DOI:
10.1039/C6RA25229J
(Paper)
RSC Adv., 2017,
7, 8467-8473
Effect of molten carbonate composition on the generation of carbon material
Received
14th October 2016
, Accepted 9th January 2017
First published on 26th January 2017
Abstract
Carbon dioxide, CO2, is thought to be a main culprit leading to global climate change and a wide variety of strategies have been proposed to reduce atmospheric CO2 levels. Here, CO2 is captured and subsequently electrochemically split into carbon materials in an electrolyzer comprising a eutectic mixture of carbonates, an Fe cathode and a Ni anode, at 600 °C and current densities of 50, 100, 200 mA cm−2. SEM, EDS, XRD and BET are employed to analyze the morphology, elemental composition, crystal structure as well as the BET surface area of the synthetic cathodic products. In addition, coulomb efficiency under different electrolytic conditions is measured via the comparison between moles of formed carbon product and the Faradays of charge passed during the electrolysis reaction. This paper investigated the effect of molten carbonate compositions on carbon product generation, and confirmed the visible dependence of produced carbon on the electrolytes.
1 Introduction
The increased carbon dioxide (CO2) emission that stems from human over-reliance on limited fossil fuels as the principal energy source has been cited as a major contributor to the greenhouse effect and global warming.1–4 The shocking growth rate of CO2 emission in turn has motivated researchers all over the world to seek feasible methods to effectively control the vast generation of CO2. Currently, the technology of carbon capture and storage (CCS) involving chemisorption5,6 and physisorption7–10 is being considered and developed, but objections also exist for its costly venture.11 Note, that CO2 is not only a greenhouse gas needed to be captured and stored, but an ideal C1 feedstock that can be applied in the field of synthesizing carbonaceous products.12,13 Therefore, the preparation of value-added, carbonaceous chemicals from CO2 emitted by industry is both an economic and environmental-friendly option that could alleviate the pressure of environmental degradation and the energy crisis simultaneously.
In recent years, researches on CO2 capture and conversion via electrolysis in molten salts have made great progress, where appropriate electrode materials, electrolyte, and reaction conditions are demonstrated to be of vital significance.14 Molten salts, and in particular molten carbonates, used as the electrolyte for this electrochemical conversion is due to their natural properties, including excellent heat and ion conduction, fast reaction kinetics as well as a wide electrochemical window, etc.15,16 With the aid of eutectic salts electrolyte, CO2 can be captured and subsequently converted into carbon monoxide (CO),17 solid carbon18,19 and even valuable chemicals of carbon nanofibers (CNFs) or their hollow form of carbon nanotubes (CNTs)20,21 that are widely applied in the manufacture of reinforced composites due to outstanding mechanical properties. Presently, carbonates, especially alkali carbonates of Li2CO3, Na2CO3 and K2CO3, are widely recognised as the optimum choice for CO2 capture and conversion, but to the best of our knowledge, rare work is dedicated to investigate the effect of carbonates composition on carbon production. In this work, Li2CO3, Na2CO3 and K2CO3 are mixed to form a binary or ternary molten carbonates electrolyte system to serve as an efficient site for electroreduction of CO2 into carbon powder; a 10 cm2 Fe wire and a 10 cm2 Ni wire are employed as cathode and anode, respectively. This paper highlights the effect of electrolytes with diverse alkali carbonates composition on the morphology and structure of the carbon product. Besides, charge and energy consumption at different electrolytes is also measured to see whether this electrochemical conversion can proceed at a relatively high coulomb efficiency.
2 Experimental
2.1 Methods and materials
An electrolysis system comprising a Fe cathode (10 cm2), a Ni anode (10 cm2), and a corundum crucible (Al2O3, purity > 99%) used to contain alkali carbonates (Li2CO3, Na2CO3 and K2CO3) with diverse mass ratio is established to allow the continuous adsorption and electrochemical conversion of CO2 into solid carbon. The electrodes of Fe and Ni are purchased from Hebei steady metal products co., LTD and need to be polished and stored in oil before electrolysis to avoid the surface oxidation. Li2CO3, Na2CO3 and K2CO3 are all of analytical purity, provided by Tianjin BA Chemical Reagent Factory.
The corundum crucible filled with mixed carbonates is set on a ceramic heating jacket heated by electricity. Before electrolysis, temperature of the corundum crucible is kept at >700 °C for several hours to ensure that the mixed carbonates can reach to molten state and moisture of the carbonates is removed. Then the temperature is cooled down to presupposed electrolytic temperature of 600 °C for electrolysis. The experiments are carried out at current densities of 50 mA cm−2, 100 mA cm−2, 200 mA cm−2 for 2 h, 1 h and 0.5 h, respectively, and then, cathode covered with carbon product is pulled out from the molten salts and carefully uncoiled. The extracted carbon product is subsequently subject to a series of post-electrolysis treatments, including acid picking (HCl), ultraphonic (10 min), bath heating (60 °C), suction filtration (0.4 μm filter membrane), abstersion (deionized water) and desiccation (80 °C, 6 h). Except experiment for GC analysis conducted at Ar atmosphere (to avoid effect of air components on the analysis of gaseous product), other experiments are performed at air atmosphere. Fig. 1a depicts the experimental conditions in detail. Pictures of helical electrodes (Ni anode, Fe cathode) before electrolysis are shown in Fig. 1b.
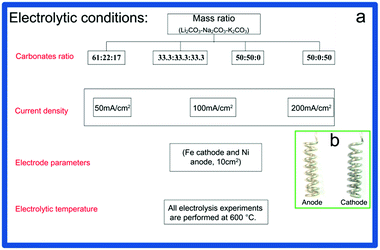 |
| | Fig. 1 (a) Specific experimental conditions and (b) electrodes before electrolysis. | |
2.2 Characterization
Elemental composition of purified cathodic product is studied by Energy Dispersive Spectroscope (EDS, X-Max, the British Oxford Instruments co., LTD). Micromorphologies and microstructures of the carbon sample are studied by Scanning Electron Microscope (SEM, SIGMA, Zeiss company) at working voltage of 10 kV, X-ray diffraction (XRD, step of 0.02, X-ray of 40 kV/40 mA, D/MAX 2200) as well as Brunauer–Emmett–Teller analysis (BET, Tristar-3020, Micromeritics Instrument Corporation).
3 Results and discussion
3.1 Reaction mechanism
CO2 removal proceeds via the electrolytic transformation of tetravalent carbon in proximity to the Fe cathode into solid and zerovalent carbon. Electrochemical reduction of CO2 into solid carbon in molten carbonates is generally thought to occur in 3 different reaction mechanisms,14 which are corresponding to eqn (1)–(3), respectively. However, it seems that more authors tend to approve the theory that CO2 is degraded into elemental carbon via the reduction of CO32− in molten melts (eqn (1)). Expounded by this mechanism, CO32− ions are directly reduced into solid carbon (lower electrolytic temperature, T < ∼850 °C) or CO (higher electrolytic temperature, T > ∼850 °C) and O2− during the electrolysis reaction, and the produced O2− ions besides being oxidized into oxygen (eqn (5)) as a by-product, further act as an absorbent for atmospheric CO2 (eqn (4)), allowing the regeneration of molten carbonates electrolyte.21 Thus, a perfect circulation of CO2 → C/CO + O2 is formed, wherein electricity consumed by this process can be replaced by sustainable energy, including solar power, wind energy, tidal energy, etc. Scheme 1 illustrates the carbon formation mechanism set forth by eqn (1), (4), and (5). Note, electrochemical conversion from CO2 to solid carbon in such molten carbonates sites is quite a complicated process, and eqn (1), (4), (5) are merely a simplification of the overall reaction, with many other complex electrochemical reactions proceeding at the same time.| | |
CO32− + 4e− → C + 3O2−
| (1) |
| | |
CO22− + 2e− → C + 2O2−
| (2) |
| | |
4M + M2CO3 → C + 3M2O
| (3) |
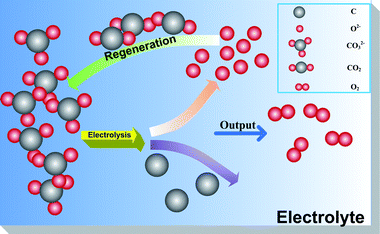 |
| | Scheme 1 Indirect conversion of CO2 into solid carbon at cathode and regeneration of the carbonate eutectic. | |
It has been addressed that electroreduction of CO32− ions into solid carbon merely occurs at electrolyte containing lithium and carbonate (Li+, CO32−), and therefore, no carbon powder is deposited in eutectic electrolyte of Na–K (Li-free) carbonates.14 The required electrolytic potential as a function of temperature calculated from relevant thermodynamic data (taken from website of NIST) can be invoked to explain why Li2CO3 reduction to carbon is more favorable than Na2CO3 and K2CO3. Relative to Na2CO3 and K2CO3, potential required by Li2CO3 to generate carbon is more positive than that to form Li metal via reduction of Li+ ions, evincing a preference for carbon deposition. As for eutectic of Na2CO3 and K2CO3, potential of carbon generation is comparative or even more negative than that of metal production, resulting in no carbon product and instead alkali metal is the preferred reduction product. However, a carbon product is observed from each of the following two Li-free molten carbonate electrolytes: a binary Na2CO3/BaCO3 mix or a ternary Na2CO3/K2CO3/CaCO3 mix electrolyte.22
In an electrolyte of Li–Na–K carbonates mixed by 33.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33.3:33.3 wt% (600 °C, 100 mA cm−2, a 10 cm2 Fe cathode, a 10 cm2 Ni anode), the obtained carbon product comprises five elements (Fig. 2a): C (93%), O (5%), Na (0.7%), K (1.1%), Au (0.2%), and all content is mole ratio. It should be noted that the peak of Au is from coating for SEM characterization. We can see that C occupies an overwhelmingly larger mole fraction (93%) in comparison to that of O, only ∼5%, denoting that the cathodic product is likely to be elemental carbon with bits of oxygen-containing functional groups.23 The presence of alkali metals, Na and K, in cathodic product verifies that carbon deposition is accompanied by very little co-deposition of alkali metals, which serves as an enabler for the decrease in coulomb efficiency. Additionally, small amounts of CO generation is also confirmed by gas chromatography (GC, Agilent 7890B) equipped with thermal conductivity detector (TCD), displayed in Fig. 2b. In this process, reaction is carried out at a sealed steel reactor, Ar atmosphere, and a vacuum pump is utilized to collect gas product every 15 minutes to avoid produced gas atmosphere inside the reactor. TCD signal displays distinctive, separated CO2, O2, and CO peaks at retention time of 2.786, 10.263, 12.585 min respectively, indicating the formation of CO2 (thermal decomposition of carbonate, such as Li2CO3 → Li2O + CO2), O2 (anodic product), and CO (cathodic by-product). No other peaks are observed at GC results, implying that there is no other composition in the gaseous product. Under ambient molten conditions the carbonate electrolyte is stable. However (in addition to O2 product), the vacuum removal of the gas favors the low pressures decomposition of the molten electrolyte as: carbonate (molten) ˗ oxide (dissolved) + CO2 (gas). Different from co-electrolysis of CO2 and H2O system,24 no methane is detected due to the lack of hydrogen source. The low, but important formation of CO demonstrates that CO2 splitting into zerovalent carbon isn't the exclusive degradation pathway at temperature of T < ∼850 °C, accompanied by very little CO generation that is presumably to occur at T > ∼850 °C.
:33.3:33.3 wt% (600 °C, 100 mA cm−2, a 10 cm2 Fe cathode, a 10 cm2 Ni anode), the obtained carbon product comprises five elements (Fig. 2a): C (93%), O (5%), Na (0.7%), K (1.1%), Au (0.2%), and all content is mole ratio. It should be noted that the peak of Au is from coating for SEM characterization. We can see that C occupies an overwhelmingly larger mole fraction (93%) in comparison to that of O, only ∼5%, denoting that the cathodic product is likely to be elemental carbon with bits of oxygen-containing functional groups.23 The presence of alkali metals, Na and K, in cathodic product verifies that carbon deposition is accompanied by very little co-deposition of alkali metals, which serves as an enabler for the decrease in coulomb efficiency. Additionally, small amounts of CO generation is also confirmed by gas chromatography (GC, Agilent 7890B) equipped with thermal conductivity detector (TCD), displayed in Fig. 2b. In this process, reaction is carried out at a sealed steel reactor, Ar atmosphere, and a vacuum pump is utilized to collect gas product every 15 minutes to avoid produced gas atmosphere inside the reactor. TCD signal displays distinctive, separated CO2, O2, and CO peaks at retention time of 2.786, 10.263, 12.585 min respectively, indicating the formation of CO2 (thermal decomposition of carbonate, such as Li2CO3 → Li2O + CO2), O2 (anodic product), and CO (cathodic by-product). No other peaks are observed at GC results, implying that there is no other composition in the gaseous product. Under ambient molten conditions the carbonate electrolyte is stable. However (in addition to O2 product), the vacuum removal of the gas favors the low pressures decomposition of the molten electrolyte as: carbonate (molten) ˗ oxide (dissolved) + CO2 (gas). Different from co-electrolysis of CO2 and H2O system,24 no methane is detected due to the lack of hydrogen source. The low, but important formation of CO demonstrates that CO2 splitting into zerovalent carbon isn't the exclusive degradation pathway at temperature of T < ∼850 °C, accompanied by very little CO generation that is presumably to occur at T > ∼850 °C.
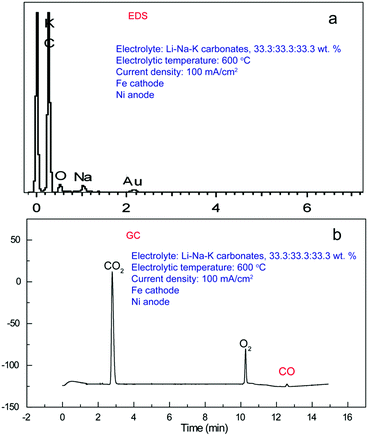 |
| | Fig. 2 (a) EDS of the solid product and (b) GC analysis of the gas product, obtained from Li–Na–K (33.3:33.3:33.3 wt%), 100 mA cm−2, 600 °C. | |
3.2 Micromorphology and microstructure characterization
SEM images of the carbon product prepared in diverse alkali carbonates are given at Fig. 3 that exhibits a dramatic morphological evolution. It has been proved that the microscopic morphology and structure of the deposited carbon product are remarkably affected by the composition of the mixed molten carbonates,21 and here, this dependence is verified again. Carbon product obtained from a binary mixture of carbonates (Li–Na, 50:50 wt%) possesses a very interesting microstructure that is formed by an accumulation of carbon spherical particles with a wide diameter distribution. Observed from SEM image (Fig. 3a), diameters of these spherical particles range from tens of nanometers to a few micrometers, which maybe dependent on electrolytic temperature as well as applied cell voltage/current density. However, in a system of Li–K binary carbonates mixed by 50:50 (wt%), the electro-deposited carbon present a noticeably different morphology of nanoflakes with thickness ranging from 50–200 nm (Fig. 3b) rather than a spherical structure from Li–Na system. Further morphological changes occur at a ternary electrolyte of Li–Na–K carbonates. Micromorphology of the carbon product obtained at electrolyte of 33.3:33.3:33.3 (wt%) presents a nanoflake-like structure with sharp edges (Fig. 3c). As composition of the molten carbonates electrolyte (Li–Na–K) is according to 61:22:17 (wt%), analogous morphology (shown in Fig. 3d) to that prepared in electrolyte of 33.3:33.3:33.3 (wt%) is observed rather than a mixture of nanoparticles and nanotubular structure prepared at ∼600 °C with Fe as cathode and Ni–Cr alloy as anode,21 indicating that electrode material is also one of the influential factors on morphology of the deposited carbon product. Corrosion resistance and conductive ability of anode material could indirectly change the voltage on electrolysis system, resulting in different deposition rate and various microstructure formations sequentially. Minor difference between the synthesized carbon product at 33.3:33.3:33.3 (wt%) and 61:22:17 (wt%) alkali carbonates is that a curly flake-like micromorphology with rough surface is formed at 61:22:17 (wt%), while a thinner and more smooth-surfaced flake-like structure is deposited at 33.3:33.3:33.3 (wt%).
 |
| | Fig. 3 SEM of carbon product prepared at 100 mA cm−2, 600 °C and diverse electrolytes. | |
We have previously noted the noteworthy increase in BET surface area at elevated current density,21 and in this work, this finding is proved again. As an example, the BET surface area of carbon product prepared at Li–Na carbonates of 50:50 wt% increases to 80.3 m2 g−1 at 200 mA cm−2, nearly twice over of that at 50 mA cm−2 (39.9 m2 g−1). This proven increase could be attributed to that larger applied current density facilitates formation of incompact structure, and in this manner, carbon product with higher BET surface area deposits at cathode. Moreover, through the comparison and evaluation among these 4 electrolyte systems, it can be found that Li–Na carbonates system favours generation of carbon product with smaller particle size and higher BET surface area, reflected on the primary micromorphology of carbon spheres, rather than irregular carbon flakes and amorphous carbon. Another, as far as we know, BET surface area of as-prepared carbon is also significantly dependent on electrolytic temperature, that is, lower temperature prefers carbon deposition with higher BET surface area. Taking Tang's work as a supporting example, BET surface area increases from 101.89 m2 g−1 of 650 °C to 212.39 m2 g−1 of 550 °C at the same voltage, and this conspicuous increase is likely due to the slower deposition rate at lower applied temperature.23 Carbon electrodeposition in molten salts electrolyte is constrained by electrons arriving from the cathode at the electrolyte interface and thus, microstructure is significantly affected by electrolytic parameters, including molten salts composition, applied current density, electrolytic temperature, etc. (Table 1).
Table 1 BET surface area of the carbon sample prepared at diverse electrolytic conditions
| Electrolyte composition (wt%) |
Current density (mA cm−2) |
BET surface area (m2 g−1) |
| Li–Na of 50:50 |
50 |
39.9 |
| 100 |
44.9 |
| 200 |
80.3 |
| Li–K of 50:50 |
50 |
14.5 |
| 100 |
19.5 |
| 200 |
47.2 |
| Li–Na–K of 33.3:33.3:33.3 |
50 |
34.5 |
| 100 |
46.5 |
| 200 |
69.1 |
| Li–Na–K of 61:22:17 |
50 |
13.2 |
| 100 |
25.7 |
| 200 |
59.6 |
As seen from Fig. 4a–d, massive spherical carbon particles with diverse diameters (micron grade) stack together irregularly. Formation of these deposited carbon sphere aggregates is likely owing to the nucleating effect of the initially generated carbon powder in this electrolyte environment, acting as a substrate for further carbon deposition. Presently, due to the potential utilizations as reinforcement materials or matrix for rubber, catalyst supports, lubricating materials, anodes in second Li-ion batteries, hydrogen storage media, etc., carbon spheres as a new reinforcement agent have drawn extensive attention and interests from carbon scientists worldwide.25–30 Especially, for some sophisticated applications, monodisperse and smooth-surfaced carbon spheres are shown to be required and advantageous.31,32 Particle size, structure as well as surface chemistry are demonstrated to be three vital influencing factors on their practical properties.33 Therefore, particular attention will be focused on the dispersion and characterization of the above carbon spheres to realize their valuable application on industry. In addition to carbon spheres, flake-like morphology is observed in carbon samples as well (red mark in Fig. 4b and d), growing under/mixed with carbon spheres. Throughout the previous reports, flake-like electrodeposited carbon samples are shown to be a common micromorphology,21,23 whose thickness is also visibly affected by electrolytic temperature and applied cell voltage/current density.
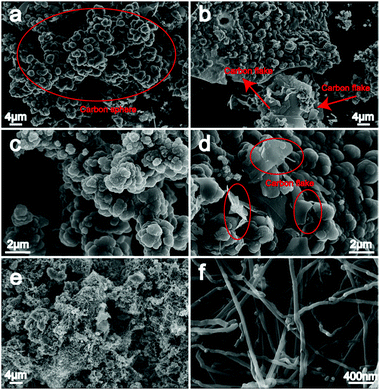 |
| | Fig. 4 (a)–(d) are lower magnification SEM images of carbon spheres obtained at Li–Na (50:50 wt%), 600 °C; (e) and (f) are carbon product synthesized at Li–Na (50:50 wt%), 750 °C. | |
Li–Na mixed carbonates are considered to be an efficient, economic, stable electrolyte for CO2 capture and electrochemical conversion as it can simultaneously decrease required energy to reach molten state than pure Li2CO3 electrolyte and doesn't contain K2CO3 that is speculated to be the most corrosive to nickel anode among Li–Na–K carbonates.34–36 Additionally, electrolysis in Li–Na eutectic carbonates of 50:50 wt% at 600 °C provides a cost effective pathway to achieve the transition from CO2 into carbon sphere subsequent to electrolysis for 1 h at 100 mA cm−2 with a coulomb efficiency of ∼85.4%. Recently it has been introduced that21 electrolysis in Li–Na carbonates electrolyte mixed by 50:50 wt% at 750 °C, 200 mA cm−2, generates a carbon product comprising over 85% carbon nanotube (Fig. 4e and f), strongly incentivizing to degrade atmospheric CO2 level by introduced molten salts electrolysis. Carbon nanotube, seamless cylinders of one or more layers of graphene, has shown promising performance for various applications involving supercapacitors, actuators, lightweight electromagnetic shields, etc., due to its outstanding mechanical strength and electrical or thermal conductivities.37,38 Gas phase CO2 dissolves and this has been isotopically traced as the source of the carbon nanotubes formed by molten carbonate electrolysis.39 As summarized by Licht group,20,21 formation factors of carbon nanotube at above mentioned electrolyte could be concluded as: (1) the utilization of a Zn-coated steel cathode (Zn can effectively lower the needed energy during carbon formation at the starting stage), (2) a constrained nickel concentration in electrolyte from anodic oxidation. An initial low current density subsequent by a higher current density is shown to be necessary for the controlled growth of carbon nanotube in pure Li2CO3 and Li–Na carbonates mixed by ∼90:10 wt%, but in electrolyte of Li–Na carbonates mixed by 50:50 wt%, carbon nanotube growth can proceed without the step of gradual increase in applied current density. As further presented by the Licht group, straight, rather than tangled, carbon nanotube is formed in pure Li2CO3 electrolyte, while electrolysis in Li–Na mixed carbonates tends to generate tangled carbon nanotube, ascribed to sp3 defects in the sp2 graphene layered structures.40 Furthermore, wall thickness of the synthesized carbon nanotube can also be controlled by regulating electrolyte composition: electrolysis in Li2CO3 mixed with sufficient CaCO3 prefers synthesizing thin-walled carbon nanotubes, while in pure Li2CO3 electrolyte, thicker carbon nanotube becomes the preferable synthetic product.40 According to recent reports,41 optimized storage capacities of the as-prepared, low-cost, CO2-derived carbon nanotube are measured over 370 mA h g−1 and 130 mA h g−1 owing to synthetic control of sp3 content in the synthetic nanostructures, with no capacity fade under durability tests up to 200 and 600 cycles in Li-ion and Na-ion batteries, respectively. These findings based on experiments strongly demonstrate the fact that ambient CO2 could act as raw feedstock in industrial production, and this transformation provides a path to eliminate carbon dioxide emissions from power or cement plants.39,42,43
XRD characterization (Fig. 5) was performed to reveal the crystal characteristics of the carbon product prepared at different molten salts composition, wherein no obvious sharp diffraction peak of carbon appears, signifying that the obtained carbon product in this work is mostly amorphous. This crystal characteristic could be ascribed to the conjecture that the crystal defects formed by disordering and irregular hexagonal annular plane make it challenging to pinpoint the location of the characteristic peaks of carbon. Whereas, an earlier application of a Zn coated (galvanized) steel cathode and a nickel anode to a pure Li2CO3 electrolyte seems to be an excellent work for generating carbon nanofiber possessing a much better crystal.20 We should note that the presence of some microstructures with preferable crystal form in above carbon samples can't be denied, and maybe it’s the existence of massive amorphous carbon that results in an overall crystal defect.
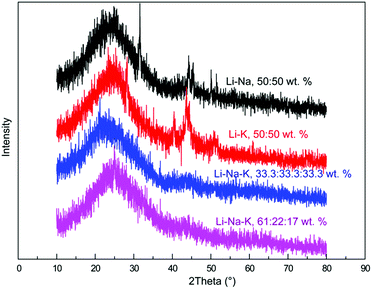 |
| | Fig. 5 XRD diffraction patterns of the carbon material electro-deposited at diverse carbonates, 100 mA cm−2, 600 °C. | |
3.3 Energy consumption
Cell-voltage plots in Fig. 6 are recorded during electrolysis reaction at diverse electrolyte composition and current densities with electrochemical workstation. Obviously, cell-voltage values increase at elevated current densities, which is consistent with the anticipated increase in overpotential with increasing current density. In terms of cell-voltage of 50 mA cm−2, Li–Na (50:50 wt%), Li–K (50:50 wt%) and Li–Na–K (61:22:17 wt%) steady at 2.2–2.3 V, while Li–Na–K (33.3:33.3:33.3 wt%) has a minor increase, floating at 2.3–2.4 V. Similarly, cell voltage of Li–Na (50:50 wt%), Li–K (50:50 wt%) and Li–Na–K (61:22:17 wt%) at 100 mA cm−2 is in the range of 2.4–2.5 V, and an amplification of 0.1–0.2 V is also observed at Li–Na–K (33.3:33.3:33.3 wt%). As the applied current density increases to 200 mA cm−2, compared to the lowest cell-voltage at Li–Na (50:50 wt%), within the range of 2.7–2.8 V, Li–K (50:50 wt%) and Li–Na–K (61:22:17 wt%) have an obvious and significant increase of 0.2–0.3 V, steadying at 2.8–3.0 V, while Li–Na–K (33.3:33.3:33.3 wt%) presents a stable cell-voltage of ∼3 V. Though the minor fluctuations probably caused by the stability of electrolyte system, electrode corrosion, variations in electrolytic temperature etc., are found to occur at constant current density electrolysis, the overall trend is relatively stable, signifying that this system tends to provide a mild and stable site for CO2 capture and carbon deposition.
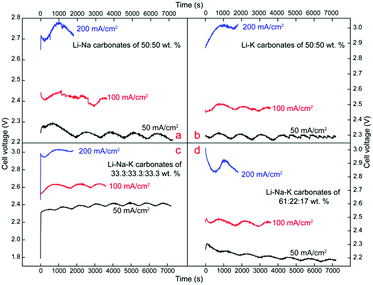 |
| | Fig. 6 Cell-voltage plots during 600 °C electrolysis in different electrolytes. | |
Coulomb efficiency under different electrolytic conditions is measured in term of the moles of carbon deposited at cathode and electricity consumed by electrolysis process (4F per mole of carbon). As shown in Fig. 7a, coulomb efficiency for carbon-generation ranges from ∼61.8% to ∼94.9%, which is considered to be a very satisfying result. In Li–Na (50:50 wt%) and Li–Na–K (61:22:17 wt%), electrolysis at different current densities, the most ideal coulomb efficiency is obtained at 200 mA cm−2, while in Li–K (50:50 wt%) and Li–Na–K (33.3:33.3:33.3 wt%), optimum coulomb efficiency is gained at 100 mA cm−2. As previously noted that solubility of nickel cation increases at sodium-containing and potassium-containing carbonates electrolyte, and predictably, pure Na/K carbonate electrolyte could be exceeding corrosive to the nickel anode, prompting the electrolyte to turns from transparent to green.20 Thus, in electrolyte containing high concentrations of Na+ and K+, a lower coulomb efficiency and a serious anode corrosion are often observed, such as in electrolyte of Li–Na–K ternary carbonates (33.3:33.3:33.3 wt%) at 200 mA cm−2. Compared with another ternary Li–Na–K system (61:22:17 wt%) and binary carbonates electrolyte (Li–Na, Li–K, 50:50 wt%), a noticeable shrinkage in coulomb efficiency occurs in Li–Na–K system (33.3:33.3:33.3 wt%) at 200 mA cm−2 (<80%), and the discussed anode corrosion above can give answers to this phenomenon. Generally, an enlarged current density leads to a violent anode corrosion, however, in spite of the weak anodic corrosion at lower applied current density, the lowest coulomb efficiency is found at 50 mA cm−2 in Li–Na (50:50 wt%), Li–K (50:50 wt%) and Li–Na–K (61:22:17 wt%), which could be attributed to the primarily slower deposition rate. In contrast, at electrolyte of Li–Na–K (33.3:33.3:33.3 wt%), anodic corrosion becomes first and foremost due to the elevated concentration of Na+ and K+, leading to the lowest coulomb efficiency at 200 mA cm−2. Anode images after electrolysis at Li–Na–K ternary electrolyte (33.3:33.3:33.3 wt%) at 200 mA cm−2 for 0.5 h are depicted in Fig. 7b. In addition to a thick layer of frozen electrolyte adhering on the surface, a serious anode corrosion is also observed (red-marked parts are after removing attached frozen electrolyte), meaning the charge passed through the electrolyzer besides achieving the reduction of CO32− into carbon deposit, and simultaneously oxidizes Ni into Ni2+ (the electrolyte turns from transparent to green), resulting in the decrease of coulomb efficiency. This is consistent with the finding that the oxygen-generating anode effects promote significant carbon product formation at cathode.20 Note, that the anodic corrosion seems to be general but inhomogeneous presumably for the uneven anode polarization current. However, in an electrolyte containing higher concentrations of Li cation (61:22:17 wt%), the Ni anode corrosion is not evident, further manifesting that the cation composition of the carbonate electrolyte determines the extent of nickel anode corrosion notably.
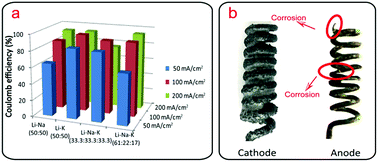 |
| | Fig. 7 (a) Coulomb efficiency at different electrolytic conditions; (b) images of electrodes after electrolysis in Li–Na–K carbonates (33.3:33.3:33.3 wt%) at 200 mA cm−2 for 0.5 h. | |
4 Conclusions
This paper for the first time presents a comparison among electrolysis in Li–Na (50:50 wt%), Li–K (50:50 wt%), Li–Na–K (33.3:33.3:33.3 wt%), and Li–Na–K (61:22:17 wt%), and highlights the effect of carbonates composition on cathodic carbon deposition (morphology and structure) as well as coulomb efficiency for carbon generation. Besides carbon flake with diverse thickness that is often observed in previous relevant publications, very interesting spherical aggregates that possess a potential to be used as elastomer filler are formed during electrolysis in Li–Na electrolyte mixed by 50:50 wt% at 100 mA cm−2. Moreover, carbon nanotube, a valuable chemical, is obtained at cathode in the same electrolyte, but higher electrolytic temperature of 750 °C. Another result concluded from this work is that molten carbonates composition could influence anode corrosion powerfully, and then recorded cell voltage and calculated coulomb efficiency (the most ideal efficiency approaching 94.9% in Li–K electrolyte at 100 mA cm−2) are indirectly affected. Energy consumption is an urgent problem to be solved and a subsequent challenging task we face is how to combine renewable energy source with this electrochemical system, to pander to environmental sustainability. In sum, converting ambient CO2 into value-added carbonaceous materials via molten salts electrolysis technology is critical for sustainable development and deserves our further investigation.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21476046 and 21306022), Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20132322120002), China Postdoctoral Science Foundation (No. 2013M540269), Postdoctoral Science Foundation of Heilongjiang Province of China (No. LBH-TZ0417), Innovative Team of Science and Technology in Heilongjiang Higher Education Institutes (No. 2013TD004) and Northeast Petroleum University (No. SJQHB201602 and YJSCX2016-018 NEPU). We are grateful to Professor Stuart Licht (The George Washington University) for valuable contributions to the research.
Notes and references
- K. E. Lonngren and E. Bai, Energy Policy, 2008, 36, 1567–1568 CrossRef.
- P. Friedlingstein, R. M. Andrew, J. Rogelj, G. P. Peters, J. G. Canadell, R. Knutti, G. Luderer, M. R. Raupach, M. Schaeffer, D. P. van Vuuren and C. Le Quéré, Nat. Geosci., 2014, 7, 709–715 CrossRef CAS.
- D. Huisingh, Z. Zhang, J. C. Moore, Q. Qiao and Q. Li, J. Cleaner Prod., 2015, 103, 1–12 CrossRef CAS.
- Y. H. Cheng, Y. H. Chang and I. J. Lu, Appl. Energy, 2015, 157, 953–973 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- T. Viktorija, S. Asbjørn and O. Espen, Energy Fuels, 2013, 27, 5373–5379 Search PubMed.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954–962 CrossRef PubMed.
- J. Byun, S.-H. Je, H. A. Patel, A. Coskun and C. T. Yavuz, J. Mater. Chem. A, 2014, 2, 12507–12512 CAS.
- A. Wahby, J. M. Ramos-Ferńandez, M. Martínez-Escandell, A. Sepúlveda-Escribano, J. Silvestre-Albero and F. Rodríguez-Reinoso, ChemSusChem, 2010, 3, 974–981 CrossRef CAS PubMed.
- B. Ashourirad, P. Arab, T. Islamoglu, K. A. Cychosz, M. Thommes and H. M. El-Kaderi, J. Mater. Chem. A, 2016, 4, 14693–14702 CAS.
- Z. Zhang and D. Huisingh, J. Cleaner Prod., 2016, 30, 1–10 Search PubMed.
- X. Gao, B. Yu, Z. Yang, Y. Zhao, H. Zhang, L. Hao, B. Han and Z. Liu, ACS Catal., 2015, 5, 6648–6652 CrossRef CAS.
- G. P. S. Lau, M. R. Schreier, D. Vasilyev, R. Scopelliti, M. Grätzel and P. J. Dyson, J. Am. Chem. Soc., 2016, 138, 7820–7823 CrossRef CAS PubMed.
- H. V. Ijije, R. C. Lawrence and G. Z. Chen, RSC Adv., 2014, 4, 35808–35817 RSC.
- Y. Kanai, K. Fukunaga, K. Terasaka and S. Fujioka, Chem. Eng. Sci., 2013, 100, 153–159 CrossRef CAS.
- Z. Shi, W. Deng, X. Song, X. Hu, B. Gao and Z. Wang, J. Chem. Eng. Data, 2016, 61, 3020–3026 CrossRef CAS.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- H. Yin, X. Mao, D. Tang, W. Xiao, L. Xing, H. Zhu, D. Wang and D. R. Sadoway, Energy Environ. Sci., 2013, 6, 1538–1545 CAS.
- B. Deng, Z. Chen, M. Gao, Y. Song, K. Zheng, J. Tang, W. Xiao, X. Mao and D. Wang, Faraday Discuss., 2016, 190, 241–258 RSC.
- J. Ren, F. F. Li, J. Lau, L. González-Urbina and S. Licht, Nano Lett., 2015, 15, 6142–6148 CrossRef CAS PubMed.
- H. Wu, Z. Li, D. Ji, Y. Liu, L. Li, D. Yuan, Z. Zhang, J. Ren, M. Lefler, B. Wang and S. Licht, Carbon, 2016, 106, 208–217 CrossRef CAS.
- J. Ren, J. Lau, M. Lefler and S. Licht, J. Phys. Chem. C, 2015, 119, 23342–23349 CAS.
- D. Tang, H. Yin, X. Mao, W. Xiao and D. H. Wang, Electrochim. Acta, 2013, 114, 567–573 CrossRef CAS.
- H. Wu, D. Ji, L. Li, D. Yuan, Y. Zhu, B. Wang, Z. Zhang and S. Licht, Advanced Materials Technologies, 2016, 1, 1–10 CrossRef.
- L. Mao, J. Qiu and L. Xing, Mater. Lett., 2008, 62, 581–583 CrossRef CAS.
- L. Xu, W. Zhang, Q. Yang, Y. Ding, W. Yu and Y. Qian, Carbon, 2005, 43, 1090–1092 CrossRef CAS.
- L. Tosheva, J. Parmentier, V. Valtchev, C. Vix-Guterl and J. Patarin, Carbon, 2005, 43, 2474–2480 CrossRef CAS.
- J. Martin-Gil, F. J. Martin-Gil, E. Moran, M. Miki-Yoshida, L. Martinez and M. José-Yacaman, Acta Metall. Mater., 1995, 43, 1243–1247 CrossRef CAS.
- Q. Wang, F. Cao, Q. Chen and C. Chen, Mater. Lett., 2005, 59, 3738–3741 CrossRef CAS.
- W. Li, D. Chen, Z. Li, Y. Shi, Y. Wan, G. Wang, Z. Jiang and D. Zhao, Carbon, 2007, 45, 1757–1763 CrossRef CAS.
- B. Friedel and S. Greulich-Weber, Small, 2006, 2, 859–863 CrossRef CAS PubMed.
- Q. Lin, M. Zheng, T. Qin, R. Guo and P. Tian, J. Anal. Appl. Pyrolysis, 2010, 89, 112–116 CrossRef CAS.
- A. A. Deshmukh, S. D. Mhlanga and N. J. Coville, Mater. Sci. Eng., R, 2010, 70, 1–28 CrossRef.
- S. Mahammadunnisa, E. L. Reddy, D. Ray, C. Subrahmanyam and J. C. Whitehead, Int. J. Greenhouse Gas Control, 2013, 16, 361–363 CrossRef CAS.
- I. A. Novoselova, N. F. Oliinyk, S. V. Volkov, A. A. Konchits, I. B. Yanchuk, V. S. Yefanov, S. P. Kolesnik and M. V. Karpets, Phys. E, 2008, 40, 2231–2237 CrossRef CAS.
-
(a) F. F. Li, S. Liu, B. Cui, J. Lau, J. Stuart, B. Wang and S. Licht, Adv. Energy Mater., 2015, 5, 1–7 Search PubMed;
(b) S. Licht, Stabilization of STEP electrolytes, 2012, accessed Jan 2017, http://arxiv.org/pdf/1209.3512.pdf Search PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- M. F. L. D. Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef PubMed.
- J. Ren and S. Licht, Sci. Rep., 2016, 6, 27760–27760 CrossRef CAS PubMed.
- S. Licht, M. Lefler, J. Ren and J. Vicini, arXiv preprint arXiv:1607.02220, 2016, in press.
- S. Licht, A. Douglas, J. Ren, R. Carter, M. Lefler and C. L. Pint, ACS Cent. Sci., 2016, 2, 162–168 CrossRef CAS PubMed.
- S. Licht, Co-Production of Cement and Carbon Nanotubes with a Carbon Negative Footprint, 2016, accessed Jan 2017, https://arxiv.org/pdf/1608.00946v1.pdf Search PubMed.
- J. Lau, G. Dey and G. S. Licht, Energy Convers. Manage., 2016, 122, 400–410 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Zhida Li
a,
Zhida Li






