
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isotope exchange reaction in tritium-contaminated vacuum pump oil: mechanism and HTO effect†
Liang Donga,
Na Yanga,
Yong Yanga,
Weiyi Lic,
Yi Quana,
Bing Denga,
Dan Menga,
Yang Du*a,
Shuo Li*b and
Zhaoyi Tan*ad
aInstitute of Nuclear Physics and Chemistry, China Academy of Engineering Physics, Mianyang, 621900, P. R. China. E-mail: zhyitan@126.com; tanzhaoyi@caep.cn
bSchool of Chemical Engineering, Chongqing University of Technology, Chongqing, 400054, P. R. China
cSchool of Physics and Chemistry, Xihua University, Chengdu, 610065, P. R. China
dSchool of Radiation Medicine and Protection (SRMP), School for Radiological and Interdisciplinary Sciences (RAD-X), Soochow University, Suzhou, 215000, P. R. China
First published on 4th January 2017
Abstract
As radioactive wastes, contaminated vacuum pump oils from tritium-processing facilities are challenging to dispose of. In this study, we investigated the mechanism of the tritium–hydrogen (T–H) exchange reaction between the tritiated water (HTO) and many typical organic molecules in vacuum pump oil using a density functional theory (DFT) method. The T–H exchange reaction could proceed with two mechanisms, the direct T–H exchange mechanism and the addition–elimination T–H exchange mechanism. The calculation indicates that the direct T–H exchange mechanism is kinetically favored. The HTO molecule is not only a reactant, but also acts as the proton shuttle in the transition state. The number of HTO molecules participating in the reaction is determined by the formation of the stable six-membered ring proton migration transition state. The T–H exchange reaction is selective, and molecules with COOH and OH groups preferentially undergo hydrogen exchange with HTO. In the case of multiple possible products, the calculations correctly predicted the predominant product. The results here will provide guidance for improving the associated decontamination process and reducing the environmental impact.
1 Introduction
In tritium-related facilities, a large amount of tritium-contaminated pump oil is produced by vacuum pumps,1 mainly through the tritium–hydrogen (T–H) isotope exchange reaction.2 The main components of vacuum pump oil are hydrocarbons, such as alkanes, cycloalkanes, and aromatic compounds,1 plus small amounts of water. When tritium interacts with the water, highly toxic tritiated water (HTO) would be formed in pump oil. The formed HTO then easily reacts with the organic components to generate more toxic organic tritium-containing compounds. The resulting radioactive waste is expensive and very difficult to dispose. Therefore, it is essential and urgent to develop economic and efficient methods to dispose/decontaminate these tritium-containing wastes.After exposure to pump oil, tritium may also react with the hydrocarbons to directly form tritiated hydrocarbons.3 In the presence of oxygen, tritium and hydrocarbons may further undergo radiochemical oxidation to produce peroxide and hydroperoxide radicals. These radicals then react with other organic molecules to form stable tritiated oxides, such as alcohols, carbonyl compounds and carboxylic acids. In the presence of HTO, various organic tritium compounds would be formed between HTO and the organic species through the T–H exchange reaction. In order to develop scientific and effective treatment methods for the tritiated waste pump oil, it is crucial to confirm the chemical forms of tritium in the oil following T–H exchange.
Several groups have made great contributions regarding the interaction of tritium with oil. Takeishi et al.1 demonstrated that pump oil can be contaminated by bubbling with trace tritium gas or tritiated water vapor, and that bubbling with gases containing non-tritiated water vapor is an effective decontamination method. Sazonov and Magomedbekov et al. established the chemical forms of tritium in vacuum pump oil and proposed the effective treatment of tritium-contaminated oil through isotope exchange.3,4 In contrast to these experimental investigations, however, theoretical modeling of the T–H isotope exchange reaction in tritium-contaminated pump oil is rare in the literature.
Previously, we reported theoretical investigations concerning the mechanism of the T–H exchange reaction between T2 and several typical organic molecules in vacuum pump oil and calculated the reaction rates to form different tritiated products.5 However, the mechanism of the T–H exchange reaction with HTO is still poorly understood. Furthermore, HTO creates hydrogen bonding interaction in the system, therefore, the reaction of organic molecules would be different and likely more complex from that with T2.
While there are considerable experimental studies of the T–H isotope exchange process, molecular level knowledge of the T–H exchange reaction between HTO and the components of pump oil is still lacking. Furthermore, previously investigations have indicated that such isotope exchange is selective for the different functional groups of the organic compounds. However, the reactivity of the organic molecules with different functional groups with HTO is unclear. A better understanding of the T–H exchange process involving HTO, and the chemical states of tritium in pump oil after this reaction would give guidance for the detritiation of the contaminated oil.
Herein, we used density functional theory (DFT) to study the gas-phase T–H exchange reaction between HTO and several organic molecules representative of the main constituents of pump oil. We examined the reactivity of these organic molecules and the selectivity of the products. Furthermore, we clarify the impact of HTO for the T–H exchange process in this paper.
2 Computational details
Among the correlated functional theories, the hybrid meta exchange–correlation M06-2X functional performs well for systems with weak interactions,6 and previous investigations indicate that M06-2X method could provide highly accurate energies.7 All calculations were carried out using the Gaussian 09 program.8 All stationary points and transition states (TSs) on the potential energy surface (PES) were optimized using M06-2X method with the 6-311++G(d,p) basis set.9 Zero-point vibrational energy (ZPVE) corrections were applied to the relative energies. The vibrational frequencies were obtained at the same level, and the characterized species were either a minimum (no imaginary frequency) or a transition state (unique imaginary frequency). Intrinsic reaction coordinate (IRC) calculations were performed to further confirm that the optimized transition state (TS) correctly connects the relevant reactant and product.An energy decomposition analysis (EDA) of the T–H exchange reaction with one or two HTO molecules presented was performed.10 The activation barrier of one step is divided into two parts: the deformation energy ΔEd and the interaction energy ΔEint. The deformation energy ΔEd is the energy required to deform the fragments into the geometries they have in the TS. The interaction energy between the fragments is obtained by the activation barrier of that step to minus the deformation energy. All the EDA energies were obtained by frequency calculations at the M06-2X/6-311++G(d,p) level of theory on the geometries optimized.
3 Results and discussion
Our previous study suggests that the ability of T–H exchange for aromatic compounds is similar to that for non-aromatic ones.5 Thus, we only choose a variety of non-aromatic molecules that are representative of the main components in pump oil. Our calculations indicate that the T–H exchange between the organic molecules and HTO mostly proceed through the direct T–H exchange mechanism. For molecules with unsaturated double bonds, the reaction may proceed through the addition–elimination mechanism.Effect of HTO
Numerous studies have proposed that the water molecule could facilitate hydrogen transfer reactions through relaying proton via the six-membered ring hydrogen migration TS.11 In the case of HTO, the T–H exchange TS could be four- or six-membered ring containing one or two HTO, respectively.We first use methane, the simplest hydrocarbon, to investigate whether one or two HTO molecules participate in this reaction. The optimized structures and their relative energies calculated at the M06-2X/6-311++G(d,p) level for the T–H exchange reaction of CH4 are given in Fig. 1.
 | ||
| Fig. 1 Reaction paths of T–H exchange of CH4, and the corresponding optimized structures along the reaction path. Red lines: the path involving one HTO; blue and black lines: the paths involving two HTOs. TSCH4–2HTO-1 and TSCH4–2HTO-2 are TSs positioned in the cis- and trans-conformations, respectively. Relative energies (kcal mol−1) of the species with regard to the reactants are shown in parentheses. | ||
The reaction starts with HTO attacking the C atom of CH4. Subsequently, T–H exchange between HTO and CH4 occurs via hydrogen migration, generating the products CH3T and H2O. The hydrogen migration processes could involve either one or two HTO molecules. For the first case, the transition state (labeled TSCH4–HTO) is a four-membered ring containing one HTO. The vibrational mode of TSCH4–HTO involves the T1 atom moving toward the C1 atom, while H1 atom simultaneously moves toward the O1 atom. When there are two HTO molecules in the system, the hydrogen migration occurs via a six-membered ring TS. The six-membered ring TS has two possible configurations, TSCH4–2HTO-1 and TSCH4–2HTO-2, with the hydrogens in the two HTO positioned in the cis- and trans-conformations with respect to the six-membered ring. The whole hydrogen migration process includes the migration of T1 to C1, T2 to O1 and H1 to O2. The net result of this three proton-transfer process is the generation of CH3T, together with H2O and HTO.
The relative energy of TSCH4–2HTO-2 (51.3 kcal mol−1) is slightly lower than that of TSCH4–2HTO-2 (52.2 kcal mol−1), suggesting the trans-conformation has less steric hindrance. The relative energy of TSCH4–HTO (75.5 kcal mol−1) is much higher than those of TSCH4–2HTO-1 and TSCH4–2HTO-2. Therefore, having another HTO molecule in the reaction system could facilitate the H–T exchange reaction through relaying the proton via a six-membered ring hydrogen migration TS containing two HTO molecules, instead of a four-membered ring involving one HTO. In other words, the H–T exchange reaction preferentially occur through the six-membered ring hydrogen migration TS with two participating HTO molecules. Moreover, in the six-membered ring, the configuration with the hydrogens in the two HTO positioned in the trans-conformation is favored.
Previous investigations concerning the role of water in the hydrogen migration process proposed that such a remarkable decrease in the relative TS energy could be ascribed to the reduced ring strain upon changing from four- to six-membered ring TS.11,12 In order to discuss the differences with one or two HTO molecules present in the reaction system and interpret the impact of HTO, the energy decomposition analysis (EDA) of this process with one or two HTO molecules was performed.10 The contributions to the interaction energy and deformation energy of the three fragments (CH4, HTO1 and HTO2) during the formations of the TSs are listed in Table 1. Compared with the four-membered ring transition state TSCH4–HTO, the reaction with two HTO molecules involves a less strained six-membered ring transition state TSCH4–2HTO-2. The more relaxant orientations of the fragments in TSCH4–2HTO-2 give enough chances for the fragments to interact with each other, resulting in stronger interaction between them. Therefore, the interaction energy between the fragments in TSCH4–2HTO-2 is 45 kcal mol−1 lower than that in TSCH4–HTO. The deformation energy of CH4 and HTO1 in TSCH4–HTO are 8.8 kcal mol−1 lower and 14.4 kcal mol−1 higher than those in TSCH4–2HTO-2, respectively. However, the presence of the second HTO molecule (HTO2) has a 22.4 kcal mol−1 deformation energy, resulting in the higher deformation energy of TSCH4–2HTO-2 than that of TSCH4–HTO (61.4 vs. 82.2 kcal mol−1). Although the deformation energy of TSCH4–HTO is lower than that of TSCH4–2HTO-2, the much lower interaction energy overwhelms this unfavorable deformation energy, resulting in the higher activation barrier of the reaction with one HTO molecule. Therefore, the presence of the second HTO molecule makes the orientations of the fragments in TSCH4–2HTO-2 more suitable for them to interaction with each other, which can overwhelm the deformation energy it brings, and it may be the main reason for the lower activation barrier of this pathway.
| Process | ΔrE≠ | ΔEd of fragment | ΔEd | ΔEint | ||
|---|---|---|---|---|---|---|
| CH4 | HTO1 | HTO2 | ||||
| CH4 + HTO1 → TSCH4–HTO | 75.5 | 32.6 | 28.8 | — | 61.4 | 14.1 |
| CH4 + HTO1 + HTO2 → TSCH4–2HTO-2 | 51.3 | 41.4 | 18.4 | 22.4 | 82.2 | −30.9 |
For molecules with adjacent carbonyl and alkyl groups, such as CH3COCH3, CH3CHO, and CH3COOH, it is known that the six-membered ring TS could only contain one HTO, since the carbonyl group would also participate in the TS. We chose to study CH3COCH3, in which all the hydrogen atoms have the same bonding. For the T–H exchange reaction path involving one HTO molecule, the calculated potential energy surface at the M06-2X/6-311++G(d,p) level and the corresponding optimized structures are given in Fig. 2.
 | ||
| Fig. 2 Reaction path of T–H exchange of CH3COCH3, and the corresponding optimized structures along the reaction path. | ||
As shown in Fig. 2, the reaction contains two steps. First, the proton migrates from the methyl group to carbonyl group via a six-membered ring TS involving one HTO molecule. A second HTO is no longer required since the carbonyl group also participates in the TS. The whole proton migration process includes the migration of H1 to O1 and T1 to O2. The net result of this one-step, two-proton-transfer process generates the intermediate CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) COT–CH3 as well as H2O. In the second step, the proton migrates from the hydroxyl group to the alkene group via a six-membered ring TS involving another HTO molecule. This process includes the migration of T1 to O1 and T2 to C1, resulting in the generation of the product CH3COCH2T. The relative TS energies of the two steps, TS1CH2COT–CH3 and TS2CH3COCH2T, are calculated to be 32.5 kcal mol−1 and 31.3 kcal mol−1, respectively.
COT–CH3 as well as H2O. In the second step, the proton migrates from the hydroxyl group to the alkene group via a six-membered ring TS involving another HTO molecule. This process includes the migration of T1 to O1 and T2 to C1, resulting in the generation of the product CH3COCH2T. The relative TS energies of the two steps, TS1CH2COT–CH3 and TS2CH3COCH2T, are calculated to be 32.5 kcal mol−1 and 31.3 kcal mol−1, respectively.
From the above two examples, it is clear that the number of HTO molecules participating in the reaction is determined by the formation of the stable six-membered ring proton migration TS. The HTO molecules are not only a reactant, but also act as the proton shuttle in the TS. For molecules without the unsaturated carbonyl group, such as CH4, C2H4, and C2H5OH, two HTO molecules would participate in the T–H exchange process via a six-membered ring TS. For those containing the carbonyl group, the carbonyl group could be part of the TS, thereby requiring only one HTO molecule in the TS.
It should be emphasized that more HTO molecules participated in the direct T–H exchange process may further decrease the transition state energy since the smaller ring strain, but too much HTO participated in would cause the chain is too long and less stable. Thus, determining the appropriate number of HTO is a very complicated problem. The further studies on how many HTO molecules are needed in the T–H exchange reaction are currently under investigation.
Direct T–H exchange mechanism
Next, we consider organic reactants containing different types of hydrogen, since they can form multiple tritiated products when the T–H exchange occurs at different positions. The T–H exchange reaction pathways of CH4, C2H6, C3H8, C2H4, C3H6, C2H5OH, CH3CHO, CH3COCH3 and CH3COOH are presented in Fig. 3, together with the optimized structures calculated at the M06-2X/6-311++G(d,p) level.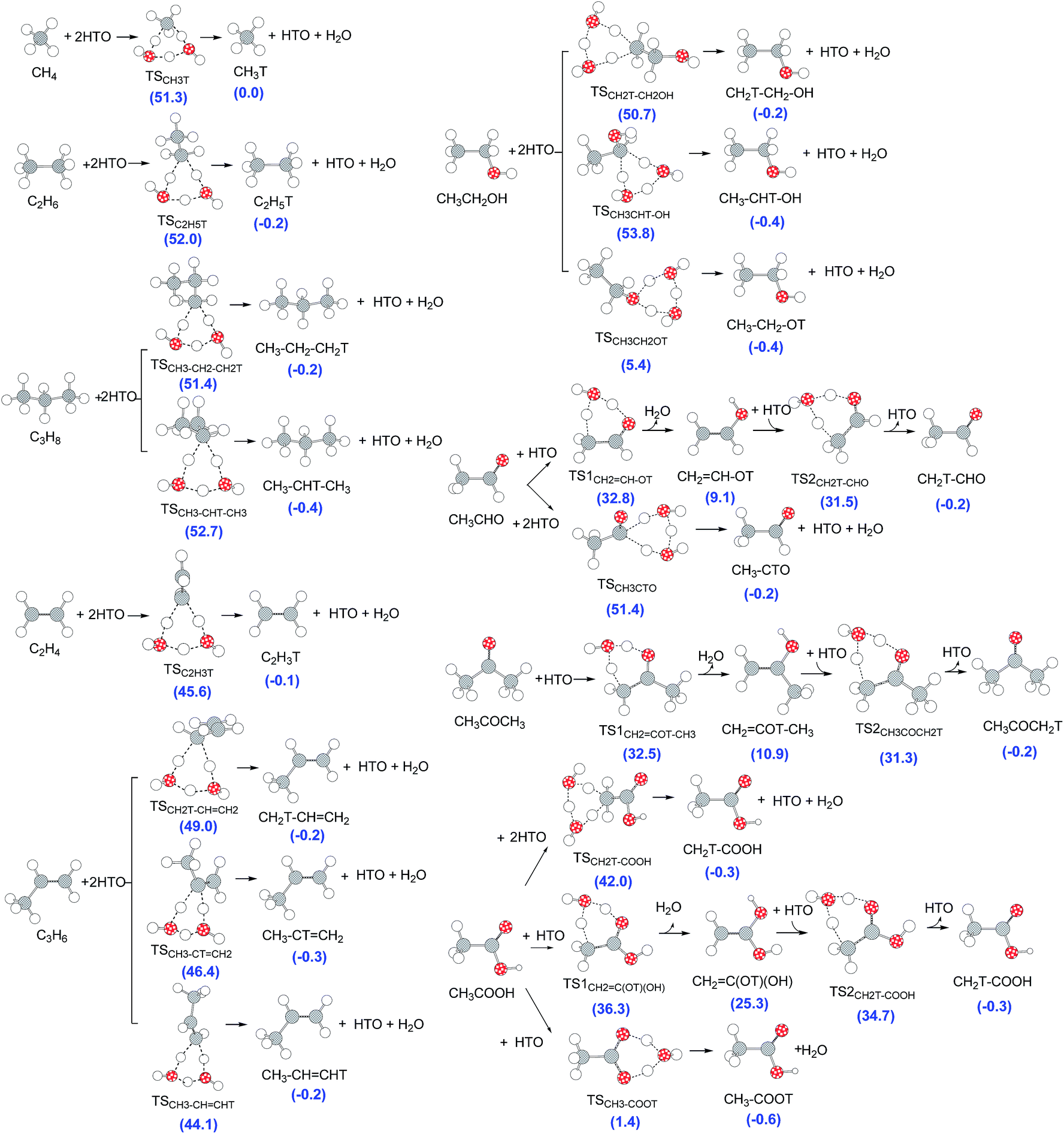 | ||
| Fig. 3 Direct T–H exchange reaction pathways of CH4, C2H6, C3H8, C2H4, C3H6, CH3CH2OH, CH3CHO, CH3COCH3 and CH3COOH. Relative energies (kcal mol−1) of the corresponding species relative to the reactants are shown in parentheses. | ||
As shown in Fig. 3, these reactions occur through a six-membered ring hydrogen migration TS assisted by two HTO molecules. For molecules with the carbonyl group (CH3CHO, CH3COCH3 and CH3COOH), the hydrogen migration would occur between the carbonyl group and neighboring group with saturated hydrogen (such as methyl or hydroxyl groups) through a six-membered ring TS assisted by one HTO molecule, forming an enol structure. Thus, there exists an additional keto–enol tautomerism TS including one HTO molecule.
The T–H exchange TS determines the reactivity of the hydrogen in the functional groups of organic molecules. In Fig. 3, all the relative TS energies for alkanes are above 50.0 kcal mol−1, while those of alkenes are below 50.0 kcal mol−1. Therefore, the activity of T–H exchange is in the order of alkene > alkane. For oxygen-containing molecules, the energies for non-oxygenated hydrogens (i.e., CH3 and CH2 in CH3CH2OH) are higher than 50.0 kcal mol−1, while that for the methyl hydrogen in CH3COOH is higher than 35.0 kcal mol−1. However, the TS energy of the COOH group (1.4 kcal mol−1) is lower than that of the OH group (5.4 kcal mol−1), and both are much lower than those of alkyl groups. Thus, T–H exchange occurs more easily in the OH and COOH groups of alcohol and carboxylic acid, respectively. This makes sense because tritium readily replace the most active hydrogen in an organic molecule.4 For the aldehyde, the T–H exchange TS of CH3 occurs through two consecutive hydrogen migrations each with a six-membered ring TS, and the relative energies of these TSs are lower than that of CHO. Therefore, the reaction of aldehydes preferentially occur in the CH3 group. Moreover, for the ketone, the relative TS energy for CH3 is 32.5 kcal mol−1, almost the same as that of aldehyde. Thus, assuming the direct T–H exchange mechanism, the functional groups are ordered as follows in terms of their ability for T–H exchange: CH3COOH > CH3CH2OH > CH3CHO ≈ CH3COCH3 > alkene > alkane.
Addition–elimination T–H exchange mechanism
For unsaturated organic molecules, there is another T–H exchange mechanism of addition–elimination. This mechanism is composed of two steps: (1) the addition of HTO to the unsaturated bond in the organic molecule, and (2) the elimination of one H2O from various hydrogen positions. If the elimination of H2O forms an enol structure, the aforementioned keto–enol tautomerism would occur assisted by another HTO. The addition–elimination T–H exchange mechanism always involves more than one TSs, and the one with the highest relative energy, namely the rate-determining step, determines the reactivity.The optimized structures of addition–elimination T–H exchange reaction pathways of C2H4, C3H6, CH3CHO, CH3COCH3 and CH3COOH are presented in Fig. 4. From the calculation results, T–H exchange through the double bond of alkene is slightly easier than through the CHO group in aldehyde or the carbonyl group in ketone, and the latter two have almost the same level of reactivity. For the aldehyde and ketone, the relative TS energies of the elimination step are much higher than those of the addition step and keto–enol tautomerism TS. Therefore, the step of H2O elimination in the addition–elimination T–H exchange mechanism determines the ability of T–H exchange in aldehyde and ketone. The T–H exchange in the COOH group is the easiest, compared to the other unsaturated groups studied here. According to the calculations, the ability of T–H exchange via the addition–elimination exchange mechanism is ordered as CH3COOH > CH3COCH3 ≈ CH3CHO > alkene. It is noted that after adding HTO to CH3COOH, the intermediate CH3–COT(OH)2 could not undergo the elimination process, since the strongly electronegative O atom retains the hydrogen. Thus, after the formation of CH3–COT(OH)2 by addition, the hydrogen exchange proceeds through relaying proton via the six-membered ring TS.
 | ||
| Fig. 4 Addition–elimination T–H exchange reaction pathways of C2H4, C3H6, CH3CHO, CH3COCH3, and CH3COOH. Relative energies (kcal mol−1) of the corresponding species relative to the reactants are shown in parentheses. | ||
It should be emphasized that peroxides could also be formed when HTO adds to molecules with oxygen-containing groups. However, the calculated reaction paths of CH3CHO, CH3COCH3, and CH3COOH in Fig. 4 indicate that the TS for forming the peroxide and the corresponding intermediate are unstable, with rather high relative energies for the conditions in experiments. Thus, these paths are no longer considered.
Compared to the direct T–H exchange mechanism, it is clear that the relative energies of the addition or elimination steps are higher since these processes have four-membered ring TS, while the direct exchange always occurs through proton relay via six-membered ring TS, facilitated by one or two HTO molecules. The remarkable energy difference is attributed to the higher strain in the four-membered ring TS.
In summary, according to the calculations involved in the direct and the addition–elimination T–H exchange mechanisms, we conclude that the ability of T–H exchange is ordered as CH3COOH > CH3CH2OH > CH3CHO ≈ CH3COCH3 > alkene > alkane, and the reaction preferentially occur through the direct T–H exchange mechanism. Tritiated water more easily exchanges the most mobile hydrogen atoms in organic molecules, in other words, molecules with COOH and OH groups most readily exchange hydrogen with the HTO. These results are somewhat different from our previous studies of T2 with organic molecules in the T–H isotope exchange reaction,5 which could proceed via either the direct or the hydrogenation–dehydrogenation mechanisms, according to the types of the organic molecules.
Products distribution
According to the Boltzmann distribution, the selectivity of the products are predicted. As mentioned above, the direct mechanism is kinetically favored (with lower relative TS energy) for T–H exchange reactions between HTO and these molecules. The corresponding product selectivities for CH4, C2H4, C2H5OH, CH3CHO, CH3COCH3, and CH3COOH were calculated. The products and the calculated selectivity from the direct T–H exchange mechanism are listed in Table 2.| Species | Product | Selectivity (%) |
|---|---|---|
| CH4 | CH3T | — |
| C2H4 | C2H3T | — |
| C2H5OH | CH2TCH2OH | ≈0 |
| CH3CHTOH | ≈0 | |
| CH3CH2OT | ≈100 | |
| CH3CHO | CH2TCHO | ≈100 |
| CH3CTO | ≈0 | |
| CH3COCH3 | CH3COCH2T | — |
| CH3COOH | CH2TCOOH | ≈0 |
| CH3COOT | ≈100 |
Reactants containing one type of hydrogen, such as CH4, C2H4, and CH3COCH3 have the single product.
Reactants containing different types of hydrogen, such as C2H5OH, CH3CHO, and CH3COOH have competitive products. In the case of C2H5OH, there are three different products. As shown in Table 1, the calculated selectivity for CH3CH2OT is almost 100.0%, while those for CH2TCH2OH and CH3CHTOH are almost zero. In other words, the CH3CH2OT + H2O formation is kinetically predominant, whereas the other two products can be ruled out.
Similarly, the reactant CH3CHO can form either CH2TCHO or CH3CTO. The selectivity of CH2TCHO is calculated to be 100.0%, while the other is almost zero. Thus, the CH2TCHO is the predominant product, while that for CH3CTO formation is so small to be negligible.
Finally, the reactant CH3COOH leads to the formation of CH2TCOOH and CH3COOT. The selectivity of CH3COOT is calculated to be 100.0%, while the others is almost zero. Thus, the CH3COOT is the predominant product.
It is helpful to handle the tritium-contaminated vacuum pump oil using more scientific and proper methods by confirming the chemical forms of tritium existed in the oil. As mentioned above, in the presence of HTO, molecules with COOH and OH groups are most readily exchanged with HTO, and CH3CH2OT and CH3COOT might be the main components in the pump oil. Using the strong acidic or basic reagents to react with the tritiated compounds might be the efficient method to recover tritium and decrease the radiation hazard.
4 Conclusions
The mechanism of the T–H exchange between HTO and many typical organic molecules in pump oil has been investigated theoretically. The reactivity of different reactants and the selectivity of the products were also examined. The major conclusions are the following:(1) T–H exchange reaction between HTO and the organic molecules in pump oil could proceed with two different mechanisms: the direct and the addition–elimination T–H exchange mechanisms. The calculations indicate that the direct T–H exchange mechanism is kinetically favored.
(2) The HTO molecule is not only a reactant, but also acts as the proton shuttle in the TS. For molecules without the unsaturated carbonyl group, the T–H exchange could easily occur via a six-membered ring hydrogen migration TS including two HTO molecules, instead of the four-membered ring TS with only one HTO molecule. For those containing the carbonyl group, the carbonyl group could be part of the TS, thereby requiring only one HTO molecule in the TS.
(3) The T–H exchange is selective. Molecules containing the COOH and OH groups exchange with the HTO more easily. The predominant products for reactants C2H5OH, CH3CHO, and CH3COOH are CH3CH2OT, CH2TCHO, and CH3COOT, respectively.
Acknowledgements
This work was supported by the Foundation of President of China Academy of Engineering Physics (No. 201501001), the Innovation Fund of Institute of Nuclear Physics and Chemistry (No. 2013CX02), the National Natural Science Foundation of China (No. 21304110 and 21402158), the Research Subject of Environmental Protection and Occupational Health, Chongqing Research Program of Basic Research and Frontier Technology (No. cstc2013jcyjA50012, and cstc2016jcyjA0508), and China Postdoctoral Science Foundation funded project (2014M562326, and 2016T90851).References
- T. Takeishi, K. Kotoh, Y. Kawabata, J. Tanaka, S. Kawamura and M. Iwata, Fusion Sci. Technol., 2015, 67, 596 CAS
; J. P. Krasznai, D. F. Mullins and R. A. Mowat, Fusion Sci. Technol., 1995, 28, 1540 Search PubMed
- E. Evans, Tritium and its compounds, Atomizdat, Moscow, 1970 Search PubMed
- A. B. Sazonov, E. P. Magomedbekov, G. V. Veretennikova, S. A. Samoilov and A. V. Zharkov, At. Energy, 2005, 98, 127 CrossRef CAS
- É. P. Magomedbekov, V. A. Shalygin, O. A. Baranova, M. Y. Isaeva and A. V. Zharkov, At. Energy, 2005, 98, 122 CrossRef
- L. Dong, Y. Xie, L. Du, W. Li and Z. Tan, J. Hazard. Mater., 2015, 287, 42 CrossRef CAS PubMed
- S. W. L. Hogan and T. van Mourik, J. Comput. Chem., 2016, 37, 763 CrossRef CAS PubMed
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc, Wallingford CT, 2010 Search PubMed
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS
- M. V. Hopffgarten and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 43 CrossRef CAS
- C. Faltin, E. M. Fleming and S. J. Connon, J. Org. Chem., 2004, 69, 6496 CrossRef CAS PubMed
- D. Roy and R. B. Sunoj, Chem.–Eur. J., 2008, 14, 10530 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25404g |
| This journal is © The Royal Society of Chemistry 2017 |