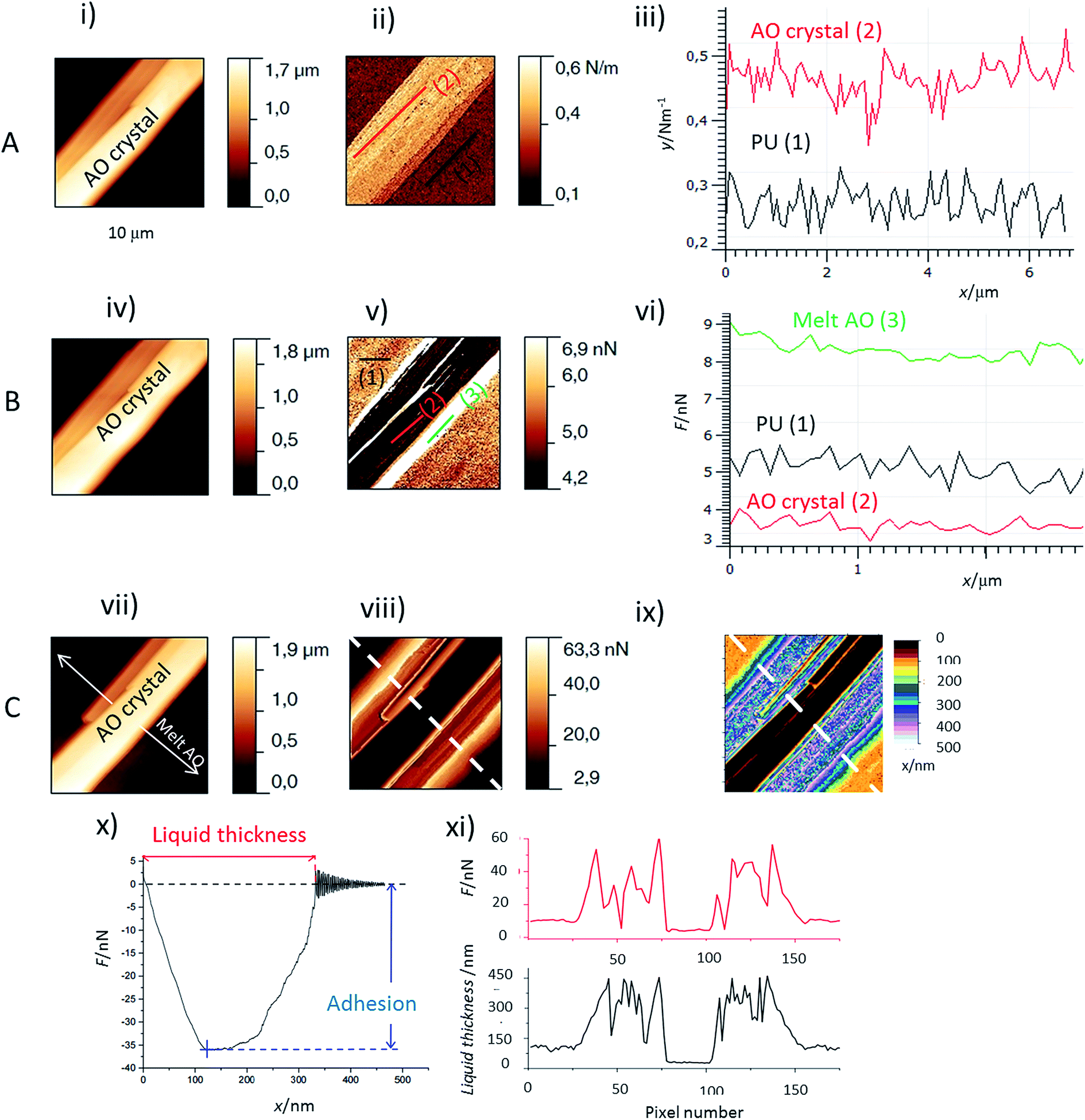 | ||
| Fig. 2 AFM images and profiles of an antioxidant needle: (A) before melting (B) at the beginning of melting (C) for an almost melt needle. Images (i), (iv) and (vii) are AFM height images (10 × 10 μm2); image (ii) is the stiffness image corresponding to image (i); (iii) values of stiffness corresponding to the lines on the stiffness image; (v) and (viii) adhesion images (retracting the tip) corresponding respectively to height images (iv) and (vii); (vi) values of adhesion corresponding to the lines respectively on the adhesion images (v); (ix) liquid thickness image which was obtained by the determination of the distance needed to retract the tip from the sample as detailed in figure (x) on the force curve in the retract range; (xi) plots of the liquid thickness and adhesion force along the dotted line drawn on the images (viii) and (ix). | ||
(i) Adhesion values. When the AFM tip is in contact with a liquid layer, the adhesion force is much higher than that for either crystallized AO or PU zones (Fig. 2v and vi). The lowest adhesion values were obtained for the crystallized AO regions. The liquid thickness (Fig. 2ix and xi, black curve) was calculated from the retract force curve as explained in Fig. 2x. We observed profiles of the adhesion perpendicular to the melting front of the AO crystal. As evidenced in Fig. 2viii and xi, red curve, the adhesion and melt liquid thickness are inversely correlated: adhesion as high/low as 60 nN/10 nN is observed for the thick/thin melted zones respectively. In comparison, the adhesion at AO crystals is around 4 nN (Fig. 2vi).
(ii) The stiffness measurements (Fig. 2ii and iii). It allows to distinguish between the crystallized AO regions from the rest of the sample: the stiffness of PU covered or not covered with melted AO is around 0.25 N m−1 but the AO crystals are stiffer (between 0.45 to 0.7 N m−1).
The behaviour of the Irganox 1076® forms I and III were studied by AFM. Our former DSC experiments revealed that the phase transitions endured by the studied samples are highly dependent on the heating rates: as mentioned earlier this was particularly evidenced in the case of form III. Moreover temperature range used for the AFM study was chosen thank to the DSC study. As a consequence, two samples with form III were evaluated by using two relevant heating rates as deduced from the DSC experiments. In the Fig. 3–6, new growing structures are marked with an arrow and disappearing structures are marked with a circle. The initial AO crystal structure is thus strongly modified during these thermal treatments. For temperatures higher than 35 °C, some structures disappeared (images 1–18 from Fig. 3 and 4 and image 2 from Fig. 5) as clearly evidenced in the slope images, by the apparition of a free PU surface, with a lower stiffness (images 13–18 from Fig. 4a and image 2–3 from Fig. 6a); meanwhile, some new structures appeared and grew with time and temperature (images 1–18 from Fig. 3 and image 2 from Fig. 5). These changes were particularly well observed in case of very slow heating ramps or isotherms performed between 42 and 43.5 °C (Fig. 3). Transformation process of the sample was observed up to 44 °C. As shown by the adhesion measurement, no liquid areas were detected during the first moments of the process (images 1–13 from Fig. 4b and image 2 from Fig. 6b). Around the temperature of 44–45 °C, the melting of AO occurred (Fig. 4b images 17–20, Fig. 6b image 3–4) and we observed the sudden growth of big solid needles with a related high value of the stiffness (Fig. 5 and 6a images 4–10); this phenomenon significantly starts at 45 °C and is very well observed when the sample is maintained a long time between 45 and 46 °C (Fig. 5 and 6). This melting/growth process was observed up to 46 °C. Then, around 47 °C, these newly evidenced big needles began to melt (Fig. 5 images 11–17): this process started by the outer side of the needle (Fig. 6b images 12–17). At around 48 °C, all the needles were melted.
 | ||
| Fig. 3 Evolution of AFM height images (10 × 10 μm2) with time and temperature for a PU film covered with form III. The sample was heated slowly. | ||
 | ||
| Fig. 4 (a) AFM stiffness images (b) AFM adhesion images. Images correspond to the height images presented on the Fig. 3. | ||
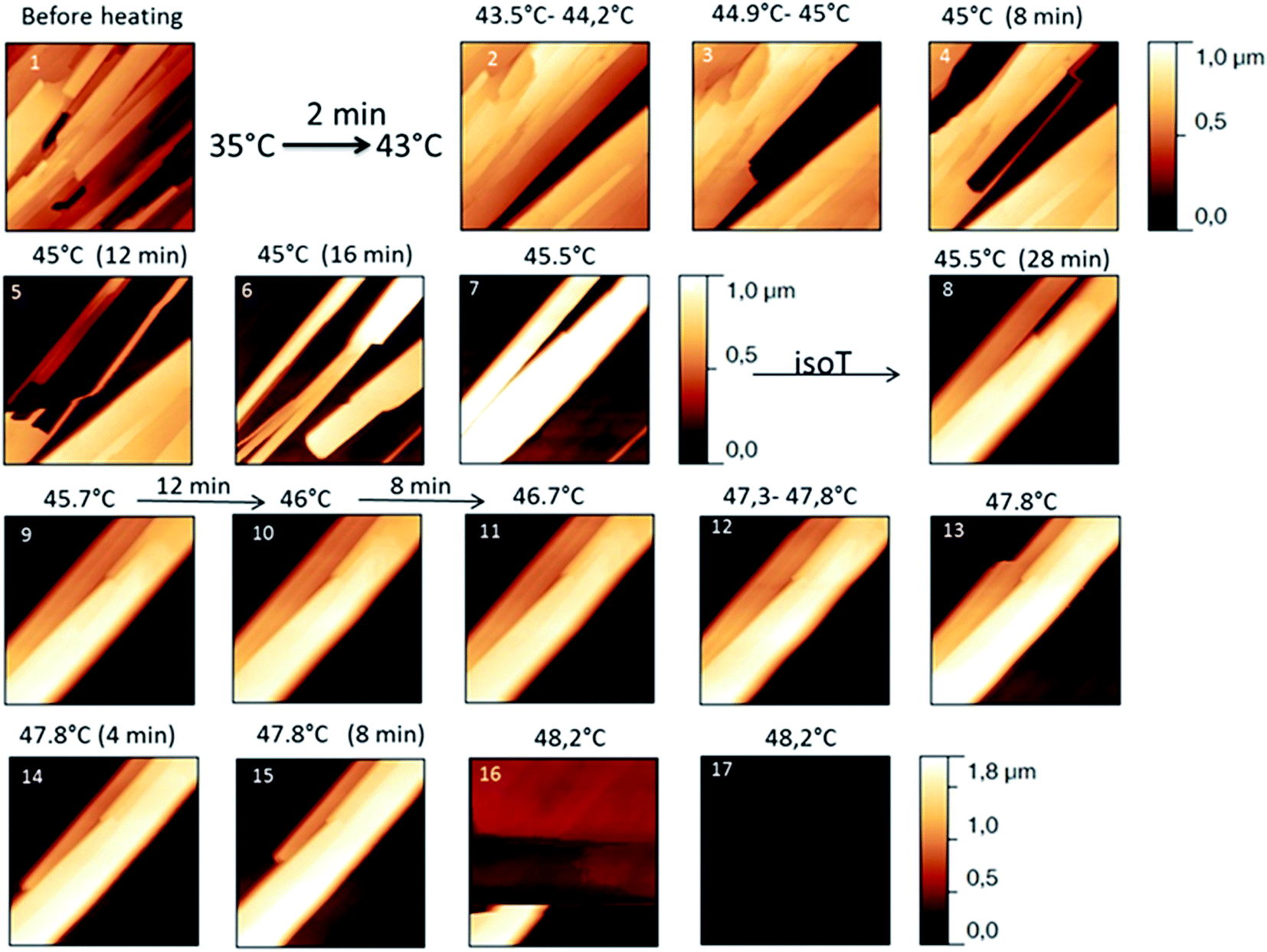 | ||
| Fig. 5 Evolution of height AFM images (10 × 10 μm2) with time and temperature for a PU film covered with form III. The heating rate used was a quick one till the form III melted. | ||
 | ||
| Fig. 6 (a) AFM stiffness images (b) AFM adhesion images. Images correspond to the height images presented on the Fig. 5. | ||
For form I (Fig. 7 and 8), slow heating was processed with long time isotherms at 43 and 43.5 °C. As for form III, some initial crystalline structures disappeared (see the circles on Fig. 7 and the related appearance of less stiff PU surface on Fig. 8a) and new structures grew (see the arrows in Fig. 7). No melting phenomenon occurred during this process as shown by the adhesion measurements (Fig. 8b).
 | ||
| Fig. 7 Evolution of AFM height images (10 × 10 μm2) with time and temperature for a PU film covered with form I for temperatures between 42 and 43.5 °C. | ||
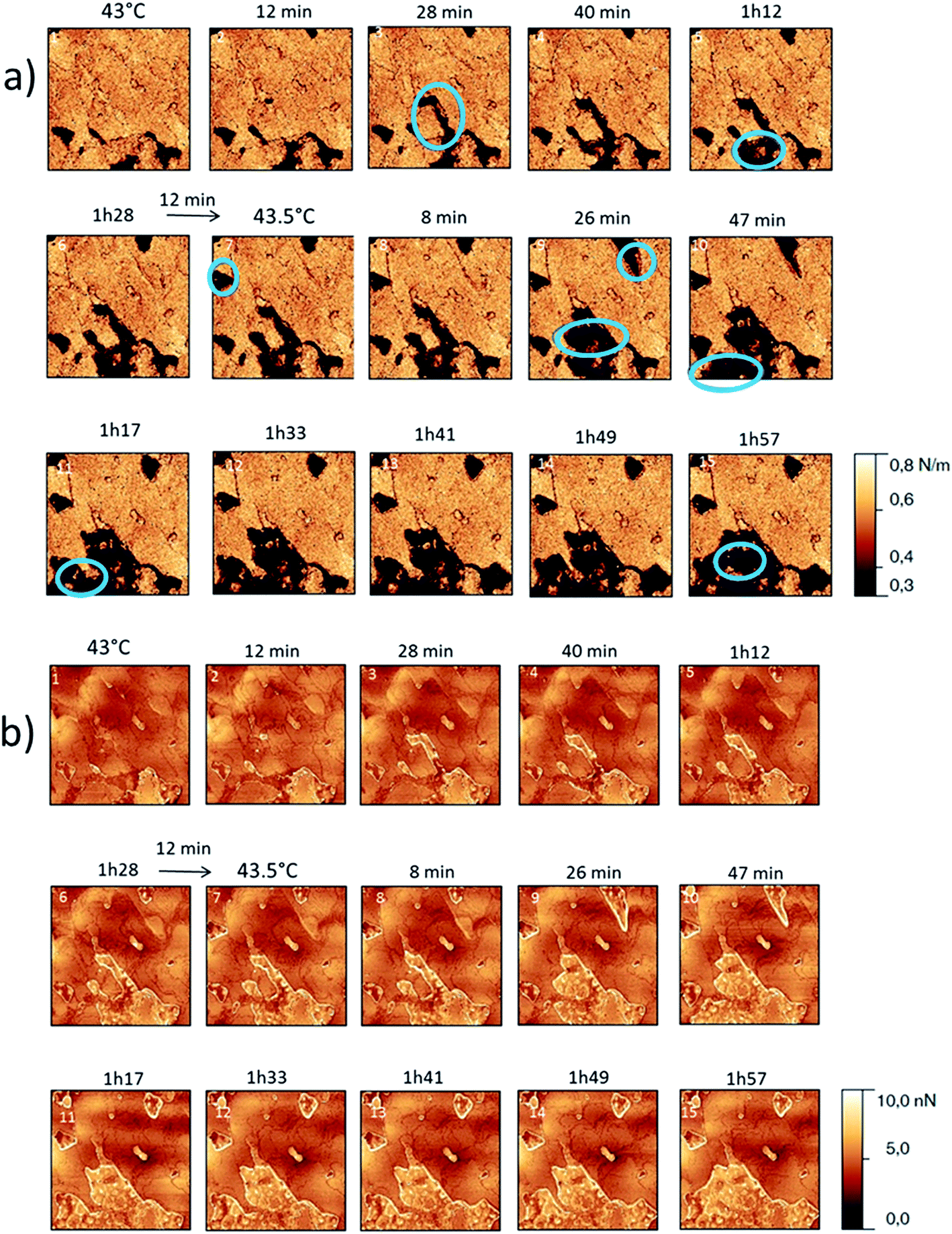 | ||
| Fig. 8 (a) AFM stiffness images (b) AFM adhesion images. Images correspond to the height images presented on the Fig. 7. | ||
FTIR
For this analysis, we used the wavenumber range between 3650 and 3550 cm−1 that corresponds to the absorbance range of the AO phenol stretching band. It has been shown that this band can be used to identify the different polymorphs:13 the form I exhibits a maximum located at 3640 cm−1, the form II at 3585 cm−1 and, the form III, at 3610 cm−1.For a quick heating (Fig. 9a and b) corresponding to a heating ratio of 1 °C min−1 up to 46 °C, the form III band decreases and then disappears without the appearance of the form II band: instead of it, a growth of the form I band was put into evidence (Fig. 9a). For higher temperature (T), the melting of this form I was observed concomitantly with the growth of the liquid form as revealed by the increase of the band absorbance at 3650 cm−1 and the decrease of the form I band absorbance (Fig. 9b).
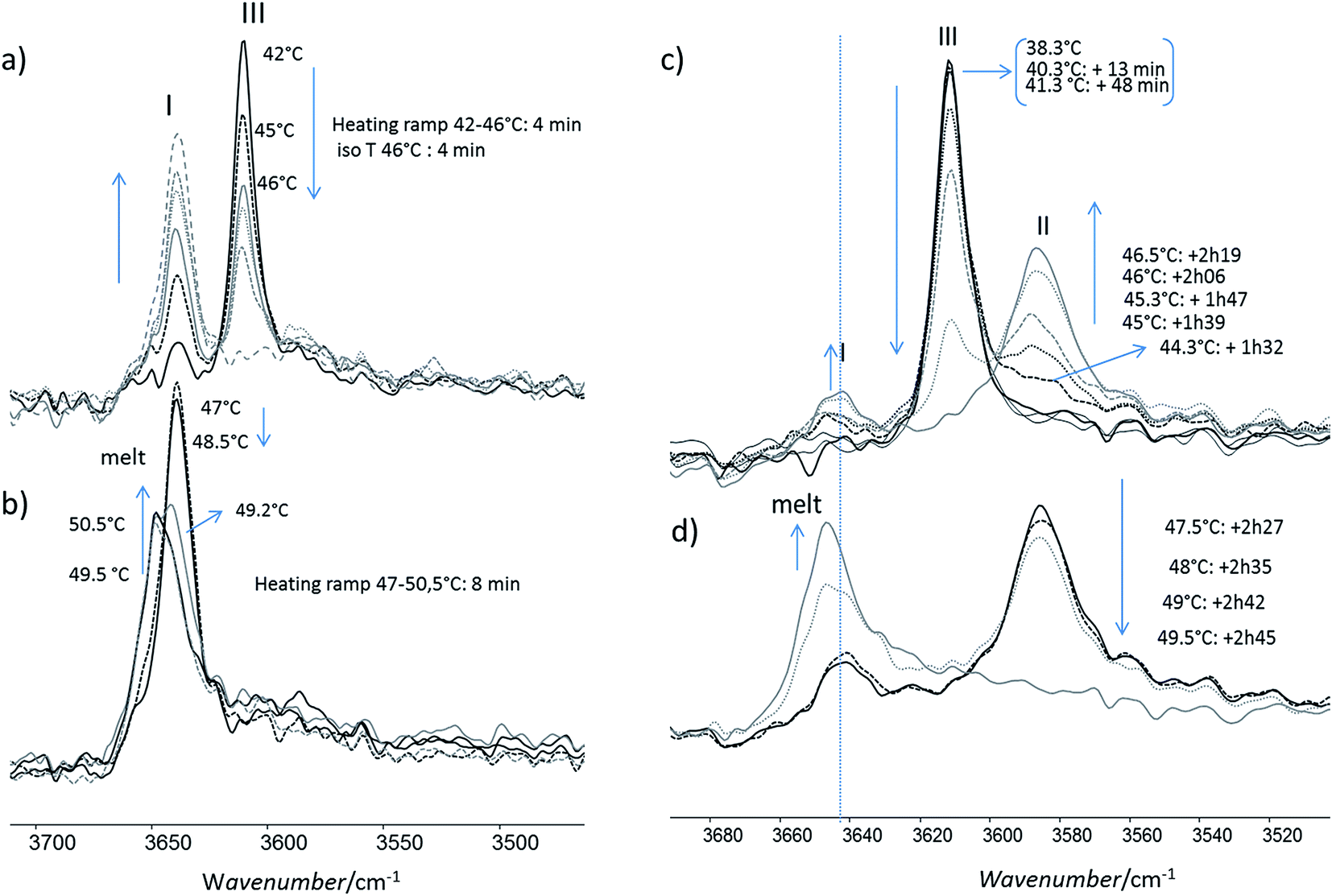 | ||
| Fig. 9 Evolution of the FTIR νOH phenol band of the form III powder: (a) the powder was heated quickly to 46 °C and maintained at 46 °C (b) the powder was then melt by heating to 50.5 °C (c) the powder was heated slowly between 42 and 46.5 °C (d) then it was melt by heating to 49.5 °C. | ||
For slow heating (Fig. 9c and d), most of the form III band disappeared as form II band grew with heating (Fig. 9c). A band characteristic of form I was detected but only with a small absorbance. By increasing the temperature from 47 °C to 50 °C, the decrease of the form II band absorbance was observed. Simultaneously a new band at 3650 cm−1 due to the liquid form (Fig. 9d) was appearing.
For isotherms at T < 45 °C, the appearance of the only form II was observed with time (Fig. 10). Increasing the temperature of several degrees had a deep impact on the phase transformation kinetics: at 37 °C, after 3 h 15, the form II band absorbance was very small (Fig. 10a). To observe an amount of form II, equivalent to that observed after 5 minutes of phase transition at 45 °C, 1 h 47 and 4 h 45 are needed at respectively 42 and 40 °C (referring to the form II/form III band intensity ratios) (Fig. 10b–d). For 45 °C isotherm (Fig. 10d), a little amount of form I is detected. For 46 °C isotherm (Fig. 10e) the form III began to melt and form III transformed into form II and form I. The same behaviour was put into evidence at 47 °C with a higher amount of form I formed during the isotherm (Fig. 10f). At 48 °C, the coexistence of liquid, form II and form I was first observed because the whole form III disappeared and melted when the powder was put in contact with the hot ATR crystal (Fig. 10g). Then the band of form I kept growing and the form II band absorbance decreased slightly with time.
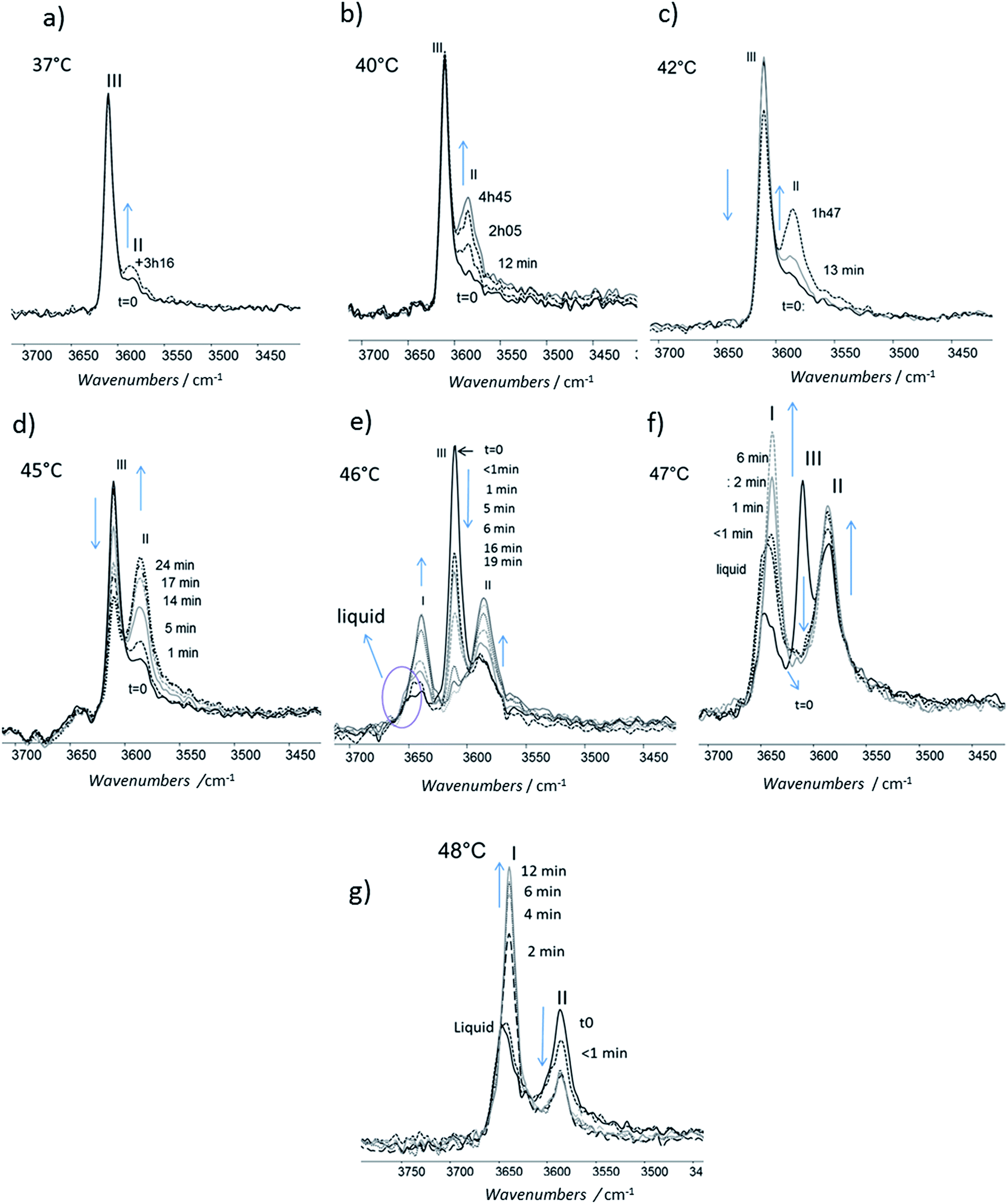 | ||
| Fig. 10 Evolution of the FTIR νOH phenol band of the form III: the powder was maintained at constant temperature and spectra were recorded with time (a) 37 °C (b) 40 °C (c) 42 °C (d) 45 °C (e) 46 °C (f) 47 °C (g) 48 °C. | ||
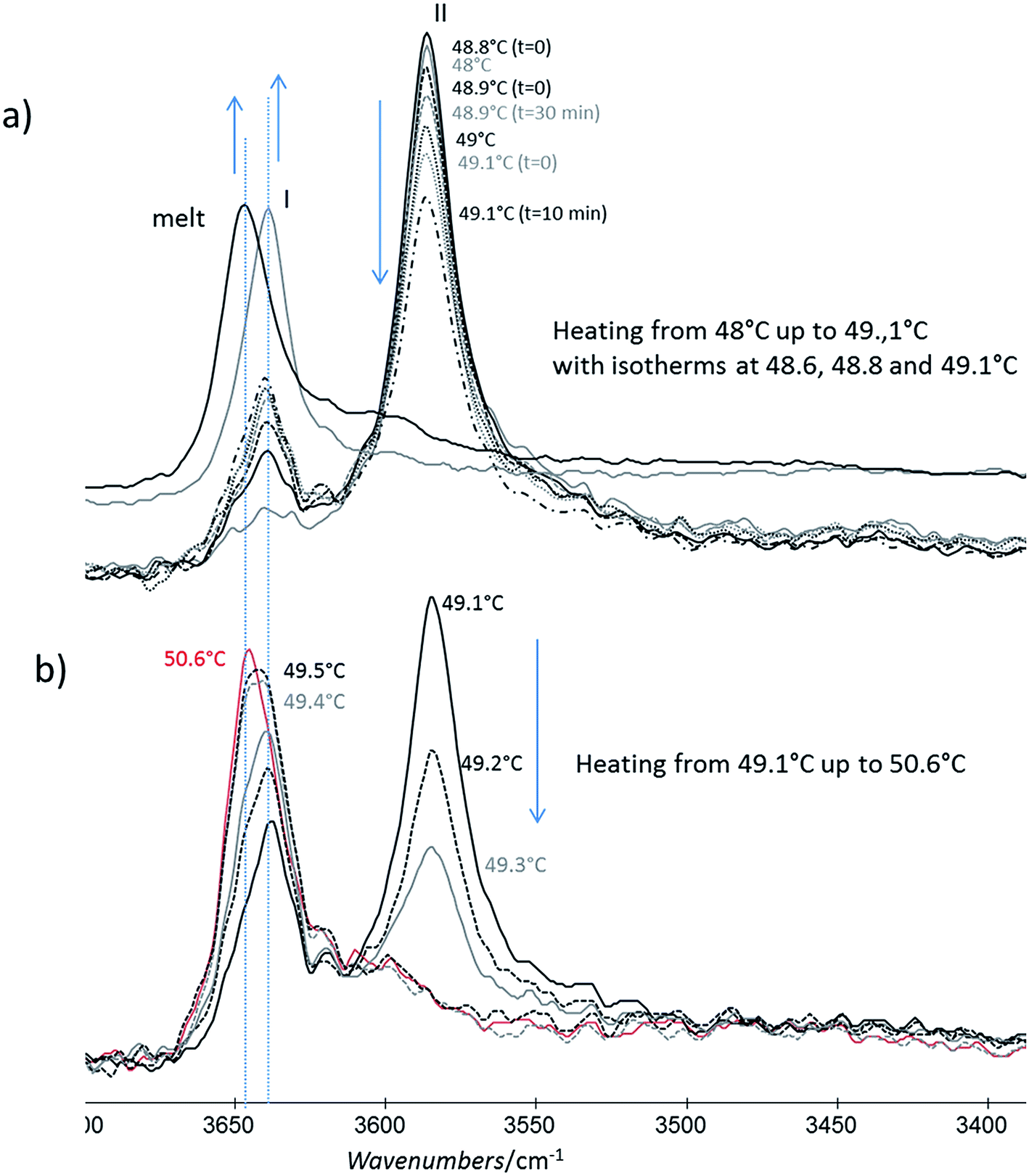 | ||
| Fig. 11 Evolution of the FTIR νOH phenol band of the form II powder with time: (a) for a slow heating between 48 and 49 °C (b) then the powder was heated slowly and melt. Spectra of form I and melt powder are given as references. | ||
Discussion
When form III was heated rapidly, the previous transition which was quite slow cannot be observed anymore by FTIR (Fig. 9a and b) even if some transformation occurred at a smaller scale (Fig. 5, image 2). When the heating was very quick (1–2 °C min−1 in DSC), the main transition was the melting of form III. When the heating rate was lower (below 0.5 °C min−1), a melting/recrystallization phenomenon was observed. This was put into evidence by FTIR, DSC and AFM. FTIR revealed the growth of form I and the transformation of form III to the liquid state even if the band characteristics of this liquid state is very close to that of form I. However this melting of form III is concomitant to an exothermic peak characteristic of a recrystallization as observed on DSC thermogram. This resulting liquid state was detected by AFM between 44 °C and 47 °C as shown in Fig. 6b (images 4–6); moreover the simultaneous growth of big needles that can be associated to form I (Fig. 5, images 6–10) was observed in the same temperature range.
In a previous paper,26 the aging behaviour of a catheter after a discharge nitrogen plasma treatment was studied. After this plasma treatment the polymer surface was more hydrophilic, but there was a hydrophobic recovery of the surface with time: at 45 °C, the hydrophobic recovery was very quick compared to 25 °C (complete recovery after 24 hours). This was explained by the presence of Irganox 1076® form III needles on the catheter surface. Indeed, tablets of Irganox 1076® form III that were exposed to the same plasma treatment showed a similar recovery phenomenon with time: 24 hour after the plasma treatment, the hydrophobic recovery was already significant at 40 °C and was complete at 49 °C. The implication of melting of form III needles and its recrystallization into form I on the surface energy was suggested, by hypothesizing that some imperfect small crystals can melt at a little lower temperature than that of the onset melting (47.2 °C) found by macroscopic DSC measurements at 0.5 °C min−1. However, by using the new results presented here, we can suggest another explanation for this aging: it could result in part from a solid/solid transition of form III into form II, especially for the lower storage temperatures (40 °C).
(i) As form III melted, the melt recrystallized into form I.
(ii) The same phenomenon was observed with the low temperature form IV (Tfus around 5 °C): as form IV melted, it recrystallized into form I.13
(iii) Ostwald rule38 states that, in a general way, the least stable polymorph crystallizes from the melt.
We might thus explain the recrystallization into form I from the melt after the melting of forms IV and III by the fact the free enthalpy of form I is below that of the liquid but higher than those of form II and form III (at least up to the form III melting temperature). Form I should be a metastable form for temperatures lower than the melting temperature of form III.
It is interesting to notice that in our case the presumed solid/solid transition proceeds with the progressive disappearance of a form and the growth of new flat structures on it. On the contrary, we observed the melting/recrystallization process gives birth to long needles growing quickly from the melt with a preferential orientation. Numerous papers have been published on the solid-state phase transitions: most of the polymorphic transformations are little understood but AFM has proved to be really a very interesting technique to follow and understand these transitions.27 The solution-mediated transformation from the α- to the γ-form of glycine was observed in situ at the crystal surface with AFM and the mechanism was discussed;39 solid state transition of caffeine crystals and caffeine–glutaric acid cocrystals from the metastable phase to the stable phase was studied too;40,41 solid–solid phase transition of octahydro-1,3,5,7,-tetrazocine was followed at sub-micrometric scale in real time;42 phase transformation process of bis(1,2-benzoquinonedioximato)platinum(II) into phase alpha was observed by AFM.43 A part of polymorphic transformations is stemming from order-disorder transition with same molecular arrangement and increased crystallographic site symmetry. For others the transition needs the forming of critical nuclei of the new phase, such as local defects: depending on the default nature, the kinetics of the solid/solid transition will be impacted.44–46 It is interesting to note that defects can be introduced by a mechanical way (surface scratching). So we cannot exclude that the AFM probe/crystal contact or local overpressure might help in creating defects on the surface and inducing the transition from form I into form II very locally at a sufficient temperature. Moreover it is known that grinding and compression can favour the phase transition process from a metastable to a stable form:47 as an example, some drugs can undergo this kind of transition during the industrial tablet compression.48 In our case, the compression stress applied on a point of the surface of the tip/substrate contact can indeed be locally strong and iterative (sample is scanned on the same zone several times): we could estimate it to 10 MPa. This might explain why changes were observed by AFM but not by FTIR measurements. Transition might be thus induced only locally on the AFM scanned zone. This kind of effect of the probe at a nanoscale was observed by Perkins et al. in the case of the local transformation of amorphous lactose to a more crystalline state.49
Summary
- Form III and form II are enantiotropes and form III is the low stable temperature form, but no information about the transition temperature was available. From AFM data presented here it can be said that this transition temperature is below 35 °C which is in concordance with DSC results.- Using Ostwald rules and AFM observations, we suggest that form I is metastable at least up to 47 °C. This may be the high-temperature stable form before melting; however, as the forms I and II have very close enthalpies of fusion, it is thus not trivial to conclude about a monotropic or enantiotropic transition between them. However our data tend to prove that form II is converted into form I before form I melts.
- No difference between all the different forms was put into evidence by analysing the force spectroscopy curves; so we cannot identified the forms on the AFM images describing the behaviour of form I with heating.
Using these results and the melting temperature of the forms, we propose an enthalpy H(T) and a free enthalpy G(T) diagram (Fig. 12) to explain the different phenomena we observed: we hypothesize that form I is the high temperature stable form.
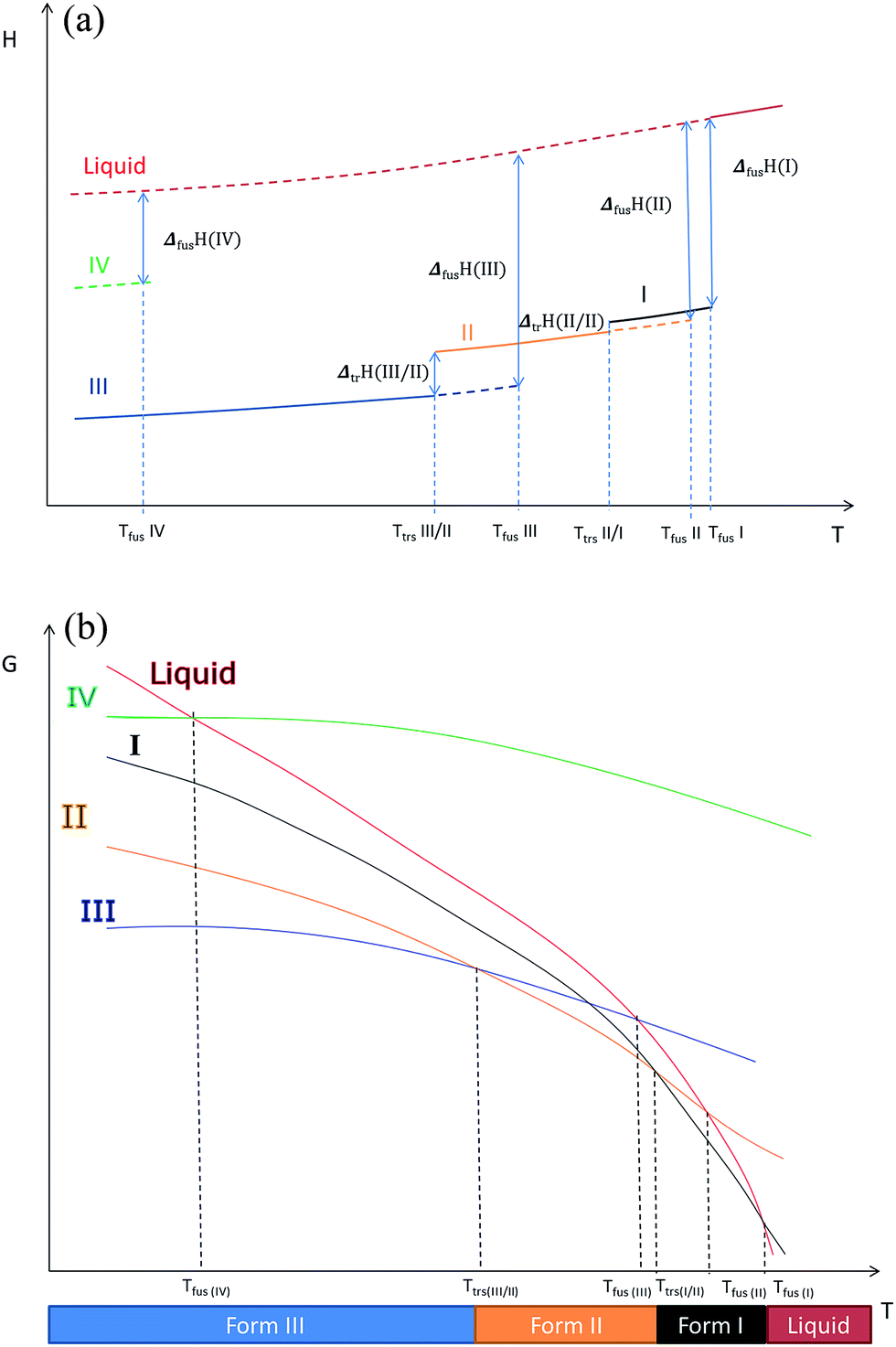 | ||
| Fig. 12 (a) Enthalpy H versus temperature diagrams for the different forms of Irganox 1076® (b) free energy G versus temperature diagrams for the different forms of Irganox 1076®. | ||
- We recall that as shown in previous paper13 form IV is metastable.
Conclusions
Transitions between the various solid phases of the Irganox 1076® antioxidant could not be fully discriminated and understood by only using either macroscopic classical DSC or FTIR during long time isotherms experiments: we proved here that AFM, by allowing the direct monitoring of time evolution of structural changes at a very small scale, enables to evidence the slow solid state transitions in their very first steps. By coupling force spectroscopy AFM to heating FTIR and DSC, we were able to better understand what happens in a complex system for which solid–solid transition and melting temperatures are so close that the transitions between these different crystallographic forms cannot be evidenced by macroscopic thermal methods. The high spatial magnification of the AFM allows discriminating between solid transitions with slow and close kinetics that could not be detected otherwise. Furthermore it may be not excluded that the local pressure induced by the AFM tip on the crystals may favour these transitions and their detection unreachable in usual conditions. However as there are no significant differences in the stiffness and adhesion values for the different solid forms of the studied phenolic compound, it was not possible to fully discriminate between the forms I, II and III in these AFM experiments based on direct mechanical measurements. New ways for enhancing contrast between these different forms in AFM data are needed.Experimental
Preparation of the samples
Irganox 1076® is a sterically hindered phenol and was provided by BASF (Plastic Additives, Switzerland) in the form of a white powder. Irganox 1076® polymorph II and III powders were obtained by recrystallization following the method given in a previous paper.13 For form I, the commercial powder was used. Spincoated films of PU films containing Irganox 1076® were realized by dissolving the AO powder and the PU (10 to 50 g L−1) in tetrahydrofuran. High amounts of AO were added in order to obtain a significant blooming of AO on the PU surface (a = mAO/mPU = 0.5 to 0.05). Substrates were Si wafers (diameter = 20 mm, thickness = 260 ± 20 μm, Siltronix, France). The spincoater was a SPIN 150 (SPS, The Netherlands). The speed of 2000 rpm was reached after 1 s of spinning and was maintained during 60 s. The used PU was a medical-grade poly(ether urethane) referenced as Pellethane 2363 80AE® (Lubrizol, USA). Low molecular weight compounds such as additives were removed by an extractive process (dissolution/precipitation: THF as a solvent and methanol as non-solvent). AFM samples were controlled by FTIR in order to check the polymorphs on the substrate.DSC
DSC was performed on a TA Instrument Q1000. The temperature range investigated was between 40 °C and 60 °C (heating rates: 0.05 °C min−1 to 2 °C min−1). AO powder (1 to 3 mg) were put in a hermetic aluminium pan and analysed under nitrogen blanketing. Enthalpy of fusion ΔfusH corresponds to the melting peak area. Calibration was performed with gallium (purity = 5 N, Tfus = 29.7646 °C, ΔfusH = 80.07 J g−1) and indium (purity = 5 N, Tfus = 156.5985 °C, ΔfusH = 28.62 J g−1). Moreover a purity analysis was realized using DSC by analysing the rate of melting.AFM
The AFM was a Nanowizard III (JPK Instruments AG, Berlin, Germany) using a Force Spectroscopy Mode (FSM) (Quantitative Imaging® (QI), JPK): at every pixel of the scanned area ((10 μm)2 with (128 pixels)2) where a force/distance curves – during the approach/retract of the AFM tip to/from the sample – was realized. The sample can be heated thanks to heating controller. With FSM local adhesion and stiffness of the sample surface are simultaneously measured. Experiments were realized in air. The probes were silicon probes CSC 37 (Micromasch, stiffness around 0.35 N m−1, measured by thermal noise). The stiffness data were calculated from the slope of the approach curves (force versus scanner elongation) at the point of the maximal applied force (setpoint 5 nN). Adhesion data on retract curves were exploited. The pixel-by-pixel extend/retract curves were done at constant speed (150 μm s−1 on a total length of 500 nm). Different heating rates were used. An image was acquired in approximately four minutes and images were registered in a continuous way during heating. Two spincoated wafers covered with a PU film on which AO has bloomed in form III were studied. The first one was heated from 28 to 45 °C in a slow way: several isotherms were used; at 42 °C for 20 min, at 43 °C for 27 min, at 43.5 °C for 54 min, and at 44 °C for 8 min. The second sample was heated from 35 to 48.5 °C, by heating the sample in a more rapid way between 35 and 45 °C: 42 °C was reached in two minutes and the increase from 42 to 45 °C was realized in 8 minutes. Next there were isotherms at 45 °C (16 min), 45.5 °C (28 min), before heating the sample to 48.5 °C in one hour. The PU film with form I blooming was analysed by heating very slowly between 42 and 44 °C and by doing isotherms at 43 °C (1 h 30) and 43.5 °C (2 h).FTIR
The spectrometer apparatus was a Perkin Elmer Spectrum 2000 using either the ATR (Attenuated Total Reflexion) mode with a heating ATR (DuraSampIR, SensIR technologies) or the transmission mode. Each spectrum was taken in the range from 4000 to 550 cm−1 (resolution 4 cm−1, 8 scans in non-heating mode, 1 when heating was applied). The Irganox 1076® form III powder was analysed during heating by FTIR. Slow or fast ways of heating were used. For the slow heating, the sample temperature was stabilized at 38 °C, and then the temperature of 46.5 °C was reached after 2 h 20. Next the powder was heated and melted in 25 minutes. For quicker heating, only 4 minutes were necessary to heat the sample from 42 to 46 °C. Then the temperature of 46 °C was maintained during 4 minutes. After this isotherm, the powder was heated and melted in 8 minutes. Moreover isotherms were realized at 37, 40, 42, 45, 46, 47 °C and 48 °C and the spectra of the powder were monitored with time. Form II was analysed by heating it very slowly between 48 and 50.6 °C. Spincoated films with blooming were analysed either in transmission or in ATR mode.Acknowledgements
The authors thank Bernard Legendre for his suggestions and remarks concerning the polymorphism study.Notes and references
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859–866 CrossRef CAS.
- S. Veesler, F. Puel and G. Fevotte, STP Pharma Prat., 2003, 13, 1–32 Search PubMed.
- L. Borka, Pharm. Acta Helv., 1991, 66, 16 CAS.
- J. Haleblian and W. McCrone, J. Pharm. Sci., 1969, 58, 911–929 CrossRef CAS PubMed.
- D. Singhal and W. Curatolo, Adv. Drug Delivery Rev., 2004, 56, 335–347 CrossRef CAS PubMed.
- E. H. Lee, Asian J. Pharm. Sci., 2014, 9, 163–175 CrossRef.
- H. Brittain and D. J. W. Grant, in Polymorphism in Pharmaceutical Sciences, Drugs and the Pharmaceutical Sciences, ed. H. Brittain, New York, 1999, pp. 279–330 Search PubMed.
- H. Kokubo, K. Morimoto, T. Ishida, M. Inoue and K. Morisaka, Int. J. Pharm., 1987, 35, 181–183 CrossRef CAS.
- J. M. Cogen and A. J. Hilner, Polym. Degrad. Stab., 2008, 93, 2193–2198 CrossRef CAS.
- J. Saunier, V. Mazel, C. Aymes-Chodur and N. Yagoubi, Int. J. Pharm., 2012, 437, 89–99 CrossRef CAS PubMed.
- H. Hasui, T. Furihata, K. Takanashi and M. Ohmori, in Patent EP 0032459 A1, 1981.
- O. Mrad, J. Saunier, C. Aymes-Chodur, V. Rosilio, S. Bouttier, F. Agnely, P. Aubert, J. Vigneron, A. Etcheberry and N. Yagoubi, Microsc. Microanal., 2010, 16, 764–778 CrossRef CAS PubMed.
- J. Saunier, V. Mazel, C. Paris and N. Yagoubi, Eur. J. Pharm. Biopharm., 2010, 75, 443–450 CrossRef CAS PubMed.
- B. J. Tyler, B. D. Ratner, D. G. Castner and D. Briggs, J. Biomed. Mater. Res., 1992, 26, 273–289 CrossRef CAS PubMed.
- B. D. Ratner, D. Briggs, M. J. Hearn, S. Yoon and P. G. Edelman, in Surface characterization of biomaterials, ed. B. D. Ratner, Elsevier, Amsterdam, 1988 Search PubMed.
- Y.-X. Wang, J. L. Robertson, W. B. Spillman and R. O. Claus, Pharm. Res., 2004, 21, 1362–1373 CrossRef CAS.
- R. E. Baier, J. Mater. Sci.: Mater. Med., 2006, 17, 1057 CrossRef CAS PubMed.
- J. Bandekar and A. Sawyer, J. Biomater. Sci., Polym. Ed., 1995, 7, 485–501 CrossRef CAS PubMed.
- M. R. Brunstedt, N. P. Ziats, M. Schubert, P. A. Hiltner, J. M. Anderson, G. A. Lodoen and C. R. Payet, J. Biomed. Mater. Res., 1993, 27, 255–267 CrossRef CAS PubMed.
- M. R. Brunstedt, N. P. Ziats, S. P. Robertson, A. Hiltner, J. M. Anderson, G. A. Lodoen and C. R. Payet, J. Biomed. Mater. Res., 1993, 27, 367–377 CrossRef CAS PubMed.
- J. Saunier, J.-M. Herry, C. Marlière, M. Renault, M.-N. Bellon-Fontaine and N. Yagoubi, Mater. Sci. Eng., C, 2015, 56, 522–531 CrossRef CAS PubMed.
- B. J. Tyler and B. D. Ratner, J. Biomed. Mater. Res., 1993, 27, 327–334 CrossRef CAS PubMed.
- D. Giron, Thermochim. Acta, 1995, 248, 1–59 CrossRef CAS.
- D. Giron, Eng. Life Sci., 2003, 3, 103–112 CrossRef CAS.
- J.-O. Henck and M. Kuhnert-Brandstatter, J. Pharm. Sci., 1999, 88, 103–108 CrossRef CAS PubMed.
- O. Mrad, J. Saunier, C. Aymes-Chodur, V. Mazel, V. Rosilio, F. Agnely, J. Vigneron, A. Etcheberry and N. Yagoubi, Eur. Polym. J., 2011, 47, 2403–2413 CrossRef CAS.
- E. H. H. Chow, D.-K. Bucar and W. Jones, Chem. Commun., 2012, 48, 9210–9226 RSC.
- D. Musumeci and M. D. Ward, CrystEngComm, 2011, 13, 1067–1069 RSC.
- P. W. Carter, A. C. Hillier and M. D. Ward, J. Am. Chem. Soc., 1994, 116, 944–953 CrossRef CAS.
- C. M. Yip and M. D. Ward, Biophys. J., 1996, 71, 1071–1078 CrossRef CAS PubMed.
- G. Mao, L. Lobo, R. Scaringe and M. D. Ward, Chem. Mater., 1997, 9, 773–783 CrossRef CAS.
- T. Jung, X. Sheng, C. K. Choi, W.-S. Kim, J. A. Wesson and M. D. Ward, Langmuir, 2004, 20, 8587–8596 CrossRef CAS PubMed.
- S. D. Durbin and W. E. Carlson, J. Cryst. Growth, 1992, 122, 71–79 CrossRef CAS.
- A. Burger and R. Ramberger, Mikrochim. Acta, 1979, 72, 259–271 CrossRef.
- A. Burger and R. Ramberger, Mikrochim. Acta, 1979, 72, 273–316 CrossRef.
- U. J. Griesser, D. Weigand, J. M. Rollinger, M. Haddow and E. Gstrein, J. Therm. Anal. Calorim., 2004, 77, 511–522 CrossRef CAS.
- D. E. Braun, T. Gelbrich, V. Kahlenberg, R. Tessadri, J. Wieser and U. J. Griesser, J. Pharm. Sci., 2009, 98, 2010–2026 CrossRef CAS PubMed.
- T. Threlfall, Org. Process Res. Dev., 2003, 7, 1017–1027 CrossRef CAS.
- A. Ito, M. Yamanobe-Hada and H. Shindo, J. Cryst. Growth, 2005, 275, e1691–e1695 CrossRef CAS.
- Y. Kishi and M. Matsuoka, Cryst. Growth Des., 2010, 10, 2916–2920 CAS.
- R. Thakuria, M. D. Eddleston, E. H. H. Chow, G. O. Lloyd, B. J. Aldous, J. F. Krzyzaniak, A. D. Bond and W. Jones, Angew. Chem., Int. Ed., 2013, 52, 10541–10544 CrossRef CAS PubMed.
- B. L. Weeks, C. M. Ruddle, J. M. Zaug and D. J. Cook, Ultramicroscopy, 2002, 93, 19–23 CrossRef CAS.
- T. Yaji, K. Yoshida, S. Isoda, T. Kobayashi, N. Sato and I. Shirotani, Proc. 4th Int. Conf. Nano-Mol. Electron., 2001, vol. 393, pp. 319–324 Search PubMed.
- Y. V. Mnyukh, J. Cryst. Growth, 1976, 32, 371–377 CrossRef CAS.
- Y. V. Mnyukh, Mol. Cryst. Liq. Cryst., 1979, 52, 163–199 CrossRef.
- Y. V. Mnyukh, Mol. Cryst. Liq. Cryst., 1979, 52, 201–217 CrossRef.
- F. P. A. Fabbiani and C. R. Pulham, Chem. Soc. Rev., 2006, 35, 932–942 RSC.
- H. K. Chan and E. Doelker, Drug Dev. Ind. Pharm., 1985, 11, 315–332 CrossRef CAS.
- M. C. Perkins, M. Bunker, J. James, S. Rigby-Singleton, J. Ledru, C. Madden-Smith, S. Luk, N. Patel and C. J. Roberts, Eur. J. Pharm. Sci., 2009, 38, 1–8 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |