
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3-Dimensional stable polyelectrolyte hollow capsules: preparation and spontaneous encapsulation†
Y. D. Chena,
J. J. Xua,
Y. Wanga,
H. Chen*a,
Q. J. Luo*b,
X. D. Lia and
W. P. Zhuc
aAffiliated Stomatology Hospital, College of Medicine, Zhejiang University, Hangzhou, 310006, China. E-mail: huic66@hotmail.com
bThe First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, 310003, P. R. China. E-mail: luoqiaojie@zju.edu.cn
cMOE Key Laboratory of Macromolecule Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
First published on 5th January 2017
Abstract
In this study, by alternatively depositing a poly(styrene)-co-poly(acrylic acid)/poly(allylamine hydrochloride) multilayer on the poly(styrene sulfonate sodium) hybrid CaCO3 spherical templates, a series of size-controlled and low-dispersed polyelectrolyte capsules were obtained after the removal of hybrid templates. These polyelectrolyte capsules can retain their 3-dimensional spheric shape after being dried at room temperature. Through investigating the structure and key factors for the formation of novel capsules, formation mechanism of the 3-dimensional stable capsule wall was discussed. Further investigation showed that the dry capsules could spontaneously entrap water-soluble positively charged substances from the bulk solution into the capsule interior, just like the wet capsules.
Introduction
Alternatively depositing oppositely charged polyelectrolyte (PE) pairs on the sacrificial cores to prepare PE hollow particles has attracted worldwide attention not only due to the facile control over particle size, geometry, wall thickness, and components, but also due to the uniformity of their constructs.1–5 In addition to the conventional sacrificial templates, some templates with a special structure and properties could also be used to prepare PE capsules with unique properties.6–8 Among these special templates, polymer built in hybrid CaCO3 microparticles were by far the most often employed sacrificial templates due to their non-toxicity, uniformity, monodispersity, and yet easy availability. Using this hybrid CaCO3 templates-assisted synthesis, biologically active polymeric capsules,9,10 multi-compartment11 and stimuli-responsive capsules,12 and capsules with other unique properties could be obtained.13–16 Since these types of templates could endow the capsules with some special properties, these templates could be called “functional templates”.8In our previous studies, the poly(styrene)-co-poly(acrylic acid) (PS–PAA) built-in hybrid CaCO3 microspheres were synthesized as functional templates to prepare “micelle-enhanced” polyelectrolyte capsules.17,18 Recently, a novel type of 3-dimensional (3-D) stable polymeric capsule was prepared via alternatively depositing PS–PAA and poly(allylamine hydrochloride) (PAH) on the PS–PAA hybrid CaCO3 spherical templates.19 These capsules had a thick and robust capsule wall and could retain the spherical shape after being dried at room temperature. A study for the formation mechanism of this capsule suggested a novel fabrication pattern between the oppositely charged PE pairs. Moreover, further studies revealed that the PS–PAA hybrid template and PS–PAA/PAH multilayer were indispensable factors to obtain such capsules.
In this study, another type of hybrid templates, poly(styrene sulfonate sodium salt) (PSS) built in CaCO3 spheric particles, were employed as functional templates to prepare polymeric capsules via alternative deposition of PS–PAA and PAH. Consequently, a series of size-controlled, low-dispersed, and 3-D stable PE capsules were also obtained. Herein, structure, properties, and formation mechanism of these capsules were investigated. Moreover, it was found that the dry capsules could spontaneously entrap the water-soluble positively charged rhodamine B into the capsule interior with a high efficiency, just like the fresh wet capsules.
Results and discussion
Synthesis of the hybrid templates
Using 1 mg mL−1 PSS, three batches of almost monodispersed hybrid CaCO3 microparticles were prepared. The size of the microspheres was modulated with the stirring speed, and the size range extended from 1 to 10 μm. Three batches of the particle sizes of 7.0 μm, 4.9 μm, and 2.3 μm were shown in Fig. 1. The thermogravimetric analysis (TGA) curve was used to determine the content of PSS in the hybrid microspheres of the batch a–c. The hybrid microspheres contained about 8–10% PSS (8, 8, and 10% for a, b and c, respectively).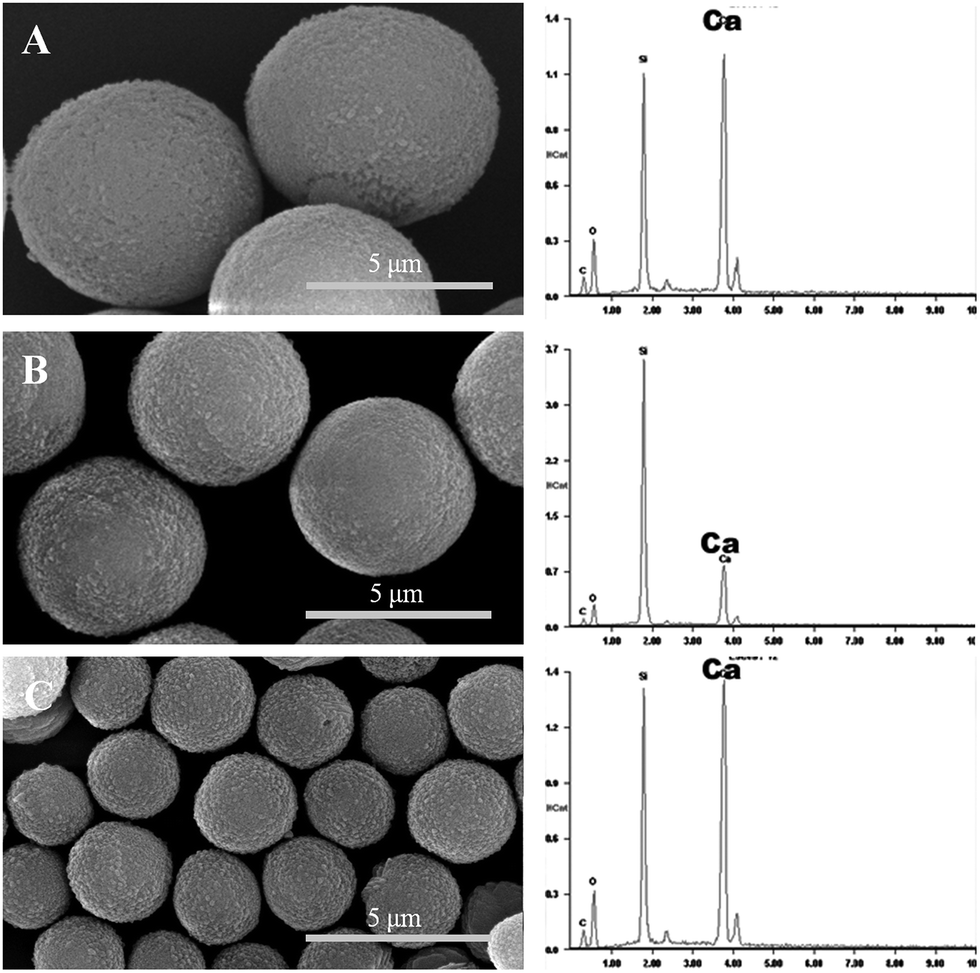 | ||
| Fig. 1 SEM images (left column) and EDAX spectra (right column) of the three batches. (A) Batch a; (B) batch b; and (C) batch c. EDAX results showed high levels of Ca in the PSS hybrid CaCO3 templates. | ||
Fabrication of the polyelectrolyte capsules
PS–PAA and PAH were alternatively deposited on the hybrid microspheres, and the templates were dissolved in trisodium EDTA after the deposition of twelve layers. Fig. 2 shows that the template-removed microparticles were spherical in shape. EDAX was used to detect whether the templates were completely removed. All of the PE coated templates had a high content of calcium; however, calcium could not be detected in the template-removed microparticles. These results suggested that the complete removal of the hybrid CaCO3 templates was successful. | ||
| Fig. 2 SEM images (left column) and EDAX spectra (right column) of the template-removed particles. (A) Batch a; (B) batch b; (C) batch c. EDAX results showed that in the template-removed microparticles, Ca could not be detected. | ||
The mean sizes of the three template-removed microspheres in the wet state, measured with a particle-size analyzer, were 7.6 μm, 5.4 μm, and 2.6 μm, whereas the mean sizes of the dry template-removed microspheres were 6.8 μm, 4.8 μm, and 2.1 μm. The drying process led to a decrease in the capsule size to some extent. However, the size measurements were not consistent with the SEM results. As observed in the SEM images, the mean sizes of the three batches were 4.2 μm, 3.1 μm, and 1.2 μm.
TEM was expected to find the hollow interior structure of the template-removed microparticles of batch a (Fig. 3); however, it did not show the hollow interior structure of the template-removed microparticles. Interestingly enough, once being exposed under the strong electron beam, the microspheres size quickly decreased. The reason might be that the strong electron beam could heat up the capsules, which led to the shrinkage of the capsules. This similar phenomenon was previously reported.20–22 It was difficult to obtain the original images of the template-removed microspheres due to their quick shrinkage. The two images for batch a in Fig. 3 were obtained while they changed in size. The size of the microparticles changed from 5.0 μm to 2.7 μm (Fig. 3A and B). Clearly, this type of microparticle was sensitive to a strong electron beam, which could also account for the difference in size between the SEM observation and size measurement. Moreover, these results strongly suggested that the templates were completely removed, and the resultant microparticles were hollow.
 | ||
| Fig. 3 (A) TEM images of the 3-D stable capsules of batch a; (B) TEM images of the decreased 3-D stable capsules of (A) under a higher electron beam. | ||
CLSM was used to observe the real time change from the fresh wet capsule to the dry capsule at room temperature. As shown in Fig. 4, most of the dry polyelectrolyte capsules still retained their 3-D spherical shape. The fluorescence intensity of the dry capsule wall weakly changed as compared to that of the wet capsule wall, and the capsule size seemed to decrease to some extent. The drying process produced aggregation of the fluorescence substances, leading to autobleaching to some extent, which should account for the weakened fluorescence intensity of the dry capsule wall. According to the results obtained from FESEM-EDAX, TEM, and CLSM analyses, the template-removed microparticles should be hollow polyelectrolyte capsules with 3-D stability.
 | ||
| Fig. 4 CLSM images of the 3-D stable capsules of batch a. (A) The fresh wet 3-D stable capsules; (B) the dried 3-D stable capsules. | ||
The structure of the 3-D stable polyelectrolyte capsules
Because the capsule wall was sensitive to a strong electron beam, a cold field emission scanning electronic microscope (Hitachi S-4800, Japan) was used to further observe the wall structure of the crushed hollow microspheres (Fig. 5). The size and morphology of the capsules under cold FESEM observation almost did not change, which further confirmed that these types of PE capsules were sensitive to heating. Statistical analysis based on a series of SEM images suggested that the thickness of the capsule wall of the batch a was about 200 nm (Fig. 5B). The capsule wall of this type was far thicker than the wall of the PE capsules with the deposition of 6 bilayers, which was about 30 nm. The wall of these capsules seemed to be extremely robust, which made the dried capsules retain their 3-D spherical shape. | ||
| Fig. 5 FESEM images of (A) the inner morphology of the crashed hollow sphere; (B) a typical image shows the wall thickness of a hollow sphere. | ||
UV-vis analysis showed that the thickness growth of the PS–PAA/PAH film had an approximately linear profile with the number of PS–PAA/PAH bilayers (Fig. S1†), which was similar to our previous report.19 The thickness of each layer was about 2 nm. Six PAH/PS–PAA bilayers could not account for more than a 200 nm thickness of the capsule wall. PSS released from the template removal should be involved in the reconstruction of the capsule wall, which increased the thickness of the capsule wall to 200 nm.
The mean mass of PSS in a template was 8% of the total mass of the template based on the TGA curve (3.66 × 10−11 g) and its corresponding capsule was about 3.13 × 10−11 g, according to eqn (1)–(3) mentioned hereinafter. The mass of PAH and PS–PAA in the capsule wall can be considered to be nearly in proportional with the thickness of PAH and PS–PAA. In combination of the thickness of each layer and the thickness of the capsule wall based on SEM, the total content of PS–PAA and PAH from the assembly procedure was evaluated to be about 15% of the capsule mass according to eqn (4). Correspondingly, PSS from the templates accounted for the other 85% percent of the mass of the capsule wall. Based on the abovementioned results, it could be calculated that about 86% PSS in the template was remained in the resultant hollow particle, and the other 14% PSS was released into the bulk solution through the capsule wall according to eqn (5). No substance could be observed in the interior of the dried hollow particles (Fig. 5). The PSS remained in the resultant hollow particle should be all infused into the capsule wall rather than existing as an encapsulated polymer evenly dispersed in the capsule interior. As a result, the PE capsule had a robust capsule wall with the thickness of 200 nm and could retain its spherical shape after being dried at room temperature.
| Vtemplate = (4/3)πr3 = 1.79 × 10−10 cm3 | (1) |
| WPSS = 8%Wtemplate = 8%ρtemplateVtemplate = 8% × (2.56 g cm−3 × 1.79 × 10−10 cm3) = 3.66 × 10−11 g | (2) |
| Wcapsule = 4πr2dρcap = 3.69 × 10−11 g | (3) |
| CPSS = (1 − do/d) × 100% = 1 − 30/200 × 100% = 85% | (4) |
| PPSS = [(Wcapsule × CPSS)/WPSS] × 100% = 86% | (5) |
The formation mechanism of the 3-D stable polyelectrolyte capsules
Based on the abovementioned results and our previous study,19 a possible mechanism for the formation of the capsules could be deduced. Once the PS–PAA/PAH multilayer was formed on the hybrid particles, a thorough rinsing process was carried out with deionized water. According to the previous report,19 the ionization of PAA segments of PS–PAA were strongly constrained, which produced a strong aggregate tendency of the PS–PAA molecules. Consequently, the interaction between PAH and PS–PAA was weakened, and this weakened interaction in turn could provide a higher degree of freedom for the PAH molecules in the PAH/PS–PAA multilayer.The inner capsule wall was a positively charged PAH layer, which was coarse because of the roughness of the template surface. As reported,23,24 the coarse surface could absorb more oppositely charged PEs and produce a thicker complex layer. As a result, the released PSS was first adsorbed onto the inner multilayer to form an additional PAH/PSS multilayer, which made the total capsule wall even thicker.
With the increasing PSS concentration in the capsule interior during the continuous templates dissolution, PSS had a strong tendency to move outward to the bulk solution through the hole filled with water in the PE multilayer.23 Consequently, a great deal of released PSS was further stuffed in the multilayer and led to an even thicker multilayer. The content of PSS in the multilayer was far more than the content of PAH, which made the (PS–PAA + PSS)/PAH multilayer unstable. In general, to make the multilayer stable, PSS would leak out of the unstable multilayer into the bulk solution to decrease the PSS content in the multilayer. However, due to the nature of the PS–PAA/PAH multilayer,19 PAH molecules with a high degree of freedom could be stretched in a conformational state to interact with more PSS molecules, which could decrease the negative potential of the whole multilayer to the greatest extent. As a result, each PAH molecule could interact with many PSS molecules to fabricate a novel (PS–PAA + PSS)/PAH PE membrane. After the template dissolution, a majority of the released PSS was rebuilt into the capsule wall and the novel 3-D stable PE capsules were obtained.
The spontaneous deposition of the wet capsules and dry capsules
The PE capsules based on the PSS hybrid CaCO3 templates could spontaneously induce positively charged substances into the capsule interior.18 Rhodamine B (MW = 479 Da) was used as the positively charged model to study the encapsulation properties of these types of capsules.18 Induced deposition of rhodamine B into the wet capsules was studied by CLSM, as shown in Fig. 6. After the addition of rhodamine B, the fluorescence intensity inside the capsules gradually increased. Selected images of the capsules at 0, 30, 90, and 120 s during the loading process are presented in Fig. 6A–D. It can be observed that the fluorescence intensity inside the capsule interior was significantly higher than that in the environment, indicating the even higher rhodamine B concentration inside the capsule than that in the bulk solution. The dynamics curve of the induced deposition showed that the fluorescent intensity in the selected capsule approached the maximum value after about 165 s (Fig. 8). | ||
| Fig. 6 Spontaneous deposition of rhodamine B for the fresh wet 3-D stable capsules of batch a. The images at selected times. (A) 0 s; (B) 30 s; (C) 90 s; and (D) 120 s. | ||
Induced deposition of rhodamine B into the dry capsules was studied by CLSM, as shown in Fig. 7. The fluorescence intensity of rhodamine B inside the capsule was not detected until 45 s. Selected images of capsules at 45 s, 810 s, and 1350 s during the loading process are presented in Fig. 7B–D. The fluorescence intensity inside the capsule interior was significantly higher than that in the environment, indicating the higher rhodamine B concentration in the capsule interior. The dynamics curve of the induced deposition showed that the fluorescent intensity in the selected capsule reached the maximum value after 1410 s (Fig. 8). For the dry 3-D stable capsules, the spontaneous deposition of rhodamine B was even slower than that of the fresh wet 3-D stable capsules.
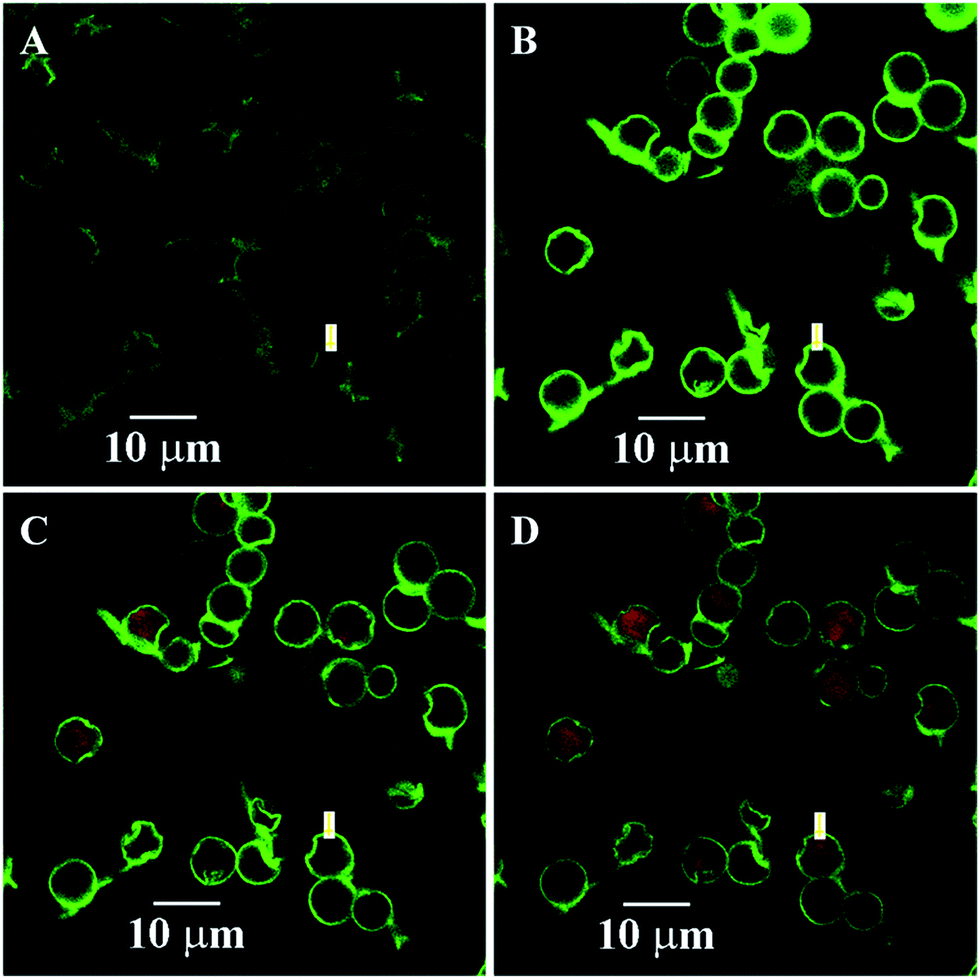 | ||
| Fig. 7 Spontaneous deposition of rhodamine B for the dry 3-D stable capsules of batch a. The images at selected times. (A) 0 s; (B) 45 s; (C) 810 s; and (D) 1350 s. | ||
 | ||
| Fig. 8 Encapsulation dynamics curve of rhodamine B for the fresh wet 3-D stable capsules and for the dry 3-D stable capsules. | ||
Although almost all of the PSS released from the templates were involved in the reconstruction of the capsule wall, there might still be a small part of the PSS left inside the capsule interior as encapsulated substances, which could serve as a driving force to entrap rhodamine B into the capsule interior. For the dry 3-D stable polyelectrolyte capsules, such encapsulated PSS was physically adsorbed onto the inner layer of the dry capsule wall. When the dry 3-D stable capsules were reimmersed in water, the physically adsorbed PSS could be redissolved into the capsule interior, which still served as a driving force to entrap rhodamine B into the capsule interior. At the beginning of the dissolution of the physically adsorbed PSS, spontaneous deposition could not be detected due to too little content of PSS inside the capsule. This could be explained by the lack of spontaneous deposition during the initial 45 s. With the PSS concentration increasing inside the capsule, the spontaneous deposition could be detected. Moreover, the drying process produced an even tighter construction of the 3-D stable capsule wall, which inevitably led to a slower speed of the spontaneous deposition of the dry 3-D stable capsule than that of the fresh wet 3-D stable capsule.
Experimental
Materials
PAH (MW = 70 kDa) was from Aldrich. PSS (MW = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) and PAA (MW = 15000 Da) were purchased from Sigma. PS–PAA (MW = 12500 Da), acid number = 210 mg g−1, fv = 2:1, Rohm and Hass company, USA) was received as a gift from Prof. Linhai Yue (Zhejiang University, P. R. China). Rhodamine B was purchased from Aladdin reagent database Inc. PAH was labeled with FITC in our lab (the molar ratio of PAH to FITC is 1:4). All the other reagents were commercially available and used as received.
000 Da) and PAA (MW = 15000 Da) were purchased from Sigma. PS–PAA (MW = 12500 Da), acid number = 210 mg g−1, fv = 2:1, Rohm and Hass company, USA) was received as a gift from Prof. Linhai Yue (Zhejiang University, P. R. China). Rhodamine B was purchased from Aladdin reagent database Inc. PAH was labeled with FITC in our lab (the molar ratio of PAH to FITC is 1:4). All the other reagents were commercially available and used as received.
Preparation of the hybrid CaCO3 microparticles
The hybrid CaCO3 microparticles were synthesized according to the previous studies.25,26 In brief, PSS was completely dissolved in 200 mL of 0.025 M calcium nitrate solution in a beaker under magnetic agitation at room temperature, into which an equal volume of 0.025 M sodium carbonate solution in another beaker was rapidly poured. The final PSS concentration was 1 mg mL−1. At the end of the reaction, the precipitated CaCO3 particles were collected and washed using a membrane filtration apparatus equipped with a 0.45 μm cellulose filter. The precipitate was dried at 50 °C in a blowing oven. The dried sample was kept for further experiments.Preparation of the PE microcapsules through LbL technique
PS–PAA was dissolved into a Na2CO3 solution (5 mg mL−1) as the negatively charged polyelectrolyte solution, and the final concentration of PS–PAA was 2 mg mL−1. PAH was directly dissolved in deionized water to be a 2 mg mL−1 solution as the positively charged polyelectrolyte solution. The concentration of NaCl in the two solutions was 0.5 M. The first deposition layer on the hybrid CaCO3 template was PAH. After 10 min of adsorption, the coating templates were centrifuged and rinsed three times with pure water. The oppositely charged PS–PAA was then added. Each adsorption step was followed by triple rinsing with pure water. After the deposition of twelve layers, the hybrid cores were dissolved in trisodium EDTA for 30 min (pH about 7.28). After centrifugation, the supernatant was removed, and the capsules were resuspended in a fresh trisodium EDTA solution. This procedure with trisodium EDTA was repeated several times, and the resultant suspension of the formed microcapsules was washed three times with deionized water and stored in the centrifuge tubes for further experiments.Encapsulation dynamics of rhodamine B for wet or dry capsules
The experimental methods were performed according to our previous reports.17 The concentration of rhodamine B in the loading solution was 2 μg mL−1. Induced loading of the positively charged substances and the loading kinetics of the hybrid capsules were studied with real-time confocal microscopy. CLSM images were obtained using a Zeiss laser scanning system equipped with a 40× objective. Using constant laser intensity and PMT gain, images were auto-collected every 15 s. The capsule suspension (20 μL, pH 6.4) was dropped onto the glass slide. After the capsules were attached onto the glass slide, 3 μL of probe solution (pH 6.4) was gently dropped into the suspension and the CLSM images were immediately obtained. The fluorescence intensity from the regions of interest inside the optically sectioned capsule was plotted versus time.As for the encapsulation of rhodamine B for dry capsules, 20 μL of capsule suspension was dropped onto the piranha treated glass slide, and then dried at the room temperature. A 20 μL aliquot of rhodamine B solution (50 ng mL−1) was gently dropped onto the right place with the dry capsules and the CLSM images were immediately obtained. The fluorescence intensity from the regions of interest inside the optically sectioned capsules was plotted versus time.
Characterization
Scanning force microscopy (SFM) images were obtained in air at 20–25 °C by using a nanoscope III multimode scanning force microscope (tapping mode, Digital Instruments, Santa Barbara, CA). The glass slides were dried with a N2 blower and directly used for SFM observation. Thermogravimetric analysis (TGA) was conducted using a TA Instrument SDT 2960. Samples were heated at 10 °C min−1 from room temperature to 1000 °C under a dynamic nitrogen atmosphere (flow rate = 70 mL min−1). The size of different particles was determined by a Coulter LS-230 laser particle size analyzer (Miami, USA). FESEM-EDX was carried out using a field emission scanning electronic microscope (FESEM, SIRION-100, Phillips) with elemental analysis using energy-dispersive X-ray spectroscopy (Genenis4000, EDAX) after a gold–palladium layer was sputtered by the E-1020 ion sputter for 120 s. For general FESEM samples, they were prepared by applying a drop of capsule suspension onto the freshly cleaved mica, and then drying at room temperature. EDX was used to detect the presence of calcium after the dissolution of the templates. Transmission electron microscopy (TEM) was performed using a JEM 1230 microscope at an acceleration voltage of 80 kV. Samples were deposited from the aqueous solutions onto the copper EM grids precoated with a thin film of Formvar. Water was allowed to evaporate from the grids at room temperature. The size of the polymeric aggregates was measured via laser light scattering detector (90Plus, Brookhaven).The hybrid capsule suspension was dropped onto a clean mica and dried at room temperature. The other clean mica was used to crush the dry capsules. The samples were coated with gold before FESEM observation.
Conclusions
By fabricating a PS–PAA/PAH multilayer on the PSS hybrid templates, a series of size-controlled, monodispersed, and 3-D stable PE capsules were obtained. By investigating the structure of the capsule, a possible formation mechanism of the 3-D stable capsule was proposed, which suggested a novel fabrication between the oppositely charged polyelectrolyte pairs. Moreover, the dry 3-D stable polyelectrolyte capsules could spontaneously entrap the water-soluble substances into its interior.Acknowledgements
The author would like to thank Prof. Jiacong Shen (Zhejiang University) and Prof. Xiaoyin Hong (Tsinghua University) for their helpful discussion and suggestions for this study. This work was supported by the Nature Natural Science Foundation of China (81600835, 51603180) and China Postdoctoral Science Foundation (2015M580517, 2016M601952).Notes and references
- Y. Kashcooli, K. Park, A. Bose, M. L. Greenfield and G. D. Bothun, Biomacromolecules, 2016, 14, 3838–3844 CrossRef PubMed.
- J. E. Wong, A. M. Diez-Pascual and W. Richtering, Macromolecules, 2009, 42, 1229–1238 CrossRef CAS.
- C. S. Peyratout and L. Dahne, Angew. Chem., Int. Ed., 2004, 43, 3762–3783 CrossRef CAS PubMed.
- A. D. Price, A. N. Zelikin, Y. J. Wang and F. Caruso, Angew. Chem., Int. Ed., 2009, 48, 329–332 CrossRef CAS PubMed.
- W. X. Song, H. Mohwald and J. B. Li, Biomaterials, 2010, 31, 1287–1292 CrossRef CAS PubMed.
- N. Feoktistova, J. Rose, V. Z. Prokopovic, A. S. Vikulina, A. Skirtach and D. Volodkin, Langmuir, 2016, 32, 4229–4238 CrossRef CAS PubMed.
- Y. J. Wang and F. Caruso, Adv. Mater., 2006, 18, 795–800 CrossRef CAS.
- X. D. Li, Q. L. Hu, L. H. Yue and J. C. Shen, Chem.–Eur. J., 2006, 12, 5770–5778 CrossRef CAS PubMed.
- B. G. De Geest, R. E. Vandenbroucke, A. M. Guenther, G. B. Sukhorukov, W. E. Hennink, N. N. Sanders, J. Demeester and S. C. De Smedt, Adv. Mater., 2006, 18, 1005–1009 CrossRef CAS.
- E. Kozhunova, Q. Ji, J. P. Hill and K. Ariga, J. Nanosci. Nanotechnol., 2015, 15, 2389–2393 CrossRef CAS PubMed.
- O. Kreft, M. Prevot, H. Mohwald and G. B. Sukhorukov, Angew. Chem., Int. Ed., 2007, 46, 5605–5608 CrossRef CAS PubMed.
- C. Dejugnat and G. B. Sukhorukov, Langmuir, 2004, 20, 7265–7269 CrossRef CAS PubMed.
- C. Y. Wang, C. Y. He, Z. Tong, X. X. Liu, B. Y. Ren and F. Zeng, Int. J. Pharm., 2006, 308, 160–167 CrossRef CAS PubMed.
- J. Li, Z. Y. Jiang, H. Wu, L. Zhang, L. H. Long and Y. J. Jiang, Soft Matter, 2010, 6, 542–550 RSC.
- T. Borodina, E. Markvicheva, S. Kunizhev, H. Moehwald, G. B. Sukhorukov and O. Kreft, Macromol. Rapid Commun., 2007, 28, 1894–1899 CrossRef CAS.
- K. Gopal, Z. H. Lu, M. M. de Villiers and Y. Lvov, J. Phys. Chem. B, 2006, 110, 2471–2474 CrossRef CAS PubMed.
- X. D. Li, T. Lu, J. X. Zhang, J. J. Xu, Q. L. Hu, S. F. Zhao and J. C. Shen, Acta Biomater., 2009, 5, 2122–2131 CrossRef CAS PubMed.
- X. D. Li, X. H. Li, J. X. Zhang, S. F. Zhao and J. C. Shen, J. Biomed. Mater. Res., Part A, 2008, 85, 768–776 CrossRef PubMed.
- X. D. Li, J. X. Zhang, Q. L. Hu and X. H. Li, J. Controlled Release, 2011, 363, 64–72 CrossRef CAS PubMed.
- K. Kohler and G. B. Sukhorukov, Adv. Funct. Mater., 2007, 17, 2053–2061 CrossRef.
- S. Leporatti, C. Gao, A. Voigt, E. Donath and H. Mohwald, Eur. Phys. J. E, 2001, 5, 13–20 CrossRef CAS.
- W. X. Song, Q. He, H. Mohwald, Y. Yang and J. B. Li, J. Controlled Release, 2009, 139, 160–166 CrossRef CAS PubMed.
- A. A. Antipov and G. B. Sukhorukov, Adv. Colloid Interface Sci., 2004, 111, 49–61 CrossRef CAS PubMed.
- D. V. Volodkin, A. I. Petrov, M. Prevot and G. B. Sukhorukov, Langmuir, 2004, 20, 3398–3406 CrossRef CAS PubMed.
- A. Jada and A. Verraes, Colloids Surf., A, 2003, 219, 7–15 CrossRef CAS.
- W. J. Tong, W. F. Dong, C. Y. Gao and H. Mohwald, J. Phys. Chem. B, 2005, 109, 13159–13165 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25613a |
| This journal is © The Royal Society of Chemistry 2017 |