
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNiO@MnO2 core–shell composite microtube arrays for high-performance lithium ion batteries†
Yuan Zhong,
Huan Huang,
Kai Wang,
Zhishun He,
Shasha Zhu,
Ling Chang,
Haibo Shao,
Jianming Wang * and
Chu-nan Cao
* and
Chu-nan Cao
Department of Chemistry, Zhejiang University, Hangzhou 310027, PR China. E-mail: wjm@zju.edu.cn; Fax: +86-571-87951895; Tel: +86-571-87951513
First published on 17th January 2017
Abstract
Tubular array structures are very attractive for electrochemical energy storage and conversion systems due to their unique physicochemical properties. Herein, a NiO microtube array is fabricated via a facile oxalic acid corrosion method followed by heat treatment. A NiO@MnO2 core–shell composite microtube array is further achieved by the anodic electrodeposition using the NiO microtube array as substrate. When applied as self-supported electrode for lithium ion batteries (LIBs), the NiO@MnO2 core–shell composite microtube array electrode shows excellent lithium storage properties. The electrode delivers a reversible capacity of 510 mA h g−1 at a high rate of 5.1 A g−1, showing its good rate capability. In particular, a reversible capacity of 1573 mA h g−1 is observed after 500 cycles at a current density of 0.53 A g−1, demonstrating the superior cycling performance of the electrode. The electrodeposited MnO2 layer as a protective shell prevents the NiO microtubes from deformation during electrochemical cycling, responsible for the superior cycle stability of the NiO@MnO2 core–shell composite microtube array electrode. The prominent lithium storage performance of the composite microtube array electrode can be attributed to its unique structure characteristics.
1. Introduction
One-dimensional (1D) micro/nanoscale tubular structures with high surface area and large porosity have attracted tremendous attention due to their unique physicochemical properties and extensive applications in energy storage, sensors, catalysts, optics, magnetics and electric transportation.1 Through various techniques such as anodization,2,3 template synthesis,4,5 and approaches based on sheets-rolling mechanism,6 Kirkendall effect,7,8 ripening effect,9,10 electrochemical dealloying11 and other methods,12,13 substantial inorganic tubular materials have been prepared. However, the complex procedures and/or special experimental conditions make most of preparation processes tedious and costly. It is still highly desirable to develop facile, tunable and template-free approaches for synthesis of 1D micro/nanoscale tubular materials.Lithium ion batteries (LIBs) have aroused extensive interest for a variety of applications, including portable electronics, hybrid electric vehicle, and renewable energy systems for intermittent energy sources (e.g., solar and wind).14–17 Intensive investigations on the anode materials with large capacities have been conducted in order to meet the ever-increasing demand for high-performance LIBs. NiO and MnO2 have been considered as promising anode materials for lithium ion batteries (LIBs) because of their high theoretical specific capacity (719 mA h g−1 for NiO, 1232 mA h g−1 for MnO2), abundant nature and environmentally benign.18,19 However, the low electric conductivity and large volume change during Li ion insertion/extraction generally result in the relatively poor lithium storage performance of NiO and MnO2, limiting their practical applications as the anode materials for LIBs. Various micro/nanostructured NiO and MnO2 materials have been fabricated to overcome the above drawbacks.20–23 It is demonstrated that tubular metal oxides materials have exhibited enhanced lithium storage performance due to their advantageous features for the Li+ ion insertion and the volume buffering during charge/discharge process.4,20,24 Furthermore, the hybrid core/shell structures have shown better electrochemical properties than the single component due to their larger surface areas and integral configurations.25,26 Therefore, developing a facile route for fabricating tubular NiO@MnO2 core–shell materials is of significant importance for their application in LIBs.
Herein, for the first time, we report the fabrication of NiO@MnO2 core–shell microtube arrays using a novel and facile strategy based on self-corrosion and electrodeposition. A possible tubular structure formation mechanism is proposed. The as-fabricated NiO@MnO2 composite microtube array electrode exhibits large lithium storage capacity, superior electrochemical cycling stability and good rate performance. The reasons that the NiO@MnO2 composite microtube array electrode shows the prominent lithium storage properties are discussed in detail.
2. Experimental
2.1. Fabrication of NiO@MnO2 core–shell microtube arrays
A nickel film was electrodeposited on the Cu foil in the electrolyte solution with 100 g L−1 NiSO4·7H2O, 35 g L−1 H3BO3, 0.1 g L−1 sodium dodecyl sulfate and 1.8 g L−1 NaOH at 60 °C. The copper foil was polished with 1000# waterproof abrasive paper and ultrasonically cleaned in deionized water for 15 minutes before electrodeposition. The electrodeposition was conducted at a constant current density of 2.5 mA cm−2 for 10 min, with nickel foil as counter electrode. After rinsed with deionized water, the copper foil with the electrodeposited nickel film was put into a 50 mL Teflon-lined stainless steel autoclave containing 10 mL 0.15 M oxalic acid in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v mixture of N,N-dimethylformamide (DMF) and water for hydrothermal reaction at 140 °C for 110 min, unless otherwise noted. After washed by ethanol and dried in air, the obtained nickel oxalate hydrate film was annealed at 350 °C for 2 h in an Ar-filled tube furnace to get the NiO film. The NiO@MnO2 composite film was prepared by an anodic electrodeposition in 0.05 M Mn(Ac)2 aqueous solution at room temperature. The anodic deposition was conducted in a two-electrode cell with the as-obtained NiO film working electrode and a platinum foil counter electrode. The electrodeposition voltage was 3 V and the deposition time was 50 s, unless otherwise noted. After thoroughly rinsed with deionized water, the as-deposited composite film was dried at 80 °C for 10 h in air.
:1 v/v mixture of N,N-dimethylformamide (DMF) and water for hydrothermal reaction at 140 °C for 110 min, unless otherwise noted. After washed by ethanol and dried in air, the obtained nickel oxalate hydrate film was annealed at 350 °C for 2 h in an Ar-filled tube furnace to get the NiO film. The NiO@MnO2 composite film was prepared by an anodic electrodeposition in 0.05 M Mn(Ac)2 aqueous solution at room temperature. The anodic deposition was conducted in a two-electrode cell with the as-obtained NiO film working electrode and a platinum foil counter electrode. The electrodeposition voltage was 3 V and the deposition time was 50 s, unless otherwise noted. After thoroughly rinsed with deionized water, the as-deposited composite film was dried at 80 °C for 10 h in air.
2.2. Material characterizations
The morphologies and microstructures of various film samples were observed by scanning electron microscopy (SEM) using a ZEISS, ULTRA™ 55 with an accelerating voltage of 5 kV, transmission electron microscopy (TEM) and selected area electron diffraction (SAED) using a Philips-FEI Tecnai G2 F30 with an accelerating voltage of 200 kV, equipped with an energy-dispersive X-ray spectroscopy (EDS). The X-ray diffraction (XRD) patterns of film samples were recorded using a Rigaku D/Max 2550 X-ray diffractometer with Cu Kα radiation at 40 kV and 300 mA. Raman spectra were recorded using a Jobin Yvon Labor Raman series HR-800 with an excitation wavelength of 514 nm. The X-ray photoelectron spectroscopy (XPS) analysis was performed by a PHI 5000C X-ray physical electronics photoelectron spectrometer with Mg Kα radiation at 15 kV and 500 W. The binding energies were calibrated with respect to the adventitious C 1s peak at 284.6 eV. The nickel(II) amount in the hydrothermal reaction solution was analyzed by inductively coupled plasma atomic emission spectroscopy (ICP, SPECTRO) on a Thermo Electron Corporation IRIS Intrepid II XSP instrument. The masses of different film samples were determined by a microbalance (Sartorius BT25S).The formation reaction of NiC2O4·2H2O was as follows:
| Ni + H2C2O4 + 2H2O → NiC2O4·2H2O + H2↑ | (1) |
According to reaction (1), the mass of NiC2O4·2H2O on the substrate could be calculated from the Ni(II) amount in the hydrothermal reaction solution and the masses of the substrate with the surface films before and after the reaction. The mass of NiO in the annealed film was determined according to the mass of NiC2O4·2H2O and the mass loss induced by the annealing treatment in Ar atmosphere. The mass loading of NiO was ∼0.2 mg cm−2. The mass of MnO2 was determined by the mass difference of the films before and after electrodeposition. The loading mass of MnO2 was ∼0.04 mg cm−2.
2.3. Electrochemical measurements
The electrochemical measurements were carried out at room temperature (25 °C) using coin cells (CR2025) with lithium metal as the counter and reference electrodes. The as-prepared films were directly used as the working electrode. 1 M LiPF6 in a 1:1 w/w mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) was used as the electrolyte, and a polypropylene membrane (Celgard 2325) was used as the separator. Cell assembly was conducted in an Ar-filled glove box. Galvanostatic charge–discharge cycles of the cells were measured between 3.0 and 0.01 V at various rates with a LAND CT2001A battery testing system. Cyclic voltammetry (CV) measurements were carried out using a CHI660A electrochemical workstation at a scanning rate of 0.1 mV s−1 between 0 and 3 V. Electrochemical impedance spectroscopy (EIS) measurements were carried out by an electrochemical analyzer (Parstat 2273), with the frequency range of 100 kHz to 0.01 Hz and a.c. signal amplitude of 10 mV.
In all the above experiments, the solutions were prepared from analytical reagents and deionized water.
3. Results and discussion
The fabrication of NiO@MnO2 core–shell microtube arrays is schematically illustrated in Fig. 1. Initially, a NiC2O4·2H2O microtube array is fabricated by the self-corrosion process of the electrodeposited Ni film in an oxalic acid solution. A NiO microtube array is achieved by the thermal treatment of the NiC2O4·2H2O microtube array film at 350 °C in Ar atmosphere. MnO2 layer are further deposited on the surface of NiO microtubes by an anodic electrodeposition, resulting in the formation of the NiO@MnO2 core–shell microtube array film.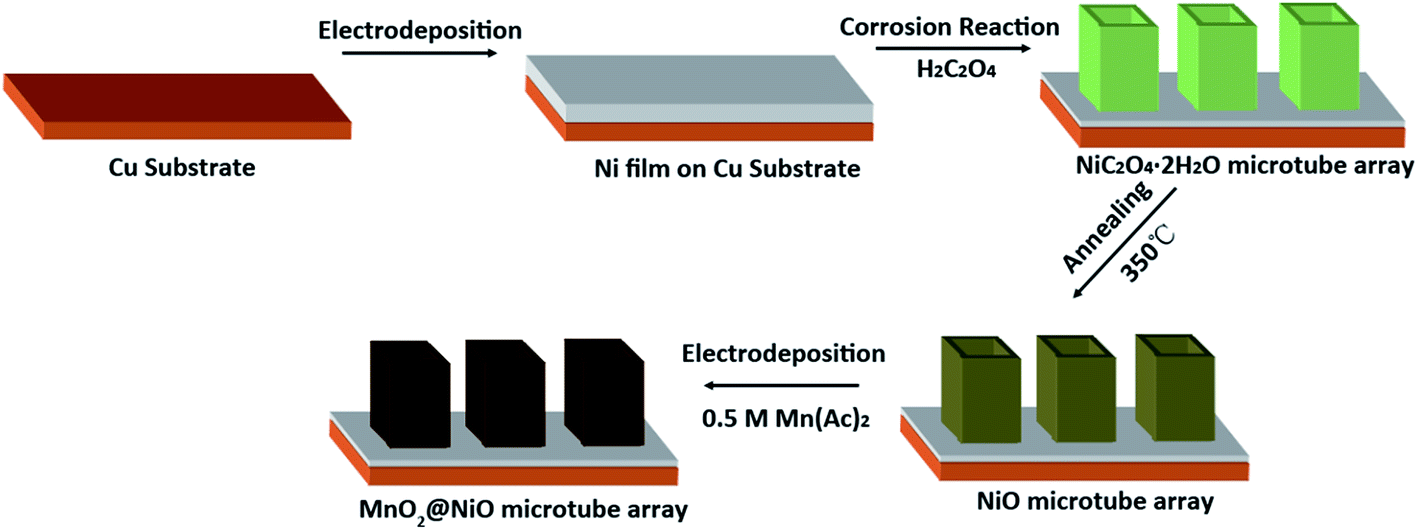 | ||
| Fig. 1 Schematic illustration for fabrication of NiO@MnO2 core–shell microtube arrays. | ||
The XRD pattern of the film fabricated by the self-corrosion process is illustrated in Fig. 2a. The XRD peaks can be attributed to NiC2O4·2H2O, Ni and Cu according to IDSC PDF 65-2865, 65-9026 and 25-0581, implying that Ni is turned into NiC2O4·2H2O by the self corrosion in the oxalic acid solution. The existence of metallic Ni indicates that the transformation from Ni to NiC2O4·2H2O is incomplete, which can result in the good combination of the corrosion product film with the substrate. The Raman spectrum in Fig. 2b confirms the formation of oxalate. The Raman peak at ∼1476 cm−1 is the characteristic peak of oxalate, which can be ascribed to the symmetric stretching mode of OCO.27 The Raman peak at ∼3334 cm−1 originates from the stretching mode of OH, indicating the existence of crystal water. It can be seen from the SEM images in Fig. 2c and d that the as-fabricated NiC2O4·2H2O film is composed of a vertically aligned array of microtubes with a square top view, and the outer diameter of the microtubes ranges from 0.5 to 1 μm. The above results of XRD, Raman and SEM show that the NiC2O4·2H2O microtube array film is formed by the self-corrosion process of the electrodeposited Ni film in the oxalic acid solution.
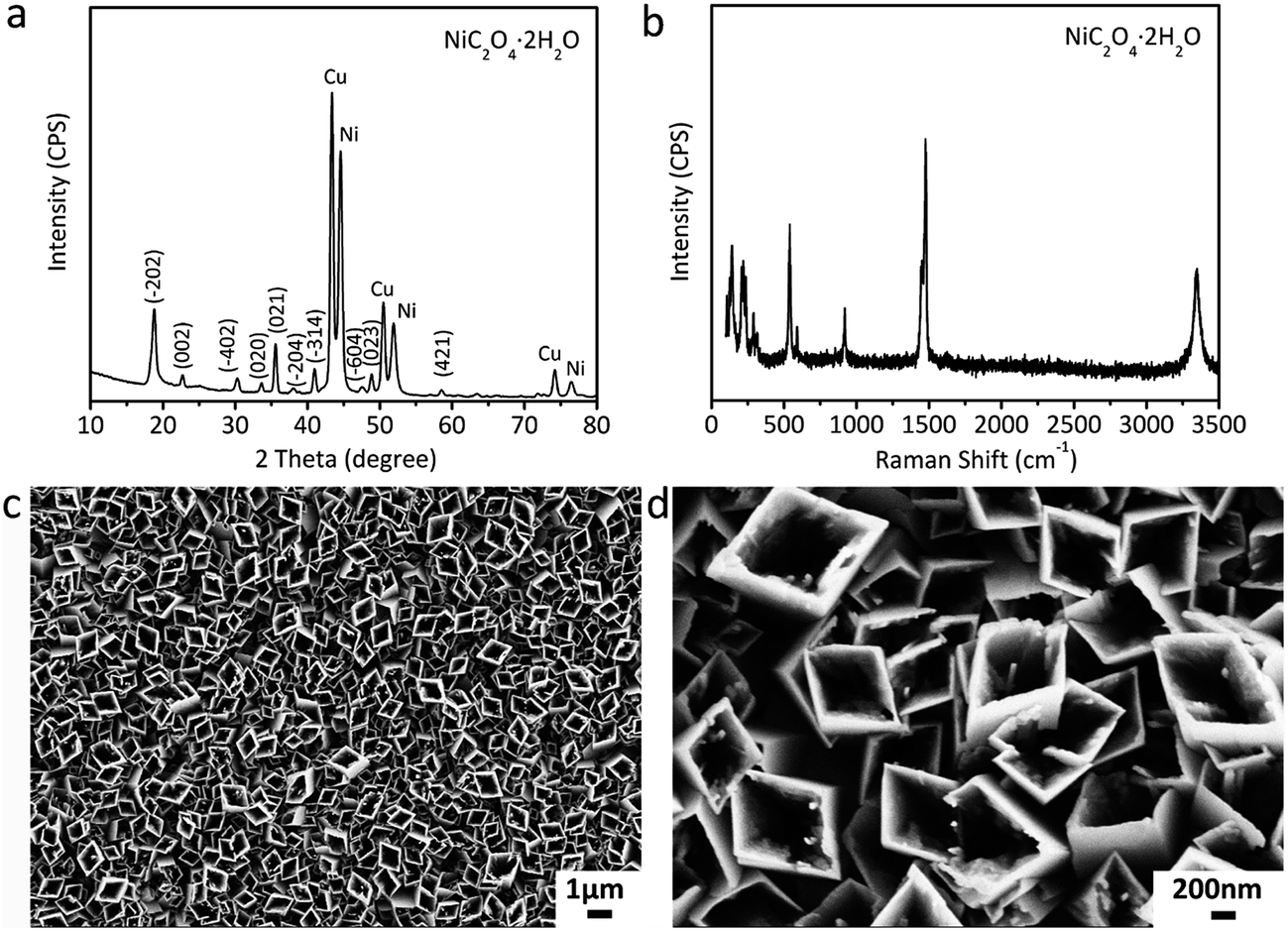 | ||
| Fig. 2 Physical characterizations of the film fabricated by the self-corrosion process. (a) XRD pattern, (b) Raman spectra, (c and d) SEM images. | ||
The morphology evolution of the NiC2O4·2H2O film during the corrosion process as well as the corresponding schematic diagram are illustrated in Fig. 3. The self corrosion reaction (reaction (1)) of nickel in the oxalic acid solution is composed of the following conjugated anodic (reaction (2)) and cathodic reactions (reaction (3)):
| Ni + C2O42− + 2H2O − 2e− → NiC2O4·2H2O | (2) |
| 2H+ + 2e− → H2↑ | (3) |
 | ||
| Fig. 3 Evolution of NiC2O4·2H2O microtube growth. (a) SEM image for 30 min, (b) SEM image for 60 min, (c) SEM image for 75 min, (d) SEM image for 110 min. (e) Schematic diagram of corresponding structure evolution. | ||
NiC2O4·2H2O crystal nuclei and H2 bubbles occur at the anodic and cathodic regions of the electrode surface, respectively. The oriented attachment of primary NiC2O4·2H2O crystals on the nucleation sites due to dipole–dipole interactions28,29 results in the formation of free-standing square micropillars on the substrate (Fig. 3a). As the micropillars grow, the hollowing process induced by the following dissolution/recrystallization reaction of NiC2O4·2H2O occurs:9,10,30
| NiC2O4·2H2O + C2O42− ↔ [Ni(C2O4)2]2− + 2H2O | (4) |
The SEM images in Fig. 3b−d exhibit that the hollowing process proceeds down to the interior from the upper surface of the micropillars. This finally leads to the formation of the NiC2O4·2H2O microtubes as schematically illustrated in Fig. 3e.
The as-fabricated NiC2O4·2H2O microtube arrays are annealed at 350 °C in Ar atmosphere to obtain NiO microtube arrays. It is noted in Fig. 4a that the XRD peaks for β-NiC2O4·2H2O are absent, and the new XRD peaks located at 2θ = 37.2°, 43.3° and 62.8° can be indexed as crystal planes (111), (200) and (220) of cubic NiO (IDSC PDF 47-1049), respectively. This indicates that NiC2O4·2H2O has transformed into NiO. Raman spectra in Fig. 4b confirm the transformation from NiC2O4·2H2O to NiO. The Raman peaks centered at ∼400 and ∼514 cm−1 can be assigned to the first-order transverse optical mode (TO) and one-phonon longitudinal optical mode (LO) of NiO, respectively.31 The Raman peak centered at ∼1060 cm−1 originates from 2LO mode of NiO.31 As illustrated by the SEM images in Fig. 4c and d, the NiO film shows a similar microtube array morphology with the original NiC2O4·2H2O film. A NiO@MnO2 composite film is further prepared by the anodic electrodeposition using the NiO microtube array as substrate. The Raman spectrum of the composite film (Fig. 4b) confirms the coating of MnO2 on the NiO film. The Raman peaks at ∼565 and ∼630 cm−1 can be ascribed to the ν2(Mn–O) symmetric stretching vibration of MnO6 groups and the ν3(Mn–O) stretching vibration in the basal plane of [MnO6] sheets, respectively.32 Fig. 5 displays the XPS spectra of the composite film. The peaks at the binding energies of 654.0 and 642.2 eV in the Mn 2p spectrum (Fig. 5a) can be attributed to the characteristic Mn 2p1/2 and Mn 2p3/2 peaks, respectively. The energy separation of 11.8 eV between the two peaks is in good coincidence with the corresponding value of MnO2.33 In the XPS Mn 3s spectrum (Fig. 5b), the two peaks at 84.4 and 89.2 eV with the separation of 4.8 eV further confirm the existence of MnO2.34 The absence of the MnO2 peaks in the XRD pattern of the composite film (Fig. 3a) indicates its amorphous nature. Compared to the NiO microtubes (Fig. 4c and d), the microtubes in the composite film show reduced diameters and thickened walls (Fig. 4e and f), demonstrating the deposition of MnO2 on the walls of NiO microtubes. This shows the formation of a core–shell structure, with each NiO microtube surrounded by the MnO2 layer. As the electrodeposition time for MnO2 increases, the inner diameter of the composite tubes decreases, and the thickness of the MnO2 layer increases, as illustrated in Fig. 4f and Fig. S1 (the ESI†). It is noted in Fig. S1b† that the composite film obtained by the electrodeposition time of 90 s exhibits the smallest inner diameter and notably thickened MnO2 shell, which could be disadvantageous to the improvement of its electrochemical performance.
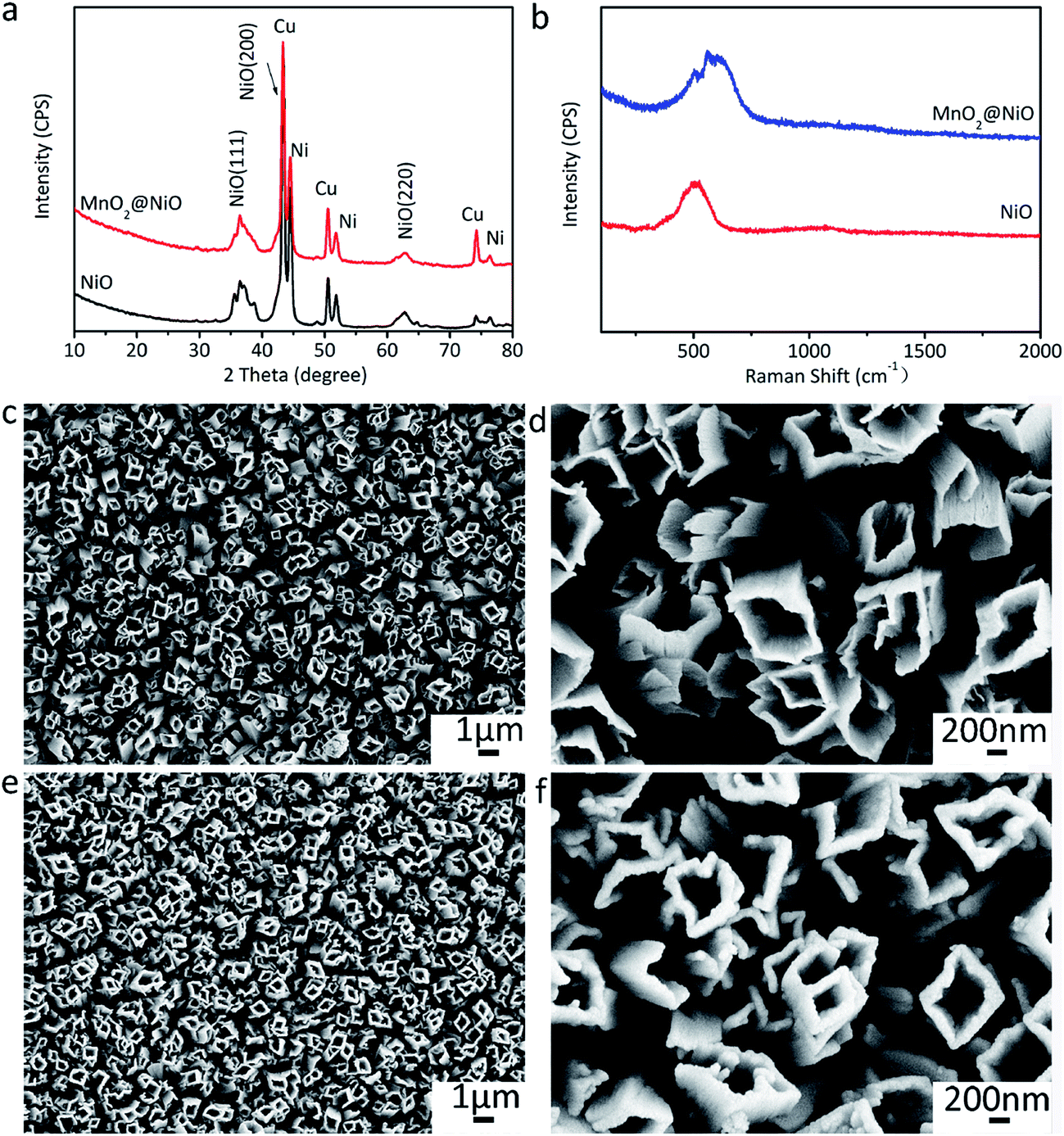 | ||
| Fig. 4 Physical characterizations of the annealed film and composite film. (a) XRD patterns of the annealed film and composite film, (b) Raman spectra of the annealed film and composite film, (c and d) SEM images of the annealed film, (e and f) SEM images of the composite film. | ||
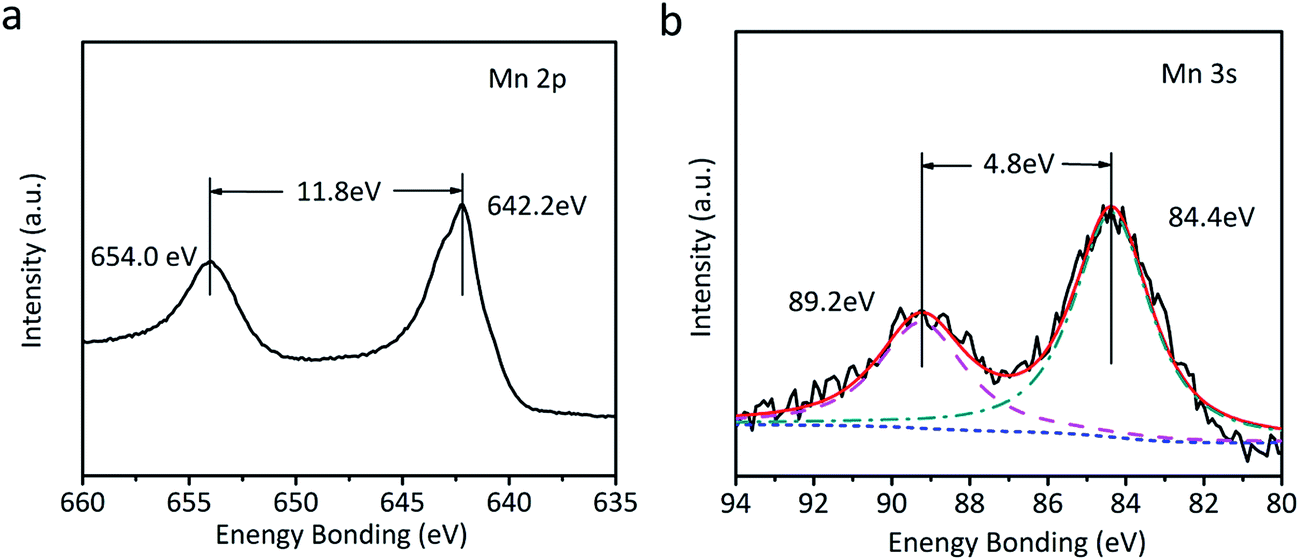 | ||
| Fig. 5 XPS spectra of the composite film. (a) Mn 2p, (b) Mn 3s. | ||
The microstructures of the NiO and NiO@MnO2 tubes are further probed by TEM, STEM and EDS, and the results are illustrated in Fig. 6. Fig. 6a shows a typical NiO tube with an inner diameter of ∼1 μm and a length of ∼2.5 μm. The HRTEM image in Fig. 6b exhibits well-resolved lattice spacings of 0.24 and 0.21 nm, corresponding to the (111) and (200) planes of cubic NiO, respectively. The SAED pattern in the inset of Fig. 6b confirms the polycrystalline nature of cubic NiO. The TEM images in Fig. 6c and d reveal that the surface of the NiO microtube is coated by the MnO2 layer, showing the core–shell structure of the composite microtubes. Meanwhile the good combination between MnO2 layer and NiO tube can be observed. No diffractive ring of MnO2 in the SAED pattern in the inset of Fig. 6d is found, confirming the amorphous nature of MnO2. The compositional line profiles in Fig. 6f exhibits that element Mn can be detected in the outside part of the tube, and element Ni is absent in the same region. This shows that the thickness of the MnO2 layer on the outside walls of the NiO microtubes is ∼35 nm.
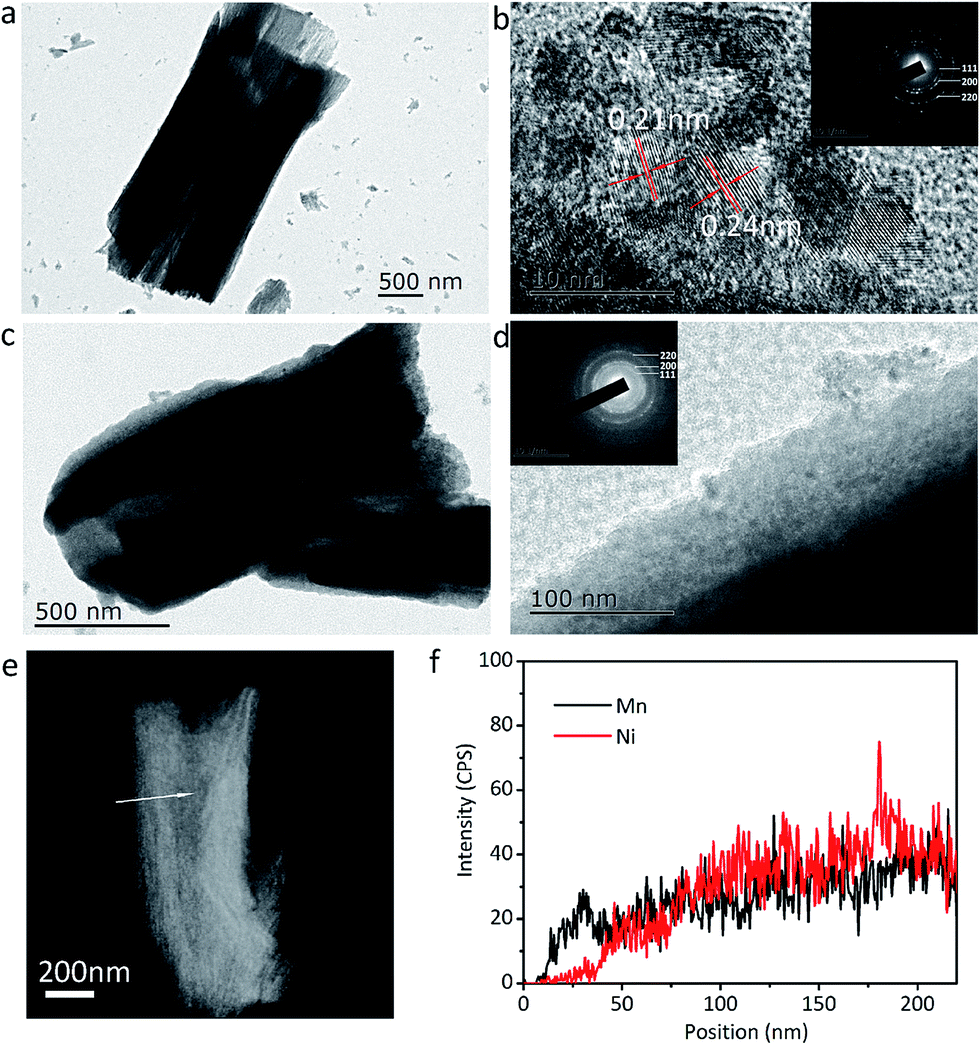 | ||
| Fig. 6 Microstructure characterizations. (a) TEM, (b) HRTEM images and SAED pattern (inset in (b)) of single NiO microtube, (c) TEM, (d) HRTEM images and SAED pattern (inset in (d)) of single NiO@MnO2 microtube, (e) STEM image of single NiO@MnO2 microtube, (f) compositional profile along the line across the tube in (e). | ||
The as-prepared microtube array films are directly applied as the electrodes for the tests of lithium storage properties. Fig. 7a shows the CV curves of the NiO@MnO2 microtube array electrode. Two peaks are observed in the first cathodic process. The reduction peak located at 1.05 V can be ascribed to the conversion of MnO2 to Mn(II), and the strong reduction peak at ∼0.4 V mainly corresponds to the conversions of Mn(II) to Mn and NiO to Ni along with the formation of SEI film.20,22 In the anodic process, the peak located at 1.30 V corresponds to the oxidation of Mn to Mn(II), and the peaks at ∼2.25 V are assigned to the oxidation of Mn(II) to Mn(IV) and Ni to Ni(II).20,22 Note that the CV curves almost overlap in subsequent cycles, demonstrating the stability of the electrode. Fig. 7b illustrates the initial charge/discharge profiles of the NiO@MnO2 microtube array electrode at 0.53 A g−1. The two plateaus at ∼1.0 and 0.4 V in the discharge profiles are consistent with the CV results in Fig. 7a. Little difference in the initial charge profiles is observed, suggesting the reversible lithium storage properties. The capacity loss in the first cycle can be ascribed to the irreversible formation of SEI film. The cycle performances of the electrodes are displayed in Fig. 7c. The reversible capacity of the NiO@MnO2 microtube array electrode gradually increases from 781 mA h g−1 at the first cycle to 1573 mA h g−1 at the 500th cycle, demonstrating the superior cycling performance. The theoretical capacity of the NiO@MnO2 microtube array electrode is calculated to be 805 mA h g−1 in terms of the mass ratio of NiO and MnO2. The extra capacity of the cycled composite electrode can be ascribed to the reversible formation of polymer/gel film resulting from electrolyte decomposition, which is common for transition metal oxide anodes for LIBs.35,36 This may be confirmed by the fact that the main contribution to the capacity increase occurs at low voltage (below 0.8 V) as illustrated in the charge–discharge profiles at the selected cycles of the NiO@MnO2 composite electrode (Fig. S2†).35 In comparison, the reversible capacity of the NiO microtube array electrode progressively increases till the 150th cycle, and henceforth starts to decrease dramatically, showing a relatively poor cycle performance. The electrochemical cycle performances at a current density of 0.53 A g−1 of the NiO@MnO2 microtube array film electrodes obtained by different electrodeposition times for MnO2 are compared in Fig. S3.† The composite film electrode obtained by the electrodeposition time of 10 s shows poorer cyclic performance. The thin MnO2 shell cannot effectively protect the NiO microtubes from deformation during the cycling, thus the capacity fading occurs after 180 cycles. For the composite film electrode achieved by the electrodeposition time of 90 s, the reduced inner diameter and thickened shell of the composite tubes may decrease the transportation rate of liquid electrolyte, which is mainly responsible for its relatively low capacity. The composite film electrode prepared by the electrodeposition time of 50 s demonstrates better lithium storage performance such as larger capacity and markedly enhanced electrochemical cycling stability, resulting from its appropriate inner diameter size and medium MnO2 shell thickness.
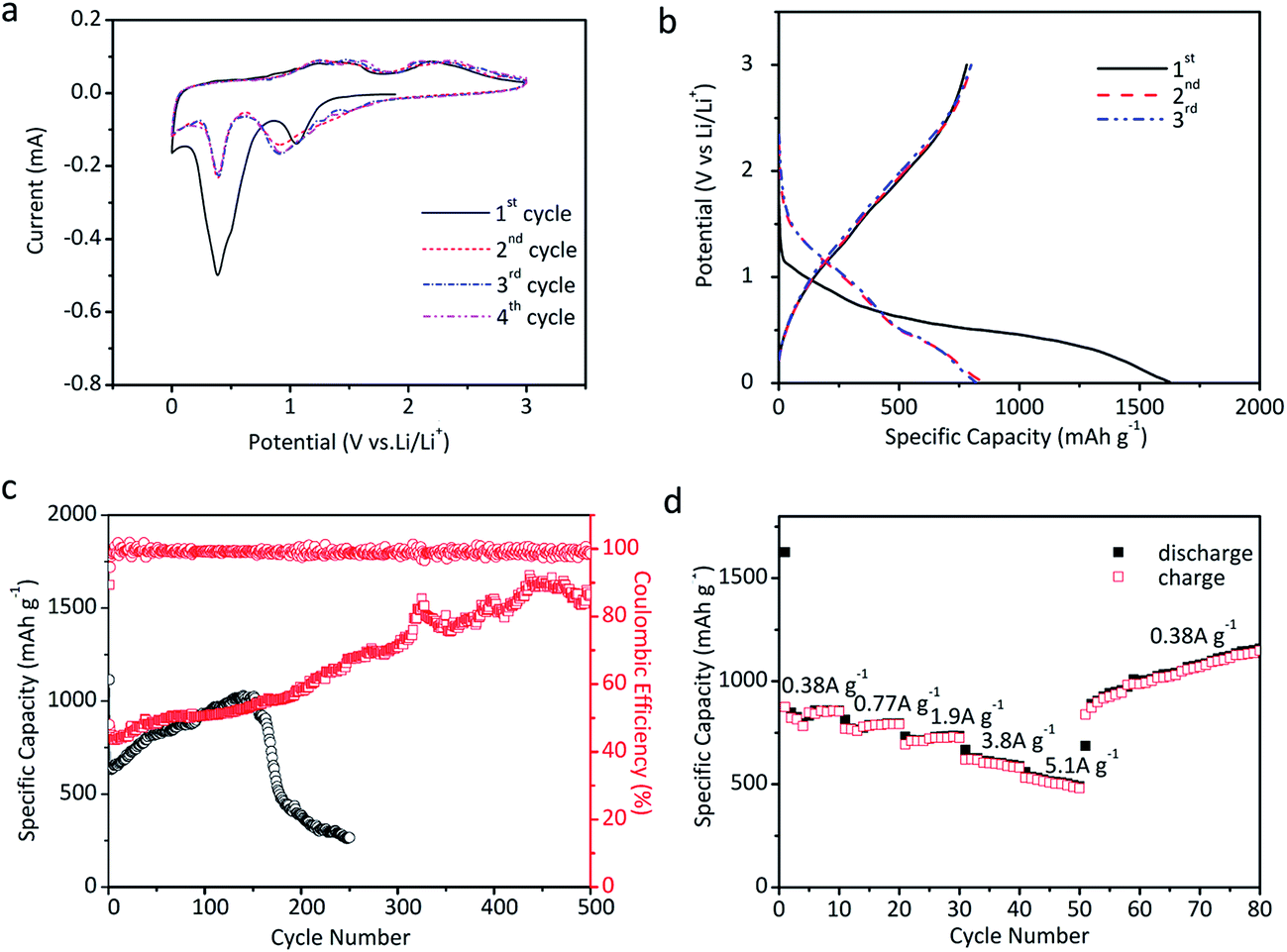 | ||
| Fig. 7 Electrochemical properties. (a) Cyclic voltammetry (CV) curves of the NiO@MnO2 microtube array electrode at a scanning rate of 0.1 mV s−1, (b) initial charge–discharge profiles of the NiO@MnO2 microtube array electrode at a current of 0.53 A g−1, (c) cyclic performances of the NiO microtube array electrode and the NiO@MnO2 microtube array electrode at a current density of 0.53 A g−1, (d) rate capability of the NiO@MnO2 microtube array electrode. | ||
Fig. 7d gives the rate capability of the NiO@MnO2 microtube array electrode. The electrode presents an average capacity of 850, 780, 720, 610 and 510 mA h g−1 at current densities of 0.38, 0.77, 1.9, 3.8 and 5.1 A g−1, respectively. The reversible capacity of the electrode at 5.1 A g−1 is as much as 60% of that at 0.38 A g−1, revealing a good rate capability. When the rate is reduced from 5.1 to 0.38 A g−1, the electrode delivers higher capacities than the original capacity at the same rate, consistent with the cycle performance in Fig. 7c.
The EIS measurements are conducted to investigate the lithium storage kinetics of the NiO and NiO@MnO2 microtube array electrodes. Fig. S4† shows the Nyquist plots of the NiO and MnO2@NiO microtube array electrodes at open circuit state after 25 cycles at a current density of 0.53 A g−1. A high-frequency capacitive loop and a straight line in low-frequency region are observed in the two Nyquist plots. The capacitive loop may be attributed to the faradaic charge transfer resistance (Rct) in parallel with the interface capacitance.37,38 The diameter of the capacitive loop represents the Rct value.37 It can be seen from the inset in Fig. S4† that the Rct value of the NiO@MnO2 microtube array electrode is obviously lower than that of the NiO microtube array electrode. This implies that the NiO@MnO2 microtube array electrode manifests faster faradic reaction kinetics, which is consistent with the lithium storage performance in Fig. 7.
Compared with other binary nanostructured transition metal oxides recently reported for LIBs (Table S1, the ESI†), the as-constructed NiO@MnO2 core–shell microtube array electrode shows larger reversible capacities at higher current rates and enhanced electrochemical cycling stability, which may be ascribed to its unique structural characteristics. The microtube array structure facilitates the transportation of Li+-containing liquid electrolyte inside the composite film, thus improving the ionic conductivity of the electrode and providing more active sites for the electrochemical reactions. The good combination between each microtube and the substrate increases the electronic conductivities of the electrode. These factors are mainly responsible for the large lithium storage capacity and good rate capability of the composite microtube array electrode. As illustrated in Fig. 8, the structural integrity and basic morphology of the NiO@MnO2 core–shell microtube array electrode are well preserved after 500 cycles at a current of 0.53 A g−1, while the tube structure of NiO microtube array electrode almost collapses after 250 cycles at 0.35 A g−1. This shows that the MnO2 shell effectively enhances the structural stability of the NiO core, resulting in the remarkably improved cycling performance of the NiO@MnO2 core–shell microtube array electrode. In addition, the microtube array architecture can buffer against the large volume change associated with the lithium storage reactions, partially manifesting the superior cycling stability of the composite microtube array electrode.4,18
 | ||
| Fig. 8 SEM images of (a) NiO microtube array electrode after 250 cycles at 0.53 A g−1 and (b) NiO@MnO2 core–shell microtube array electrode after 500 cycles at a current of 0.53 A g−1. | ||
4. Conclusions
The NiO@MnO2 core–shell microtube array films have been successfully prepared by a facile self-corrosion strategy and subsequent electrodeposition. A formation mechanism of NiC2O4·2H2O microtubes is proposed. The NiO microtube array film is achieved by the thermal treatment of the NiC2O4·2H2O microtube array film. The electrodeposition of MnO2 on the walls of NiO microtubes leads to the formation of a core–shell structure, with each NiO microtube surrounded by the MnO2 layer. The as-prepared NiO@MnO2 core–shell microtube array electrode delivers the excellent lithium storage performance such as large reversible capacity, superior cycling stability and good rate capability, resulting from the unique structure characteristics. The as-constructed strategy based self corrosion and electrodeposition can be applied as a facile and general route to prepare other composite shell/core micro/nanotube array film materials with excellent electrochemical performance.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21373182 and 51271168), the Zhejiang Provincial Natural Science Foundation of China (LY17B030004) and the Science and Technology Plan Project of Zhejiang Province (No. 2017C31078).References
- C. N. R. Rao and A. Govindaraj, Adv. Mater., 2009, 21, 4208–4233 CrossRef CAS.
- X. Z. Li, Y. Y. Fang, Y. P. Hu, H. F. Huo, S. L. Zhao, X. F. Long, J. T. Ma and R. Li, RSC Adv., 2015, 5, 69962–69969 RSC.
- S. S. Zhu, P. P. Zhang, L. Chang, Y. Zhong, K. Wang, H. B. Shao, J. M. Wang, J. Q. Zhang and C. N. Cao, Phys. Chem. Chem. Phys., 2016, 18, 8529–8536 RSC.
- H. Hu, L. Yu, X. Gao, Z. Lin and X. W. Lou, Energy Environ. Sci., 2015, 8, 1480–1483 CAS.
- L. Liu, H. Z. Kou, W. Mo, H. Liu and Y. Wang, J. Phys. Chem. B, 2006, 110, 15218–15223 CrossRef CAS PubMed.
- W. X. Zhang, X. G. Wen, S. H. Yang, Y. Berta and Z. L. Wang, Adv. Mater., 2003, 15, 822–825 CrossRef CAS.
- H. J. Fan, M. Knez, R. Scholz, K. Nielsch, E. Pippel, D. Hesse, M. Zacharias and U. Gösele, Nat. Commun., 2006, 5, 627–631 CAS.
- Q. Wang, B. Geng, S. Wang, Y. Ye and B. Tao, Chem. Commun., 2010, 46, 1899–1901 RSC.
- S. Liu, C. Mao, Y. Niu, F. Yi, J. Hou, S. Lu, J. Jiang, M. Xu and C. Li, ACS Appl. Mater. Interfaces, 2015, 7, 25568–25573 CAS.
- L. Zhou, W. Wang, L. Zhang, H. Xu and W. Zhu, J. Phys. Chem. C, 2007, 111, 13659–13664 CAS.
- X. Dai, D. Chen, H. Q. Fan, Y. Zhong, L. Chang, H. B. Shao, J. M. Wang, J. Q. Zhang and C. N. Cao, Electrochim. Acta, 2015, 154, 128–135 CrossRef CAS.
- M. H. Wang, L. Y. Cui, S. Y. Li, Z. X. Li, T. L. Ma, G. Y. Luan, W. Liu and F. L. Zhang, RSC Adv., 2016, 6, 58452–58457 RSC.
- L. Mohan, S. Viswanathan, C. Anandan and N. Rajendran, RSC Adv., 2015, 5, 93131–93138 RSC.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- N. Feng, X. L. Sun, H. W. Yue and D. Y. He, RSC Adv., 2016, 6, 72008–72014 RSC.
- J. J. Song, G. J. Shao, Z. P. Ma, G. L. Wang and J. Yang, Electrochim. Acta, 2015, 178, 504–510 CrossRef CAS.
- Y. Zhong, H. Q. Fan, L. Chang, H. B. Shao, J. M. Wang, J. Q. Zhang and C. N. Cao, J. Power Sources, 2015, 296, 255–260 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- X. Sun, C. Yan, Y. Chen, W. Si, J. Deng, S. Oswald, L. Liu and O. G. Schmidt, Adv. Energy Mater., 2014, 4, 1300912 CrossRef.
- Y. Zhang, W. Zhang, Z. Yang, H. Gu, Q. Zhu, S. Yang and M. Li, Angew. Chem., Int. Ed., 2015, 54, 3932–3936 CrossRef CAS PubMed.
- Z. Cai, L. Xu, M. Yan, C. Han, L. He, K. M. Hercule, C. Niu, Z. Yuan, W. Xu, L. Qu, K. Zhao and L. Mai, Nano Lett., 2015, 15, 738–744 CrossRef CAS PubMed.
- L. Li, A. R. O. Raji and J. M. Tour, Adv. Mater., 2013, 25, 6298–6302 CrossRef CAS PubMed.
- G. Tong, J. Guan and Q. Zhang, Adv. Funct. Mater., 2013, 23, 2406–2414 CrossRef CAS.
- N. Wang, Y. Zhai, X. Ma and Y. Qian, RSC Adv., 2015, 5, 61148–61154 RSC.
- Z. Sun, W. Ai, J. Liu, X. Qi, Y. Wang, J. Zhu, H. Zhang and T. Yu, Nanoscale, 2014, 6, 6563–6568 RSC.
- Q. Ma, H. He and C. Liu, Atmos. Environ., 2013, 69, 281–288 CrossRef CAS.
- J. Xu, J. Cai, J. M. Wang, L. Y. Zhang, Y. Q. Fan, N. Zhang, H. Zhou, D. Chen, Y. Zhong, H. Q. Fan, H. B. Shao, J. Q. Zhang and C. N. Cao, Electrochem. Commun., 2012, 25, 119–123 CrossRef CAS.
- R. Amutha, S. Akilandeswari, M. Muruganandham, M. Sillanpaa, B. Ahmmad and T. Ohkubo, J. Nanosci. Nanotechnol., 2011, 11, 3171–3179 CrossRef CAS PubMed.
- W. Cao, Y. Mao and X. Peng, CrystEngComm, 2014, 16, 10916–10920 RSC.
- H. Wu, G. Wu, Q. Wu and L. Wang, Mater. Charact., 2014, 97, 18–26 CrossRef CAS.
- F. Buciuman, F. Patcas, R. Craciun and D. R. T. Zahn, Phys. Chem. Chem. Phys., 1999, 1, 185–190 RSC.
- S. W. Lee, J. Kim, S. Chen, P. T. Hammond and Y. S. Horn, ACS Nano, 2010, 4, 3889–3896 CrossRef CAS PubMed.
- S. Sun, P. Wang, S. Wang, Q. Wu and S. Fang, Mater. Lett., 2015, 145, 141–144 CrossRef CAS.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dollé, L. Dupont and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A627–A634 CrossRef CAS.
- Y. Jiang, D. Zhang, Y. Li, T. Yuan, N. Bahlawane, C. Liang, W. Sun, Y. Lu and M. Yan, Nano Energy, 2014, 4, 23–30 CrossRef CAS.
- T. Y. Wei, C. H. Chen, K. H. Chang, S. Y. Lu and C. C. Hu, Chem. Mater., 2009, 21, 3228–3233 CrossRef CAS.
- P. P. Zhang, S. S. Zhu, Z. S. He, K. Wang, H. Q. Fan, Y. Zhong, L. Chang, H. B. Shao, J. M. Wang, J. Q. Zhang and C. N. Cao, J. Alloys Compd., 2016, 674, 1–8 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25463b |
| This journal is © The Royal Society of Chemistry 2017 |