
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Separation of Cu(II) from Cd(II) in sulfate solution using CaCO3 and FeSO4 based on mechanochemical activation
Xuewei Li,
Zhiwu Lei,
Jun Qu,
Xiaowen Zhou,
Zhao Li and
Qiwu Zhang *
*
School of Resources & Environmental Engineering, Wuhan University of Technology, 430070 Wuhan, Hubei, China. E-mail: zhangqw@whut.edu.cn
First published on 12th January 2017
Abstract
Cadmium and its compounds are important resources in different industries; on the other hand, cadmium is one of the most toxic heavy metals which can cause various health problems. Therefore it is important to develop effective methods for the separation of cadmium from other commonly associated metals from the stance of both resource recycling and environmental purification. Lime neutralization (Ca(OH)2) and ferrite are widely used to precipitate heavy metals. Limestone (calcium carbonate: CaCO3) is too stable to be used directly for this purpose. Mechanochemical activation was introduced to increase the activity of CaCO3. Fe(II)sulfate heptahydrate (FeSO4·7H2O) was used as a selective precipitation agent. As a result, Cu(II) was preferentially precipitated as (Fex,Cuy)O while the Cd(II) remained in the solution. The residual of Cu(II) ions in solution could be controlled at less than 0.1%, meanwhile more than 90% of Cd(II) ions remained in aqueous solution. Then, Cu(II)–Cd(II) separation was achieved by a simple solid–liquid separation.
1. Introduction
The separation of metal ions from industrial and waste solutions is extremely important in industrial applications.1,2 Efficient and economical methods for the separation and recovery of metal ions have drawn great attention due to the increasing demand on metal production.3–5 Several technologies are used for the separation and recovery of metal ions, including chemical precipitation, solvent extraction, ion exchange, liquid membranes, electrodialysis etc.5–11 Cadmium and its compounds are important resources and used in different industries such as electroplating, pigments, synthetic chemicals, ceramics, metallurgical and photographic products, electronics and other industries.12 Cadmium is a relatively rare metal, and primarily produced as a by-product from mining, smelting and refining of ore concentrates of zinc sulfide. It is also present in the metallurgical processing of other metals such as copper and lead.13 On the other hand, cadmium is one of the most toxic heavy metals, which can cause various health problems, such as choking, anemia, abdominal pain, dysfunction etc.14,15 Therefore, cadmium is both a resource and a very common heavy metal toxicant. As a resource, it is necessary to purify and remove copper impurities therein; as a heavy metal toxicant, it is necessary to remove cadmium from other metals in smelting, such as Cu, Zn and Ni. It is significant to develop effective methods for the separation of cadmium from other commonly associated metals.The separations of Zn(II)–Cd(II) and Ni(II)–Cd(II) have been studied extensively.13,16–20 However, few documents have discussed the separation of Cu(II)–Cd(II). In this regard, the Cu(II)–Cd(II) system was chosen for this investigation. The conventional chemical precipitation was effective and the formed precipitates can be separated from the water by sedimentation or filtration. Hydroxide precipitation is the most widely used chemical precipitation technique. For example, the lime milk (Ca(OH)2) neutralization is a widely practiced technique in the hydroxide precipitation.21,22 However, the process of Ca(OH)2 neutralization cannot achieve the selective precipitation of metal ions. Limestone (CaCO3) may be used as an adsorbent for the removing of heavy metals at very low concentration, due to its easily available, low-cost and insoluble in water. However, CaCO3 is too chemically stable to neutralize the salts to precipitate heavy metals at an amount of stoichiometric ratio for a complete reaction. Thus, an activation process should be introduced for the application of CaCO3 on the precipitation of metal ions. Mechanochemical activation (MA) is well-known in the expanding field of mechanochemistry and applied in many fields, such as materials syntheses, surface modification, hydrometallurgy, resource recycling and so on.23,24 In our previous studies we have investigated the separation of copper from nickel and cobalt in the sulfate solutions using CaCO3 based on MA.25,26 In that system, the activated CaCO3 reacted with copper sulfate to form precipitate of copper compounds, but not with nickel sulfate or cobalt sulfate, achieving the separation of them. However, in the effort to separate Cu from Cd, we noted that it was easy to form the co-precipitation of Cu(II)–Cd(II) compound by milling the mixture of Cu(II)sulfate, Cd(II)sulfate and CaCO3 in aqueous solution. So the selective precipitation agent should be used in Cu–Cd–Ca system for Cu(II)–Cd(II) separation.
Fe(II)sulfate heptahydrate (FeSO4·7H2O) is often used as a precipitation agent to remove heavy metals from waste solution, the so-called ferrite process (FP). In the ferrite process, Fe(II) can be oxidized to Fe3+ and transformed into iron (Fe(II)/Fe(III)) compounds, such as Fe3O4 (FeO·Fe2O3), FeOOH and Fe(OH)3.27 For yielding high quality ferrite and suppressing the formation of the other crystal phases, the solution pH needs to be controlled at 9–11 and reaction temperature over 70 °C. A dosage of Fe2+ 5–10 times higher than the gross metal content in solution is required to ensure the removal efficiency.28 The heavy metal ions can be removed by the incorporation into the structure through co-precipitation when they coexist with Fe(II).
In this work, we present a detailed study on the Cu(II)–Cd(II) separation using CaCO3 assisted by adding Fe(II)sulfate heptahydrate. MA is used to increase the reactivity of CaCO3. As the reactivity of CaCO3 increased at a level, Fe(II) is transformed into iron (Fe2+/Fe3+) compounds, at the same time, Cu(II) is preferentially precipitated by the FP while Cd(II) still exists in aqueous solution. The residual of Cu(II) ions in solution could be controlled at less than 0.1%, meanwhile more than 90% of the Cd(II) ions remained in aqueous solution, though a simple solid–liquid separation. This result is important for the development of new environment-friendly processes of various metals separation.
2. Materials and methods
2.1 Materials
Cu(II)sulfate pentahydrate CuSO4·5H2O, Cd(II)sulfate 8/3hydrate CdSO4·8/3H2O, Fe(II)sulfate heptahydrate (FeSO4·7H2O) and CaCO3 from Sinopharm Chemical Reagent Co., Ltd in analytical grade (purity > 99%) were used as received without further purification.2.2 Methods
With a separate case, 0.01 mol CuSO4·5H2O or 0.01 mol CdSO4·8/3H2O and equimolar CaCO3 with 2 mL distilled water (designated as Cu–Ca and Cd–Ca system), were put into a zirconia pot of 45 mL inner volume with 7 zirconia balls of 15 mm in diameter. A laboratory scale planetary mill (Pulverisette-7, Fritsch, Germany) was used to conduct the milling operation with a milling speed of 400 rpm and milling time of 60 min (10 min grinding and 5 min pause in turn). Then the mixture solutions containing 0.0075 mol CuSO4·5H2O, 0.0025 mol CdSO4·8/3H2O and 0.01 mol CaCO3 (designated as Cu–Cd–Ca system) or 0.005 mol FeSO4·7H2O, 0.005 mol CuSO4·5H2O and 0.01 mol CaCO3 (designated as Cu–Fe–Ca system) were conducted at same grinding operation by fixing 2 mL distilled water. And then the FeSO4·7H2O was gradually added in the above-mentioned Cu–Cd–Ca system, setting the ratio of Fe(II)/Cu(II) at 0.2, 0.3, 0.5 and 1. All the products were dried at 80 °C for 24 h for phase identification. X-ray diffraction (XRD: RU-200B/D/MAX-RB RU-200B, Japan) analysis was performed to identify the phases in the ground samples. For the separation test, a Cu–Cd–Ca–Fe system was conducted at same grinding operation with five parameters changed of Fe–Cu ratio, dosage of CaCO3, milling time, milling speed and the ratio of Cd/(Fe + Cu). Detailed information was specified in the following section of Experimental results. Then, the prepared samples were agitated in 200 mL distilled water at room temperature for 60 min by a magnetic stirrer, respectively. And 10 mL solution was centrifuged at the 8933 × g for 5 min. The concentrations of Cu, Cd and Fe in the supernatant were determined by inductively coupled plasma (ICP) spectrophotometry (ICP-AES: Optima-3300SYS, Perkin Elmer, USA). In addition, the pH of the sample from different Fe–Cu ratio was detected at different conditions by a pH meter (METTLER TOLEDO FE20-FiveEasy™, Switzerland).Mild milling experiment was conducted for pH measurement for simulating the high-energy ball milling operation. The test was carried out in a vertical type stirred media mill. The net volume of the milling chamber is 200 mL. The mixture of 0.0075 mol CuSO4·5H2O and 0.0025 mol CdSO4·8/3H2O was put into the chamber with 100 mL of distilled water. The FeSO4·7H2O and CaCO3 were added in the Cu–Cd solution. The molar ratios of Fe(II)/Cu(II) were set at 0, 0.2, 0.3, 0.5, 1 and 2. The molar ratio of Ca/(Fe,Cu) was fixed at 1. After adding CaCO3, the mixture was milled with 100 g zirconia balls of 2 mm in diameter at rotational speed of 250 rpm by a stirred mill for 30 min.
3. Results and discussion
3.1 Differences between Cu–Ca, Cd–Ca, Cu–Cd–Ca and Cu–Fe–Ca system
The metal sulfate was separately milled with CaCO3 in aqueous solution. Fig. 1(a) displays the XRD pattern of milling the mixture containing copper sulfate and calcium carbonate. CaSO4·2H2O (dihydrate gypsum) (JCPDS-ICDD card no. 33-0311) and Cu2CO3(OH)2 (malachite) (JCPDS-ICDD card no. 41-1390) were determined in the sample, indicating that the copper sulfate and calcium carbonate were transformed into basic copper carbonate and gypsum by the mechanochemical processing. Fig. 1(b) shows the case of milling the mixture of CdSO4 and CaCO3. CaCO3 (JCPDS-ICDD card no. 05-0586) and CdSO4·H2O (JCPDS-ICDD card no. 53-0841) were determined in the XRD pattern, indicating that the raw materials still existed in the sample. Although the existence of CaSO4·2H2O as a reaction product suggested a partial reaction between CaCO3 and CdSO4, the raw materials of calcium carbonate and cadmium sulfate remained as the main phases. The reaction might be as eqn (1). Since the CO32− easily hydrolyzed in water as shown in eqn (2), it was probable that the generation of CdCO3 might be hindered.| CaCO3 + CdSO4 + 2H2O → CaSO4·2H2O↓ + CdCO3↓ | (1) |
| CO32− + H2O → HCO3− + OH− | (2) |
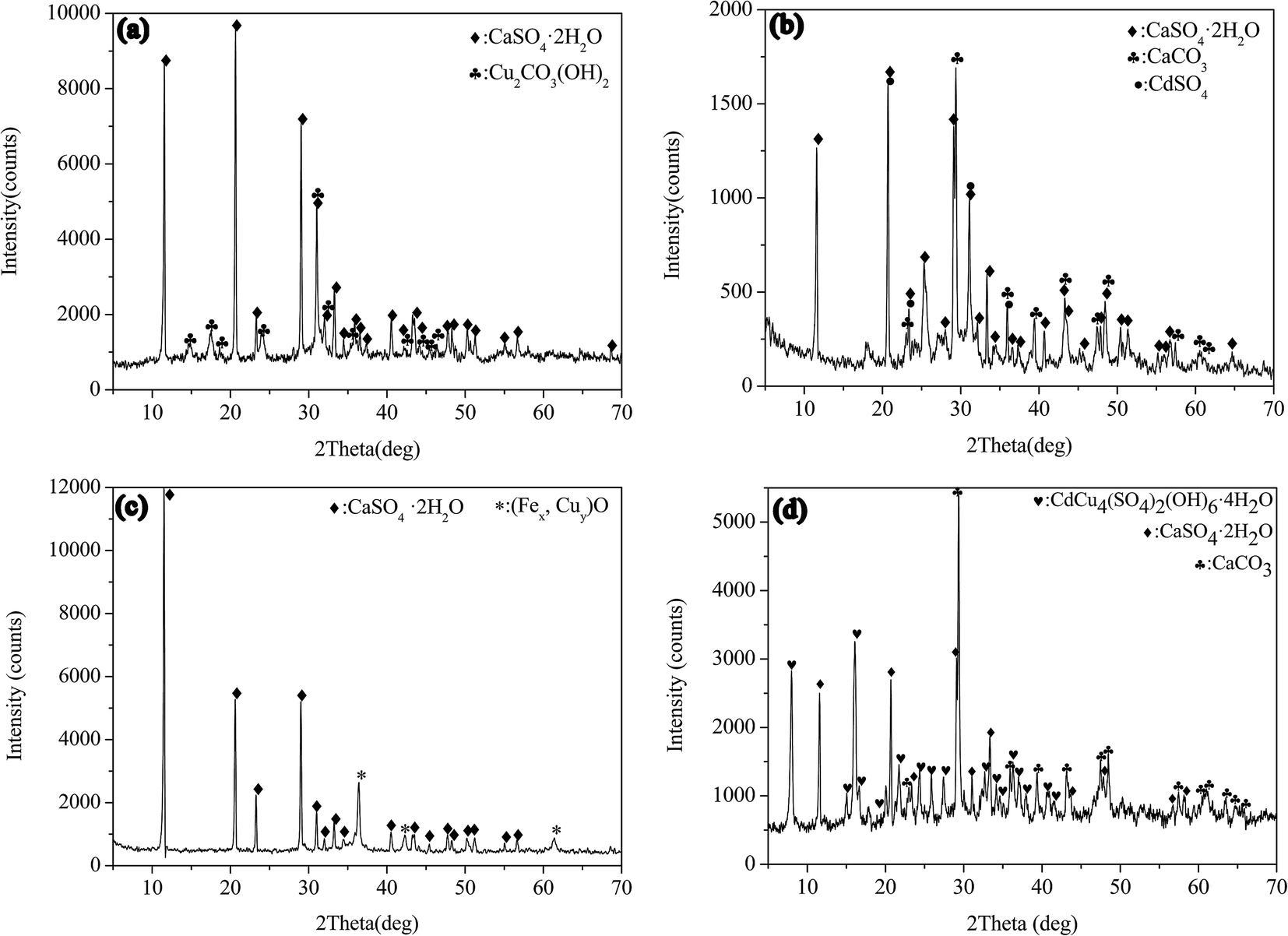 | ||
| Fig. 1 XRD patterns of the samples from milling the metal sulfates with CaCO3: (a) CuSO4 + CaCO3; (b) CdSO4 + CaCO3; (c) CuSO4 + FeSO4 + CaCO3; (d) CdSO4 + CuSO4 + CaCO3. | ||
The mixture powders of copper sulfate, ferrous sulfate and calcium carbonate were milled in aqueous solution, as shown in Fig. 1(c). The CaSO4·2H2O was clearly identified in the sample. In addition to the peaks of CaSO4·2H2O, the rest of the peaks were consistent with FeO (JCPDS-ICDD card no. 46-1312) and Cu2O (JCPDS-ICDD card no. 05-0667). In the FP, the Fe(II) can be occupied by Cu(II), leading to the incorporation of Cu(II) in FeO. So the rest of peaks might be attributed to the compound (Fex,Cuy)O. In the case of milling the mixture of copper sulphate, cadmium sulphate and calcium carbonate, the phase of hydrated basic sulfate of copper and cadmium with the molecular formula of CdCu4(SO4)2(OH)6·4H2O (niedermayrite) (JCPDS-ICDD card no. 50-1550) was identified in the XRD pattern, as shown in Fig. 1(d). It demonstrated that the co-precipitation of Cu(II) and Cd(II) occurred in the milling process. CaCO3 reacted with CuSO4 by liquid assisted milling operation, and the reactions were represented as eqn (3) and (4) as follows:
| 2CuSO4 + 5H2O + 2CaCO3 → Cu2CO3(OH)2↓ + 2CaSO4·2H2O↓ + CO2↑ | (3) |
| 4CuSO4 + CdSO4 + 3CaCO3 + 13H2O → CdCu4(SO4)2(OH)6·4H2O↓ + 3CaSO4·2H2O↓ + 3CO2↑ | (4) |
Although there was a significant difference between Cu(II) and Cd(II)sulfates when they were individually milled with CaCO3, the separation of them could not be achieved by only using the CaCO3 due to the co-precipitation of Cu(II) and Cd(II) in the forms of CdCu4(SO4)2(OH)6·4H2O.
3.2 Effect of Fe(II) on phase transition and Cu–Cd separation
The existence of Fe(II) effect on the Cu–Cd–Ca system was investigated by changing the molar ratio of Fe(II)/Cu(II) from 0, 0.2, 0.3, 0.5 to 1, with other parameters fixed: Cu(II)–Cd(II) ratio at 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; the dosage of CaCO3 equal to the mole of Fe(II) + Cu(II); the milling time at 60 min; rotating speed at 400 rpm; solid–liquid ratio at 1:1. As shown in Fig. 2, CdCu4(SO4)2(OH)6·4H2O, CaSO4·2H2O, (Fex,Cuy)O and CaCO3 were determined in the samples. From the pattern a to e, the intensity of the peaks (020) and (040) belonging to CdCu4(SO4)2(OH)6·4H2O was decreased with the increase of Fe(II) ions. In contrast, the intensity of the peaks from (Fex,Cuy)O became stronger correspondingly. As a result, the existence of Fe(II) ions had effectively prevented the generation of CdCu4(SO4)2(OH)6·4H2O.
:1; the dosage of CaCO3 equal to the mole of Fe(II) + Cu(II); the milling time at 60 min; rotating speed at 400 rpm; solid–liquid ratio at 1:1. As shown in Fig. 2, CdCu4(SO4)2(OH)6·4H2O, CaSO4·2H2O, (Fex,Cuy)O and CaCO3 were determined in the samples. From the pattern a to e, the intensity of the peaks (020) and (040) belonging to CdCu4(SO4)2(OH)6·4H2O was decreased with the increase of Fe(II) ions. In contrast, the intensity of the peaks from (Fex,Cuy)O became stronger correspondingly. As a result, the existence of Fe(II) ions had effectively prevented the generation of CdCu4(SO4)2(OH)6·4H2O.
 | ||
| Fig. 2 XRD patterns of the samples from Cu–Cd–Ca–Fe system with the increasing of Fe(II)–Cu(II) ratio: (a) 0; (b) 1/5; (c) 1/3 (d) 1/2; (e) 1. | ||
In addition, CaCO3 was activated through the milling operation. Many CO32− were released into water and hydrolysis occurred as eqn (2). Meanwhile, the Fe2+ combined with hydroxyl groups as eqn (5). After milling, the samples were stirred in beakers for duration of time. The stirring operation provided oxygen from air for chemical oxidation as eqn (6). Thus, the ferrite formation was assisted by the operation of milling and stirring, without high pH (9–11) and high temperature (over 70 °C).
| Fe2+ + 2OH− → Fe(OH)2 | (5) |
| 4Fe(OH)2 + O2 + 2H2O → 4Fe(OH)3↓ | (6) |
The Cu(II) was preferentially precipitated by the FP in the Cu–Cd–Ca–Fe system and the Cd(II) remained in aqueous solution. In this regards, Cu(II) had been separated from the Cd(II) solution by solid–liquid separation.
Fig. 3 shows the effect of Fe(II) on the residual ratio of Cd(II) in aqueous solution. The residual ratio of Cd(II) increased from 28.2% to 74.7% with the ratio of Fe(II)/Cu(II) increased from 0 to 1, clearly indicating that the existence of Fe(II) was beneficial to maintain the solubility of Cd(II). The result was consistent with the results of XRD analysis in Fig. 2, which demonstrated that the Fe(II) ions prevented the formation of CdCu4(SO4)2(OH)6·4H2O while promoting the generation of (Fex,Cuy)O.
 | ||
| Fig. 3 Effect of Fe(II) ions on the residual ratio of Cd(II) in aqueous solution. | ||
Therefore, the residual ratio of Cd(II) in aqueous solution increased with the increase of Fe(II) addition. Considering this phenomenon, the Cu(II)–Cd(II) separation might be achieved by regulating parameters.
3.3 Separation of Cu(II) and Cd(II)
:1, Fe–Cu ratio at 1:1, Cu–Cd ratio at 3:2 and Ca–(Fe + Cu) ratio at 0.9. The rotational speed of the planetary mill was set at 200, 300, 400, 500 and 600 rpm. As shown in Fig. 4, the residual ratio of Fe and Cu decreased from 22.1% and 17.3% to 0 with the speed increased from 200 to 600 rpm, while the value of remaining Cd in the solution decreased from 96.4% to 86.3%. Considering the separation efficiency, 400 rpm was an optimal rotational speed.
 | ||
| Fig. 4 Effect of speed on the residual ratio of Cd, Cu and Fe in aqueous solution. | ||
:1, Fe–Cu ratio at 1:1, Cu–Cd ratio at 3:2, milling time at 60 min and grinding rotational speed at 400 rpm. The molar ratio of Ca/(Fe + Cu), as a variable factor, was set at 0.5, 0.9, 1.3, 1.7, 2.1 and 2.5. The residual ratio of three metal ions was decreased with the increase of CaCO3. The declining tendency was relatively slow for Cd, compared with that of Fe and Cu. As the molar ratio of Ca/(Fe + Cu) increased from 0 to 2.5, the residual ratio of Cd decreased from 98% to 54% with 47.66% to 0 for Cu and 17.3% to 0 for Fe. It was noteworthy that the residual ratio of Cd was 90.7% when the molar ratio of Ca/(Fe + Cu) at 0.9, accompanying with only 5.8% Fe and 3.5% Cu remain in aqueous solution. The results revealed that the effective separation of Cu(II) and Cd(II) could be achieved using CaCO3 with Fe(II)sulfate heptahydrate.
 | ||
| Fig. 5 Effect of CaCO3 on the residual ratio of Cd, Cu and Fe in aqueous solution. | ||
:1, Fe–Cu ratio at 1:1, Cu–Cd ratio at 3:2, Ca–(Fe + Cu) ratio at 0.9, and grinding rotational speed at 400 rpm. The milling time was set at 15, 30, 60, 90 and 120 min. The results are shown in Fig. 6. It was seen that the residual ratio of Cd, Cu and Fe in aqueous solution decreased with the increasing of milling time. The residual ratio of Cd decreased from 92.4% to 76.6% as the milling time increased from 15 to 120 min, with 12% to 0 and 7.7% to 0 for Fe and Cu respectively. The results indicated that there existed an optimal grinding time for high separation efficiency.
 | ||
| Fig. 6 Effect of milling time on the residual ratio of Cd, Cu and Fe in aqueous solution. | ||
 | ||
| Fig. 7 Effect of Cd on the residual ratio of Cd, Cu and Fe in aqueous solution. | ||
3.4 Mechanism discussion
The Fe(II) addition prevents the generation of CdCu4(SO4)2(OH)6·4H2O precipitation and promotes the formation of (Fex,Cuy)O phase. The Cu(II) is precipitated in the form of (Fex,Cuy)O while the Cd(II) remains in the solution. Then the separation of Cu(II) from Cd(II)sulfate solution is achieved by a simple solid–liquid separation. The mechanism of the process can be interpreted in two aspects as the following: (a) the acidic reaction environment is conducive to prevent the precipitate of Cd(II); (b) Fe2+ is oxidized to Fe3+ and transformed into iron (Fe(II)/Fe(III)) compounds. The ferrite has an essential role in the precipitation of Cu(II).Mild milling experiment was conducted to simulate the process of high-energy ball milling. The pH was monitored as shown in Fig. 8(A). It can be seen that the pH value of Cu–Cd–Ca system is higher than that of Cu–Cd–Ca–Fe system. Acidic pH in the Cu–Cd–Ca system is derived from the hydrolysis of Cu(II). Considering the relatively weaker hydrolysis of Fe(II) than Cu(II), the pH value of Cu–Cd–Ca system should be theoretically lower than that of Cu–Cd–Ca–Fe system at the same molar metal ions. However, the hydrolysis of Fe(III) is stronger than Cu(II). The lower value from the Cu–Cd–Ca–Fe system might be caused by the superimposed effect of the hydrolysis of Cu(II) and Fe(III). The theoretical solubility of cadmium hydroxide vs. pH is shown in Fig. 8(B).29 It demonstrated that the range of pH value leading to cadmium precipitate is 8–11. So from the aspect of pH, the additional Fe(II) is not conducive to the precipitation of cadmium.
 | ||
| Fig. 8 pH values effected by the additional Fe(II) (A) and theoretical solubility of cadmium hydroxide vs. pH (B). | ||
On the other hand, the ferrite has an essential role in the precipitation of Cu(II). The XRD pattern of the sample formed by milling FeSO4 and CaCO3 in solution is shown in Fig. 9. The FeSO4 and CaCO3 were transformed into FeO(OH) (goethite) (JCPDS-ICDD card no. 29-0713) and CaSO4·2H2O. It indicated that the Fe(II) was oxidized to Fe(III) by milling with CaCO3 and transformed into Fe(III) compounds, such as FeO(OH). So in the Cu–Cd–Ca–Fe system, a part of Fe(II) also is oxidized to Fe(III) and transformed into iron compounds. The ferrite preferentially precipitate Cu(II) rather than Cd(II) may be ascribed to the difference in their ionic radii. The ionic radius of Fe(II), Cu(II) and Cd(II) are 0.77, 0.71 and 0.97 Å, respectively.16,30 The ionic radii of Cu(II) is close to that of Fe(II) but with larger difference from that of Cd(II). The smaller the ionic radius, the greater its affinity to occupy the sites of the Fe(II). Thus the Cu(II) preferentially occupies the position of Fe(II) in the process of FP. This is consistent with the description in Fig. 2. The Fe(II) ions prevented the formation of CdCu4(SO4)2(OH)6·4H2O. And the compound (Fex,Cuy)O was facilitated by the increasing of Fe(II). As a result, a part of Cu(II) is co-precipitated with iron, leaving almost all Cd(II) in solution.
 | ||
| Fig. 9 XRD pattern of the sample formed by milling FeSO4 and CaCO3 in solution. | ||
The selective precipitation of Cu(II) can be achieved by adding Fe(II) in the Cu–Cd–Ca system. FeSO4·7H2O is the most common flocculant used in the removal of heavy metals. The sources of CaCO3, such as calcite, limestone and shells, are around the world and very low-cost. In this process, the CaCO3 and FeSO4·7H2O are used to separate Cu(II) and Cd(II) and the result is feasible. The dosage of FeSO4·7H2O in this process is less than 20% that of traditional FP processing under the same processing capacity. In addition, the neutral sediments are environmentally friendly.
4. Conclusion
Mechanochemical activation technique is efficiently used to the activation of CaCO3. The FeSO4·7H2O is used as a selective precipitation agent. The process allows the reaction of calcium carbonate and ferrous sulfate with copper sulfate to form precipitate of copper compounds, but without similar reaction with cadmium sulfate. The separation of copper from cadmium is feasible by a simple solid–liquid separation as filtering, with 99% copper in solid and 90% of cadmium remaining in aqueous solution at optimized conditions. The experimental results may serve as the basis to treat various types of solutions containing Cu–Cd ions to develop an easy and environment-friendly separation process. The concept developed in this report may be also applied to other metals separation when some large differences in specific property exist as the case here.References
- S. E. Kentish and G. W. Stevens, Chem. Eng. J., 2001, 84, 149–159 CrossRef CAS.
- R. S. Juang and S. W. Wang, Water Res., 2000, 34, 3795–3803 CrossRef CAS.
- A. R. Mahdavian and M. A.-S. Mirrahimi, Chem. Eng. J., 2010, 159, 264–271 CrossRef CAS.
- F. J. Alguacil, M. Alonso and A. M. Sastre, Chem. Eng. J., 2002, 85, 265–272 CrossRef CAS.
- F. J. Alguacil, A. Cobo and M. Alonso, Chem. Eng. J., 2002, 85, 259–263 CrossRef CAS.
- M. Sadrzadeh, T. Mohammadi, J. Ivakpour and N. Kasiri, Chem. Eng. J., 2008, 144, 431–441 CrossRef CAS.
- P. Cañizares, Á. Pérez and R. Camarillo, Desalination, 2002, 144, 279–285 CrossRef.
- X. J. Yang, A. G. Fane and C. Pin, Chem. Eng. J., 2002, 88, 37–44 CrossRef CAS.
- D. Pathania, G. Sharma and R. Thakur, Chem. Eng. J., 2015, 267, 235–244 CrossRef CAS.
- R. Kumar, M. Kumar, R. Ahmad and M. A. Barakat, Chem. Eng. J., 2013, 218, 32–38 CrossRef CAS.
- M. Laatikainen and K. Laatikainen, Chem. Eng. J., 2016, 287, 74–82 CrossRef CAS.
- M. S. Safarzadeh, M. S. Bafghi, D. Moradkhani and M. O. Ilkhchi, Miner. Eng., 2007, 20, 211–220 CrossRef.
- B. Ramachandra Reddy, D. Neela Priya and J. Rajesh Kumar, Hydrometallurgy, 2004, 74, 277–283 CrossRef.
- W. Wei, J. K. Bediako, S. Kim and Y.-S. Yun, J. Taiwan Inst. Chem. Eng., 2016, 61, 188–195 CrossRef CAS.
- J. Li, C. Chen, K. Zhu and X. Wang, J. Taiwan Inst. Chem. Eng., 2016, 59, 389–394 CrossRef CAS.
- M. Horsfall Jr and A. A. Abia, Water Res., 2003, 37, 4913–4923 CrossRef CAS PubMed.
- K. Takeshita, K. Watanabe, Y. Nakano and M. Watanabe, Hydrometallurgy, 2003, 70, 63–71 CrossRef CAS.
- P. R. Danesi, R. Chiarizia and A. Castagnola, J. Membr. Sci., 1983, 14, 161–174 CrossRef CAS.
- L. B. Chaudhari and Z. V. P. Murthy, J. Hazard. Mater., 2010, 180, 309–315 CrossRef CAS PubMed.
- M. S. Safarzadeh and D. Moradkhani, Sep. Purif. Technol., 2010, 73, 339–341 CrossRef CAS.
- D. R. Jenke, G. K. Pagenkopf and F. E. Diebold, Environ. Sci. Technol., 1983, 17, 217–223 CrossRef CAS PubMed.
- J. G. Dean, F. L. Bosqui and K. H. Lanouette, Environ. Sci. Technol., 1972, 6, 518–522 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- P. Balaz, M. Achimovicova, M. Balaz, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutkova, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC.
- X. W. Li, Z. W. Lei, J. Qu, H. M. Hu and Q. W. Zhang, Sep. Purif. Technol., 2017, 172, 107–112 CrossRef CAS.
- X. W. Li, Z. W. Lei, J. Qu, Z. Li and Q. W. Zhang, Sep. Sci. Technol., 2016, 51, 2772–2779 CrossRef CAS.
- J. C. Lou and C. K. Chang, Sep. Purif. Technol., 2007, 57, 513–518 CrossRef CAS.
- Y. J. Tu, C. K. Chang, C. F. You and S. L. Wang, J. Hazard. Mater., 2012, 209, 379–384 CrossRef PubMed.
- D. M. Ayres, A. P. Davis and P. M. Gietka, Engineering Research Centre Report, 1972, pp. 518–522 Search PubMed.
- S. T. Breviglieri, É. T. G. Cavalheiro and G. O. Chierice, Thermochim. Acta, 2000, 356, 79–84 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |