
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-ions linked surface-confined molecular dyads of Zn-porphyrin–metallo-terpyridine: an experimental and theoretical study†‡
Bhawna Geraa,
Arun Kumar Mannab and
Prakash Chandra Mondal§
*a
aDepartment of Chemistry, University of Delhi, New Delhi-110007, India
bDepartment of Materials and Interfaces, Weizmann Institute of Science, Rehovot, Israel
First published on 5th January 2017
Abstract
Covalently-assembled monolayers of functionalized Zn-porphyrin complex (5,10,15,20-tetra(4-pyridyl)-porphyrin, Zn-TPyP) are employed as the template layer for fabricating heterogeneous molecular dyads composed of opto-active metallo-terpyridyl complexes on SiOx substrates. Metallo-linkers such as Cu2+ and Pd2+ are exploited to connect the Zn-porphyrin template layer with the metallo-terpyridyl complexes (M–PT, M = Fe2+, Ru2+, Os2+, while PT = 4′-pyridyl terpyridyl) using a wet chemical “layer-by-layer” (LBL) technique. Formation of both the template and dyad layers over the SiOx substrates were probed by atomic force microscopy (AFM) and UV-vis absorption techniques. The molecular films were used for comparison and to study the effect of different metallo-linkers based on the changes in the characteristics of the Soret bands of Zn-TPyP and the metal-to-ligand charge-transfer (MLCT) bands of terpyridyl complexes. Besides, detailed electronic structure calculations based on first-principles density functional theory (DFT) and time-dependent DFT (TDDFT) have been performed for understanding experimentally observed photophysical properties of the surface-confined dyads.
1. Introduction
Porphyrin derivatives are the central molecular building blocks of several enzymes and proteins like haemoglobin, cytochrome c, chlorophyll and vitamin B-12.1 Porphyrins with different central metal ions, so-called metallo-porphyrins, and peripheral substituents have been synthesized in large variety in order to mimic the numerous bio-functional processes.2 Since, these π-conjugated, aromatic systems are capable of absorbing light in the visible and near infra-red region, they act as excellent photosensitizers and show several stimulating applications such as in photodynamic therapy, catalysis, opto-electronics and others.3 Further, exterior functionality in the multifunctional porphyrins has been used to generate novel 3D architectures along with large open channels which can be achieved by coordination reaction of metallo-linkers (Cd2+/Cu+) to the pendant pyridyl ligands.4Self-assembled monolayers (SAMs) of these molecules on different solid substrates are highly ordered and thus can be used as the model system to study several surface-based properties. For instance, the porphyrin-based molecular thin films have shown several interesting electrical, optical properties and have also been used as chemical sensors.5 Due to stable and extended π-conjugated macrocyclic core, and extensive charge holding capacity, the metallo-porphyrins have also been explored for the construction of photonic and information storage devices.6 Interestingly, functional metallo-porphyrins (also well-known as metallo-ligands) have recently been assembled over ferromagnetic substrates (for example, Fe, Co, Ni and its alloys) for fundamental studies in surface coordination chemistry which is an emerging field of molecular ‘spintronics’.7 However, molecular based thin films fabricated from metallo-porphyrins with different metal centers in the core with different exterior functionalities have already been studied,8 but the solid state molecular architectures which combine both the metallo-porphyrins and metallo-terpyridyls are almost untouched. However, solution-based studies using metallo-porphyrins and metallo-terpyridyl have been well-documented.9 Since, both components offer rich photophysical and electrochemical properties, so molecular engineering of both the porphyrins and terpyridyl complexes over the solid substrate to provide a promising approach for creating a hybrid molecular architecture with potential applications in material science. In this direction, layer-by-layer (LBL), a unique method to a conglomerate molecular assembly, which can be achieved by repeating the reaction components are combined sequentially.10 Most importantly, the great advantage of this elegant technique is that film orientation and a number of layers onto the substrate can be controlled which is nearly impossible by existing solvothermal processes. As a consequence, it provides a promising route for fabricating ordered multifunctional thin films of desired components.
In our previous study,11 we have emphasized on intermolecular electronic communication between heterogeneous metallo-terpyridyl based dyads. Herein, we design and fabricate meso-[5,10,15,20-tetra(4-pyridyl)-porphyrin-Zn, see Scheme 1], (Zn-TPyP) monolayers by means of a covalent bond formation onto the flat glass and silicon substrates. The Zn-porphyrin monolayers containing free pyridyl groups employed as a template layer for fabricating heterogeneous molecular dyads with terpyridyl complexes over the same platform. In order to fabricate metallo-organic dyads, we consider the popular layer-by-layer method.10a,b The marked differences in optical properties of the molecular dyads with different metallo-linkers have been studied in details.
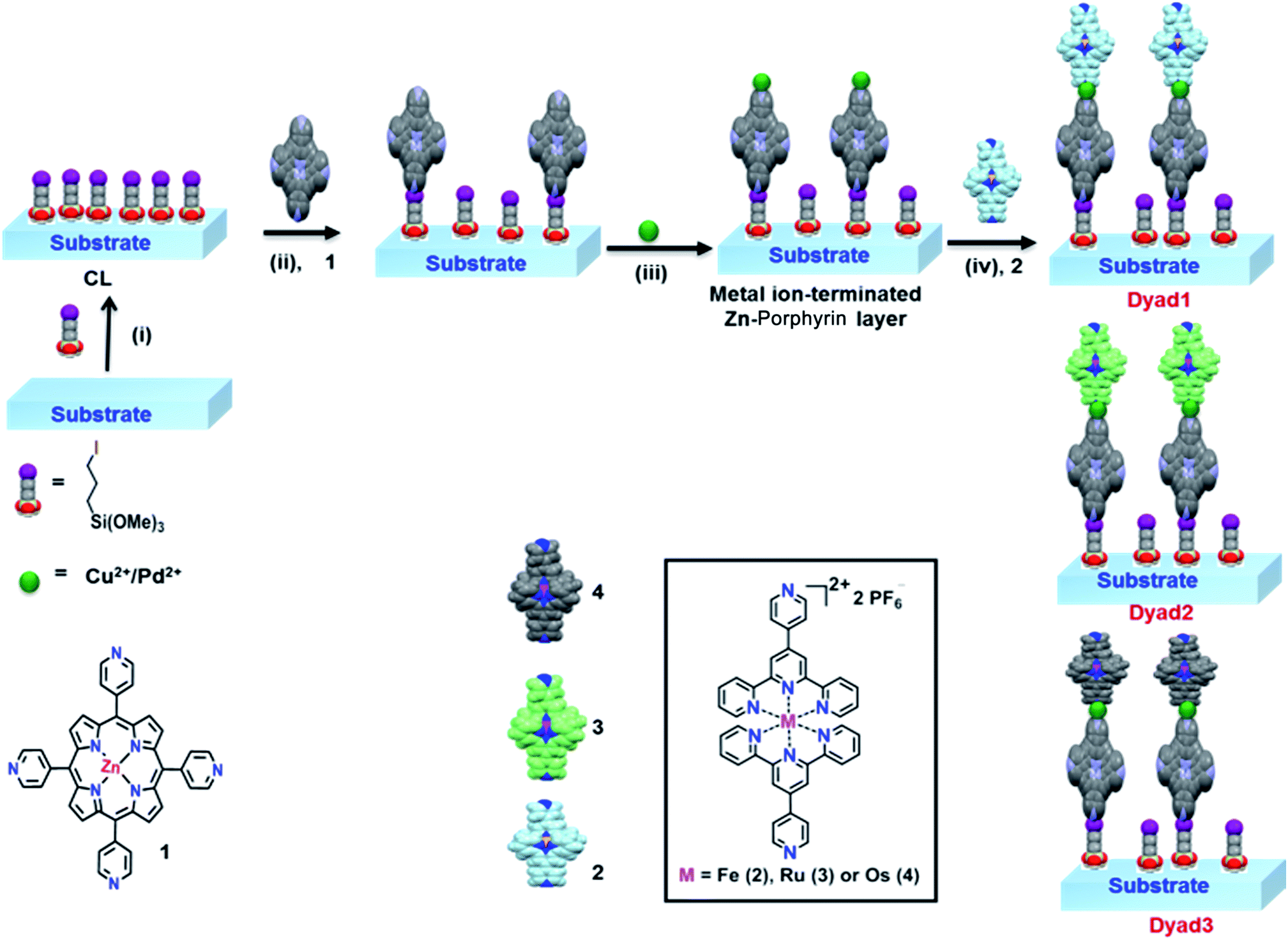 | ||
| Scheme 1 Schematic description of the perpetration method for metal-directed Zn-porphyrin–terpyridine dyads onto SiOx substrates which can be divided into four different steps: (i) immobilization of 3-iodo-n-propyl-trimethoxysilane onto a SiOx substrate to form coupling layer (CL), (ii) quaternization of pendant 4′-pyridyl group of Zn-porphyrin complex to form covalent assembled monolayers (template layers), (iii) coordination of metallo-linkers, Cu(NO3)2/PdCl2(PhCN)2 with metal-mediated pyridine-terminated monolayers, and (iv) coordination of a respective M–PT (M = Fe2+, Ru2+, Os2+) unit to fabricate the respective heterogeneous metallic molecular dyads. | ||
2. Experimental sections
2.1. Materials
Pyridine-4-carboxaldehyde, 2-acetyl pyridine, RuCl3·3H2O, (NH4)2OsCl6, FeCl2, 3-iodo-n-propyltrimethoxy-silane, pyrrole, PdCl2 and NH4PF6 were purchased from Sigma-Aldrich. Salicylic acid was purchased from Alfa Aesar and was used as received. Zinc acetate, cupric nitrate, 30% aq. ammonia, dry n-pentane, dichloromethane, n-hexane were purchased from S. D. Fine Chemicals (Mumbai, India). Solvents were purchased from Merck. All the chemicals were used as received without any further purification. Teflon-lined autoclaves (25 mL and 50 mL) were purchased from Prakash Scientific, Bangalore, India. Single-crystal silicons (100) were purchased from Georg Albert PVD-Beschichtungen (Silz, Germany). Soda lime glasses were purchased from Chase Scientific, India.2.2. Synthesis of the Zn-porphyrin and terpyridyl complexes
Synthesis of the meso-tetra-(4-pyridyl)porphyrin, Zn-TPyP and metallo-terpyridyl complexes have been performed via established method and characterization data is provided in ESI.‡Formation of the coupling layers. Smooth and native silicon substrates (100) were cut into 1 × 1 cm2 and activated by sonication in n-hexane, acetone and 2-propanol for 20 min each followed by drying under N2 stream. The glass substrates were cleaned by immersing in a freshly prepared “piranha” solution (composition of “piranha” solution is: 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (v/v) of conc. H2SO4/30% H2O2) for over 1 h [Caution: “piranha” is a dangerous oxidizing agent and must be handled with apposite self-protection]. Consequently, the glass substrates were rinsed with deionized water and then exposed to “RCA” [composition of RCA:H2O/NH4OH/30% H2O2, 5:1:1 (v/v)] for 45 min followed by rinsing with plenty of deionized water and drying under N2. In order to activate the substrates, they were kept in an oven at 110 °C for 2 h. Freshly prepared glass and silicon substrates were allowed to react with dry n-pentane solution of 3-iodo-n-propyltrimethoxy-silane (200:1, v/v) for 30 min under N2. Then the coupling layer containing substrates were thoroughly washed with dry n-pentane followed by sonication with n-pentane, dichloromethane and 2-propanol. Finally, the resulting iodo-terminated coupling layers were dried under N2 followed by drying at 120 °C for 45 min.
:3 (v/v) of conc. H2SO4/30% H2O2) for over 1 h [Caution: “piranha” is a dangerous oxidizing agent and must be handled with apposite self-protection]. Consequently, the glass substrates were rinsed with deionized water and then exposed to “RCA” [composition of RCA:H2O/NH4OH/30% H2O2, 5:1:1 (v/v)] for 45 min followed by rinsing with plenty of deionized water and drying under N2. In order to activate the substrates, they were kept in an oven at 110 °C for 2 h. Freshly prepared glass and silicon substrates were allowed to react with dry n-pentane solution of 3-iodo-n-propyltrimethoxy-silane (200:1, v/v) for 30 min under N2. Then the coupling layer containing substrates were thoroughly washed with dry n-pentane followed by sonication with n-pentane, dichloromethane and 2-propanol. Finally, the resulting iodo-terminated coupling layers were dried under N2 followed by drying at 120 °C for 45 min.
2.3. Formation of covalent assembled monolayers and molecular dyad layers
The coupling layer containing substrates were placed in a 50 mL Teflon-lined autoclave having a mixture of dry chloroform/toluene (3:7, v/v) solution of the corresponding complexes (0.5 mM) and kept at 85 °C for 56 h in a programmed oven. The autoclaves were slowly cooled to room temperature and the functionalized substrates were rinsed with acetonitrile, acetone and 2-propanol, respectively before drying it under N2. The observance of the respective absorption at specific wavelength indicated the attachment of the Zn-porphyrin complex over the substrates. However, increasing the reaction time did not show a further increase in absorption, but lower reaction time (<15 h) showed less absorption. Freshly prepared monolayers (template layers) on silicon, and glass substrates were allowed to react with 0.5 mM solution of the corresponding metallo-linkers [for example, Cu(NO3)2/PdCl2(PhCN)2] in dry acetonitrile for 30 min at room temperature under exclusion of light and without any disturbance. The metal-mediated monolayer films [SiOx/Zn-TPyP/M(Cu/Pd)] were then carefully rinsed in acetonitrile and dried under N2. Consequently, the molecular films were immersed in acetonitrile solution of different M–PT complex for 2 h resulting in the formation of hetero-metallic molecular dyad layers, [SiOx/Zn-TPyP/M(Cu/Pd)/M–PT] as shown in Scheme 1. The dyad layers were rinsed in acetonitrile first and then mild sonication with the same solvent in order to remove physisorbed materials and dried under N2 before recording AFM, PL and UV-vis spectra.
3. Results and discussion
3.1. Design and fabrication of surface-confined porphyrin–terpyridyl molecular architectures
To fabricate surface-confined heterogeneous molecular dyads, we consider well-known metallo-ligands such as metallo-porphyrin and metallo-terpyridyls which are feasible due to the linearly situated free pendant pyridine groups in the former and octahedral geometry of the latter.12 This hybrid assembly results via a combination of square pyramidal/tetrahedral geometry of the metallo-linkers, Cu2+ and Pd2+ along with trans positioning of the pyridine ligands.13 Multicomponent assemblies were fabricated through our smart design which utilizes stepwise coordination reactions of metallo-linkers and metallo-ligands (Scheme 1). Initially, an iodine-terminated coupling layer (CL) was formed, serving as a template layer for the covalent attachment of Zn-TPyP. The resulting monomolecular template layer was reacted with either Cu(NO3)2 or PdCl2(PhCN)2 in acetonitrile. Subsequently, the Cu/Pd-terminated template layer was immersed in a solution of a M–PT unit to fabricate heterogeneous molecular dyad. Different dyads layers were fabricated by combining different M–PT units as the top layers. The surface morphology of the Zn-TPyP monolayers and the dyad layers grafted over the silicon substrates were surveyed using atomic force microscopy (AFM) technique. Thereafter, photoluminescence (PL) characteristics of the covalently adsorbed Zn-TPyP on the silicon substrate were examined. UV-vis measurements of the molecular films were performed and compared with the solution of the respective metallo-ligands, monolayers and metal-directed dyad layers.3.2. AFM images of the Zn-TPyP monolayers and Zn-TPyP/Cu/Fe–PT dyad layers on silicon
Tapping-mode AFM image of the covalently assembled monolayers, Zn-TPyP onto the Si(100) substrates showed a reasonably smooth film surface almost without any islands, grains or defects. The root-mean-square roughness, Rrms measured for 500 nm × 500 nm scan area for the Zn-TPyP-based monolayer was estimated at ∼0.32 nm (Fig. 1a). Such a low Rrms value revealed the formation of a homogeneous and defect-free molecular layer over the silicon substrate. | ||
| Fig. 1 Representative AFM micrograph of (a) Zn-TPyP covalent assembled monolayer, and (b) representative Zn-TPyP/Cu/Fe–PT prepared on the silicon (100) substrates. Root mean square roughness (Rrms) was estimated for the monolayers at Rrms = 0.32 nm, while for the representative Zn-TPyP/Cu/Fe–PT dyad layer, Rrms = 0.78 nm. The scan area was 500 nm × 500 nm. | ||
However, the Rrms value for the multi-metallic based dyad layers were found to be increased. For instance, representative Zn-TPyP/Cu/Fe–PT dyad layers prepared on the silicon (100) substrates showed, Rrms ∼ 0.78 nm measured for the scan area 500 nm × 500 nm (Fig. 1b). We attribute this increase in surface roughness due to attachment of both the metallo-linker and metallo-terpyridyl layer over the metallo-porphyrin layer. Significantly, this is in line with our recently published results, where we fabricated metallo-terpyridyl based dyads on the same substrate.14 Further, the increase in roughness might be the result of the physisorbed of the metallo-linkers, and Zn-TPyP onto the silicon substrates.
3.3. Photoluminescence spectrum of the Zn-TPyP monolayers
The Zn-TPyP covalently attached to the silicon substrates showed a prominent fluorescence emission at 665 nm, while it displayed, relatively, weak signals at 610, 725 nm when excited at λ = 420 nm (Fig. 2). The strong and weak PL signals demonstrate, unequivocally, the attachment of the Zn-TPyP complex over the silicon substrate. Additionally, we have observed similar PL emissions in chloroform solution of Zn-TPyP without much deviation. Note that our surface PL characteristics are in good agreement with the previous reports on Cu(II)-porphyrin monolayers where silane containing coupling layer reacted with a single pendant group of porphyrin derivative.5d Besides, similar PL behaviour both in solution and thin films of Zn(II)-porphyrin derivatives were reported by other research groups.15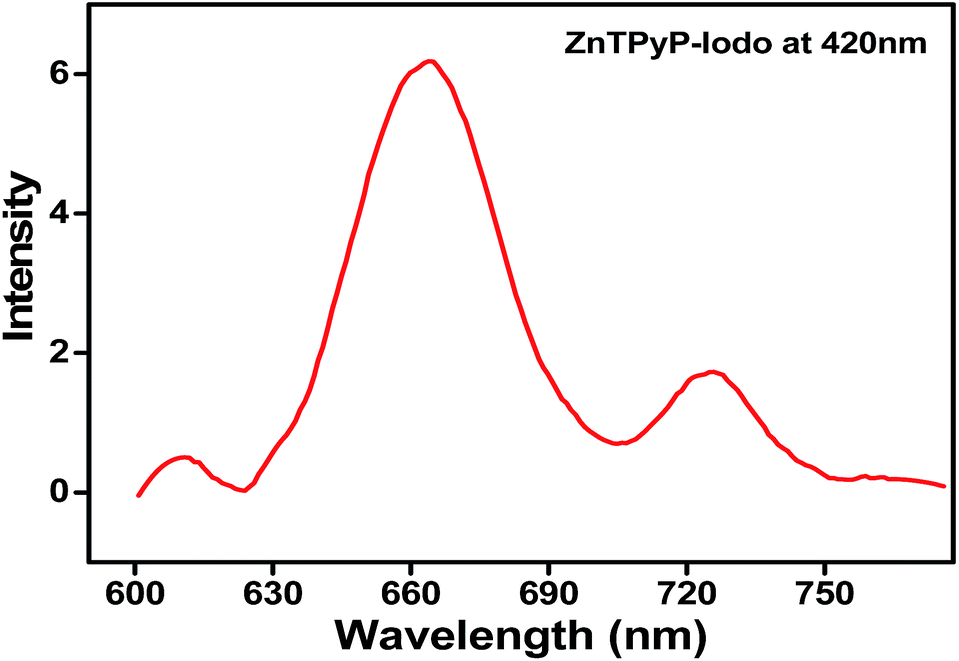 | ||
| Fig. 2 Photoluminescence spectrum of the Zn-TPyP monolayers on silicon substrate recorded at room temperature. The fluorophore, Zn-TPyP was excited at 420 nm. | ||
3.4. UV-vis spectra of the monolayers and dyad layers
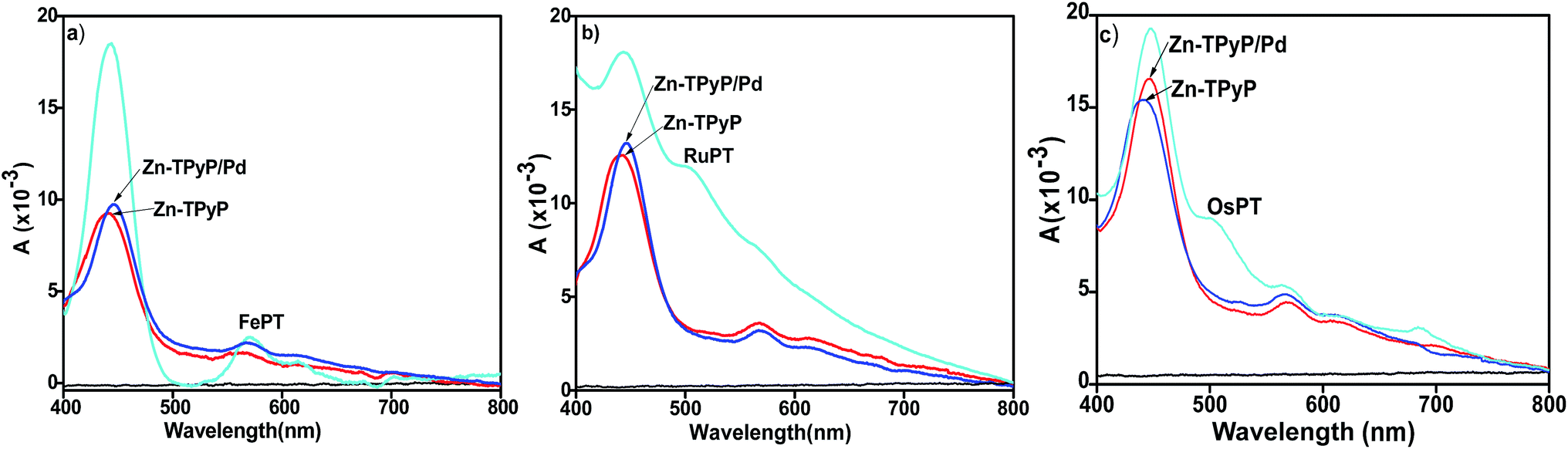
The formation of Zn-TPyP monolayers on glass substrates was monitored by conventional UV-vis spectroscopy (λ = 400–800 nm). A background UV-vis spectrum was recorded on a blank glass substrate which did not show any signal in the spectrum. On the other hand, the Zn-TPyP-based covalent monolayer on glass exhibited a prominent band at λmax = 442 nm, and another absorption band with relatively, very less intensity at λmax = 560 nm (Fig. 3). These bands are attributed to Soret and Q-band, respectively as also observed in the solution spectrum of Zn-TPyP.3c,5d Therefore, the attachment of Zn-TPyP onto the glass substrate was clearly implied. Further, upon immobilization, the peak maximum of the Soret band and the Q-band was red-shifted by 21 nm and 9 nm, respectively. This could be a consequence of quaternization of pendent pyridyl N-atoms and/or related to the cofacial orientation of the molecules within the purview of exciton theory.5a Other factor like strong electronic coupling between the optically active module and the substrate can be involved as well. From the value of this red shift, we strongly believe that coupling layer was reacted with a single pendant pyridyl group, as similar red shift was observed with H2TPyP.5a,16 Importantly, we have previously observed similar spectral shifts for polypyridyl complexes covalently attached to glass substrates.10a,b,11,14,17 In addition, we observed peak broadening of the Soret band (full-width at half-maxima, FWHM, ca. 50 nm) relative to that of Zn-TPyP in solution. This broadening of the Soret absorption band can be attributed to dense packing, and possibly strong intermolecular π–π interactions between the adjacent π-electron rich Zn-TPyP complex, which may interact in a side-on manner.5a However, this feature is less prominent in solution as demonstrated by contrasting FWHM value, 28 nm. UV-vis spectroscopy allows for estimating molecular footprint on the basis of the constancy of molar extinction coefficient values. Surface coverage of the monolayer, Γ, was calculated using the well-known equation, Γ = (NAAλ)/2ελ, where NA is the Avogadro's constant, Aλ and ελ are the absorbance and the isotropic molar extinction coefficient at the specific wavelength, respectively. In our case, the surface coverage of the Zn-TPyP-monolayer was estimated at 67(±3) × 1011 metallo-ligands per cm2, considering the equivalent molecular extinction coefficient (ε ≈ 4 × 105 cm−1 M−1) for the Soret band at λmax = 421 nm in CHCl3. This coverage further indicates formation of densely packed monolayers. The Soret band was blue shifted by 9 nm (as compared to the Zn-TPyP monolayer) upon coordination with Cu(NO3)2 when reacted with the pendant pyridyl groups. This is illustrative of strong electron-withdrawing tendency of Cu2+ from the π-electron cloud system, so that it's d-orbital can reach to maximum electron density, d9 → d10. Interestingly, we observed similar trends with d9-Cu center during the fabrication of the terpyridyl based oligomer systems.9a,b Further, the formation of heterogeneous dyad layer, Zn-TPyP/Cu/Fe–PT was confirmed by the appearance of a prominent absorption band at λmax = 579 nm, attributed to the metal-to-ligand charge-transfer [MLCT, Fe(dπ) → Pytpy(π*)] in addition to the Soret band (Fig. 3a). The MLCT band of the Fe–PT layer showed a bathochromic shift by 10 nm than that of its solution spectrum recorded in acetonitrile.16 On the other hand, Cu2+ linked dyad layer with Ru–PT as the top layer showed the typical MLCT band at λmax = 504 nm (Fig. 3b). The presence of Os–PT layer was established by the manifestation of triplet MLCT band at 680–700 nm, which is weak and broad peak, can be distinguished from Ru–PT spectrum (Fig. 3c). In all cases, a red shift in the MLCT transition was observed as compared to solution spectrum which further supported their attachment to Cu-terminated Zn-TPyP monolayer. The average molecular density of the Fe–PT, Ru–PT and Os–PT in the dyad layers was obtained at ca. 52(±2) × 1011, 55(±2) × 1011 and 57(±2) × 1011 metallo-ligands per cm2, respectively. This estimation was made by considering the molecular extinction coefficients (ε) of the M–PT solid films are similar to those observed in solution for the MLCT band, ε ≈ 23000, 29800, and 27500 cm−1 M−1 respectively.11,18
 | ||
| Fig. 3 UV-vis spectra of Cu2+-mediated molecular dyads for (a) Zn-TPyP/Cu/Fe–PT, (b) Zn-TPyP/Cu/Ru–PT, and (c) Zn-TPyP/Cu/Os–PT layers fabricated on glass substrates. Solid black, red, blue, and cyan lines represent the corresponding spectrum of the glass baseline, Zn-TPyP monolayers, Cu-terminated monolayers and the respective dyad layers. | ||
Interestingly, a red shift in the Soret band was observed when the free pyridyl groups containing Zn-TPyP-based covalent monolayer were reacted with PdCl2(PhCN)2 in CH3CN. For instance, Pd-driven Zn-TPyP layer showed a red shift of 6 nm than that of the template layer. The dyad layer made from Fe–PT unit exhibited an MLCT band at λmax = 572 nm in addition to the Soret band at λmax = 444 nm (Fig. 4a). The red shift of the Pd2+ mediated dyad layers can be attributed to its electronic configuration which is d8 and has fully filled T2g orbital and half-filled Eg orbital. Therefore, the addition of electron from pyridyl nitrogen to the Eg orbital of Pd2+ led to destabilization and consequent increase in the energy of its ground state. Hence, the decrease in the transition energy between the ground state and excited state led to observe a red shift. Similarly, the formation of the multicomponent dyad layers, Zn-TPyP/Pd/Ru–PT and Zn-TPyP/Pd/Os–PT were confirmed by the presence of the respective MLCT bands. For instance, the Zn-TPyP/Pd/Ru–PT layer showed a prominent MLCT band at 505 nm, while Zn-TPyP/Pd/Os–PT dyad exhibited two MLCT bands at 501 nm, and 685 nm (Fig. 4b and c). The UV-vis data presented in Fig. 3 and 4 are summarized in Table 1.
 | ||
| Fig. 4 UV-vis spectra of Pd2+-mediated molecular dyads for (a) Zn-TPyP/Pd/Fe–PT, (b) Zn-TPyP/Pd/Ru–PT, and (c) Zn-TPyP/Pd/Os–PT layers prepared on the glass substrates. Solid black, red, blue, and cyan lines represent the corresponding spectrum of the glass baseline, Zn-TPyP monolayers, Pd-terminated monolayers and the respective dyad layers. | ||
3.5. Theoretical calculations
To shine light for unraveling and understanding microscopic details of the observed photophysical properties we have performed detailed electronic structure calculations based on first-principles density functional theory (DFT) and time-dependent DFT (TDDFT). Geometries of all five metal complexes (1, 2, 3, 4 and 5) in their 0 (for 1), +2 (for 2 and 3) and +4 (for 4 and 5) charged states are fully optimized in gas phase considering different spin states and without imposing any symmetry constraint by using the unrestricted DFT method. We use a range-separated hybrid exchange and correlation ωB97XD functional that was developed to account for long-range charge transfer and dispersion interactions,19 with 6-31G(d) basis set20,21 for light elements (H, C, N, O) and LANL2DZ basis set22 augmented with an effective core potential (ECP) for metal atoms (Fe, Cu, Zn and Pd). Normal modes analysis was performed for each complex in order to confirm minimum energy structures on the ground state potential energy surface at the same calculation level as employed for the geometry optimization. Excited state calculations, solving for 30 low-lying singlet excited states were implemented by using TDDFT method employing 6-31+G(d,p) basis set for light atoms and LANL2DZ for the metals with an ECP for representing core electrons potential. For the sake of completeness and for computational comparison, we have also performed excited state calculations by using two other DFT exchange–correlation functionals: long-range corrected Coulomb attenuated range-separated CAM-B3LYP23 and semi-empirical B3LYP24 hybrids. All calculations were implemented by using Gaussian 09 DFT software package.25We begin by comparing the ground state DFT results (such as structures, low-energy spin state, frontier molecular orbitals) for the complexes without and with the metal ions (Cu2+ and Pd2+). DFT calculations predict broken symmetry singlet as the minimum energy spin state for the complexes 1, 3 and 5 studied here. Whereas Cu2+ containing complexes 2 and 4 show a spin doublet state as the low-energy ground state. Optimized structures and frontier molecular orbitals (HOMO: highest occupied molecular orbital and LUMO: lowest unoccupied molecular orbital) isosurfaces as calculated using ωB97XD functional for all five complexes are depicted in Fig. 5. From the optimized geometries of complexes 4 and 5, we find that two tpy ligands connected via Fe are aligned nearly perpendicularly, a structural feature is also found in the isolated Fe2+–PT26 and the central M–N (M = Cu, Pd) bond distances along the long molecular axis are slightly affected (0.02 Å reduced and increased for the complexes 4 and 5, respectively) than the M–N bond distances found in their porphyrinic monomer complexes 2 and 3 (see the numbers listed in Fig. 5). An unpaired electron is distributed on the porphyrin macrocycle for the complexes 2 and 4 (see spin-density diagram in ESI, Fig. S2‡). For complexes 3 and 5 two unpaired electrons localized at porphyrinic π molecular orbital and Pd dz2 orbital are coupled antiferromagnetically via Py bridging ligand, forming a spatially separated broken symmetry singlet state. Computed electronic HOMO–LUMO gaps as obtained using ωB97XD functional are 5.62/5.62, 5.35/4.45, 4.44/3.96, 4.64/4.44 and 4.07/4.44 eV for the α/β spin, respectively, for the complex 1, 2, 3, 4 and 5, indicating reduced gap values for complexes with metal ions (Cu2+ and Pd2+). Furthermore, complexes 4 and 5 containing Fe2+–PT result in slightly smaller gap value (see data in ESI Table S1‡). Similar findings are also obtained by CAM-B3LYP functional, whereas as expected B3LYP functional largely underestimates the gaps (see data listed in ESI Table S1‡). Here, we note that range-separated hybrid density functionals (ωB97XD and CAM-B3LYP) produces physically meaningful orbitals and also provide quantitatively accurate fundamental (Kohn–Sham HOMO–LUMO) gap by mitigating self-interactions errors and restoring missing derivative discontinuity in exchange–correlation potential. As is shown in Fig. 5, HOMO and LUMO frontier orbitals are primarily distributed at the porphyrinic moiety for complexes 1, 2 and 5. While for complexes 3 and 4 HOMO is distributed over porphyrin LUMO (β) is localized at Pd for complex 3 and the LUMO (α) of complex 4 is distributed on one tpy and on the nearby bridged Py group in the complex, clearly suggesting a charge-transfer (CT) character associated with HOMO to LUMO electronic promotion.
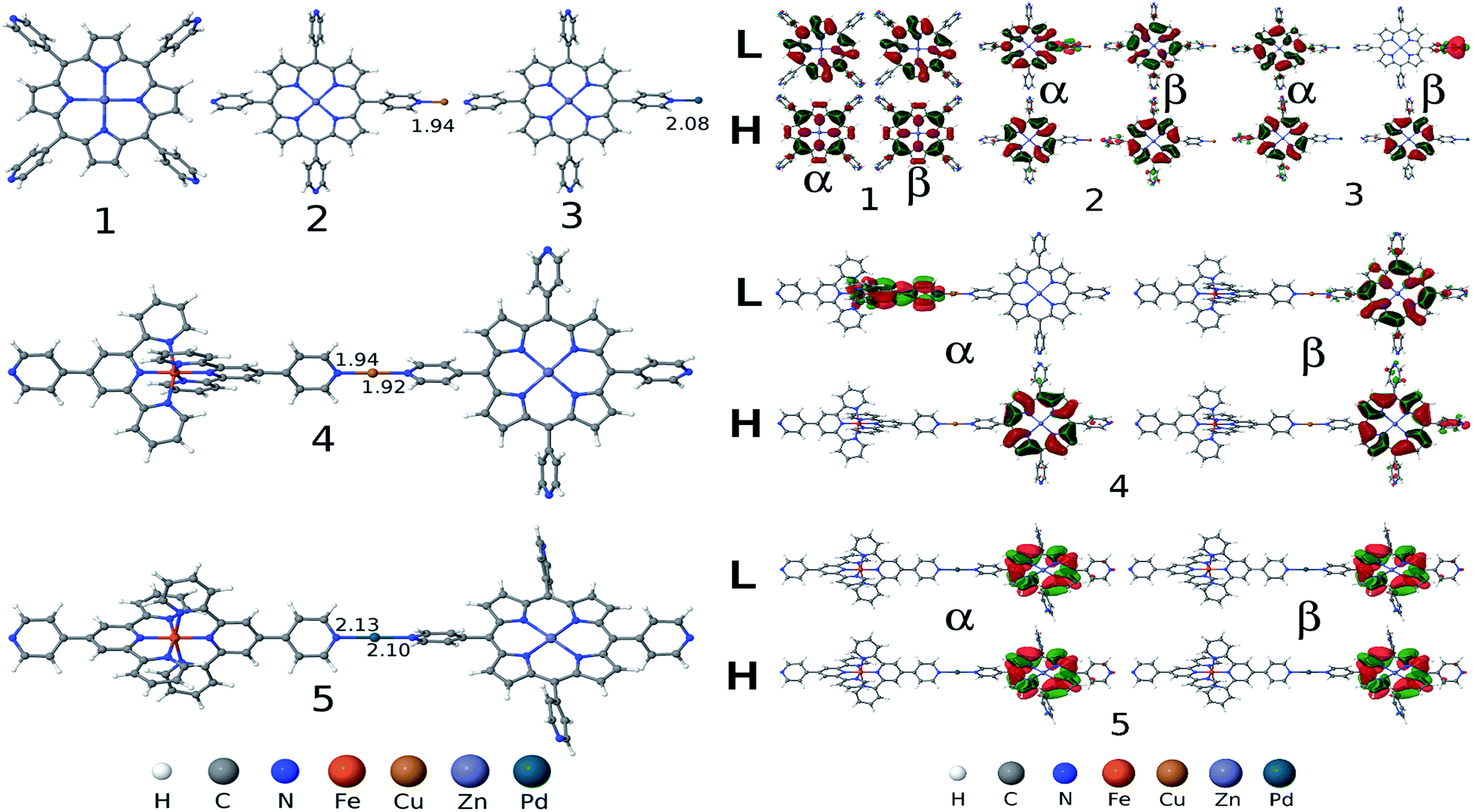 | ||
| Fig. 5 Left panel: Gas-phase optimized geometries of five metal complexes (1: Zn-TPyP; 2: Zn-TPyP/Cu2+; 3: Zn-TPyP/Pd2+; 4: Zn-TPyP/Cu2+/Fe2+–PT and 5: Zn-TPyP/Pd2+/Fe2+–PT) obtained by using DFT method employing ωB97XD functional (important central M–N bond lengths are indicated in Å). Right panel: HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) frontier orbitals isosurfaces for five complexes (1–5) as calculated employing ωB97XD functional with 6-31+G(d,p) basis set for light atoms and LANL2DZ basis set for metals with ECP for the core electrons. H and L stand for HOMO and LUMO, respectively. α and β refer the alpha and beta set of spin–orbitals, respectively. | ||
We next discuss the optical absorption characteristics of these complexes as calculated using TD-DFT method. Optical absorption spectra, a few low-lying electronic excitations and the primary orbitals replacement that are involved in the electronic excitations are provided in Fig. 6 and in ESI Table S2.‡ For complex 1 our computational results show two degenerate low-lying excited states at 560 nm with very weak absorption intensity (indicated by very small oscillator strengths). These are characterized as the Zn-porphyrin Q-band electronic states. This is in good agreement with experimentally measured Q-band peak position at 560 nm. On the other hand the calculated B-band electronic states are found at 362 nm, which is overestimated by about 0.6 eV than the corresponding experimental peak at 442 nm (see data listed in ESI Table S2‡). Both Q and B band electronic states involve electronic promotion from the HOMO (HOMO−1) to LUMO (LUMO+1) orbitals. Noticeably, we find that metal coordination at terminal pyridine N changes the absorption spectrum significantly, mainly by affecting frontier orbitals energies. Q-Band electronic states red shifted by about 46 nm due to the metal coordination (see Fig. 6 and data in ESI Table S2‡). Importantly, for both the complexes 2 and 3 B-band states are splitted into two peaks (one substantially large red shifted peak at 405 nm and a slightly blue shifted peak at 354 nm) compared to the single peak at 362 nm found for the complex 1 in its pristine form. While red shifted band involves electron promotion primarily from HOMO−8 to LUMO the blue shifted excitation involves HOMO to LUMO+2 electronic transition (see FMOs diagram in Fig. 6 and ESI data in Table S2‡). Both electronic bands show partial ligand-to-ligand charge-transfer (LLCT) as characterized by the frontier orbitals analysis. Nevertheless, a slightly larger blue shift is predicted for complex 2 than the complex 3 (compare absorption spectrum in Fig. 6). This fully supports our experimental observation that the Cu2+ containing complex 2 exhibits slightly blue shifted B-band. Additionally, we found two low-lying excited states (810 nm and 781 nm for complex 2 and 804 nm and 779 nm for complex 3) with very small oscillator strengths below the Q-band electronic states, exhibiting partial LLCT (see Fig. 6 and ESI Table S2‡). For complexes 4 and 5, a few low-lying excited states are found at around 1980–1990 nm and 778–806 nm, with very weak absorption intensities (indicated by negligibly smaller oscillator strength values, see listed data in the ESI (Table S2‡)). These states involve electronic excitation primarily localized on Zn-porphyrin moiety and also display partial LLCT character (see orbitals diagram in ESI Fig. S3‡).
 | ||
| Fig. 6 Simulated optical absorption spectra (top left) and a few low-lying electronic excitations with relevant frontier molecular orbitals replacement for the complex 1, 2 and 3 as calculated by using ωB97XD XC functional with 6-31+G(d,p) basis set for light atoms and LANL2DZ basis set for metals with ECP for the core electrons. Computed oscillator strengths are convoluted by a Gaussian function with width 0.1 eV for obtaining the optical spectrum. H and L stand for HOMO and LUMO, respectively. α and β refer the alpha and beta set of spin–orbitals, respectively. | ||
Quantitative differences between the calculated and experimentally measured low-lying peak positions can be attributed to an anticipated large electrostatic stabilization of electronically excited states possessing substantial charge transfer character in the polar environment present by solvent (such as acetonitrile) or substrate dielectrics, which is not considered by the present TDDFT implementation. Such effects can be substantial depending on the nature of CT states as reported previously.27,28 We also note that similar results are obtained using the other range-separated hybrid CAM-B3LYP XC functional, whereas traditional hybrid B3LYP largely underestimates the fundamental gaps and produces several low-energy spurious CT states (see data from ESI Table S2‡). Overall, our computational findings are in fairly good agreement with experimental observed photophysical properties of the metal-driven molecular dyads prepared on SiOx substrates.
4. Conclusions
Solid state molecular engineering with transition metal-directed multifunctional metallo-porphyrins and terpyridyl complexes have been demonstrated. It is observed that metallo-linkers play an important role in controlling the electronic interaction along the heterogeneous molecular dyads. For instance, the Cu-terminated assemblies showed a blue shift, while Pd-mediated assemblies showed a red shift, as compared to the Zn-TPyP template layer in their respective UV-vis spectrum. The experimental results were reinforced by TD-DFT calculations. Thus, the multimetallic novel assemblies are interesting in terms of their surface properties and could be potentially useful in fabricating devices having tailored opto-electronic, electrochromic behavior, for molecular logic as well as in the growing area of “on-surface magneto-chemistry” applications.Acknowledgements
BG and PCM thank University Grant Commission, and Council of Scientific and Industrial Research, New Delhi, India for JRF, and SRF, respectively. Financial support from the DST-Nano Mission, Government of India is also acknowledged. The authors thank Department of Chemistry and USIC, University of Delhi for Mass, NMR, FTIR, and Department of Physics for AFM facilities. PCM also acknowledges Alberta Innovates Technology Features, and National Research Council, Canada for financial support. AKM thanks Prof. Swapan K. Pati, JNCASR, Bangalore, India for providing computational resources.References
-
(a) S. Singh, A. Aggarwal, N. V. S. D. K. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Chem. Rev., 2015, 115, 10261–10306 CrossRef CAS PubMed
; (b) R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC
-
(a) H.-K. Liu and P. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed
-
(a) M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC
-
(a) B. F. Abrahams, B. F. Hoskins, D. M. Michail and R. Robson, Nature, 1994, 369, 727–729 CrossRef CAS
-
(a) D.-Q. Li, B. I. Swanson, J. M. Robinson and M. A. Hoffbauer, J. Am. Chem. Soc., 1993, 115, 6975–6980 CrossRef CAS
-
(a) Z. Liu, A. A. Yasseri, J. S. Lindsey and D. F. Bocian, Science, 2003, 302, 1543–1545 CrossRef CAS PubMed
-
(a) N. Ballav, C. Wäckerlin, D. Siewert, P. M. Oppeneer and T. A. Jung, J. Phys. Chem. Lett., 2013, 4, 2303–2311 CrossRef CAS
-
(a) K. Shimazu, M. Takechi, H. Fujii, M. Suzuki, H. Saiki, T. Yoshimura and K. Uosaki, Thin Solid Films, 1996, 273, 250–253 CrossRef CAS
-
(a) A. Harriman, F. Odobel and J.-P. Sauvage, J. Am. Chem. Soc., 1995, 117, 9461–9472 CrossRef CAS
-
(a) P. C. Mondal, J. Y. Lakshmanan, H. Hamoudi, M. Zharnikov and T. Gupta, J. Phys. Chem. C, 2011, 115, 16398–16404 CrossRef CAS
- T. Gupta, P. C. Mondal, A. Kumar, Y. L. Jeyachandran and M. Zarnikov, Adv. Funct. Mater., 2013, 23, 4227–4235 CrossRef CAS
- J. E. Beves, E. C. Constable, C. E. Housecroft, C. J. Kepert and D. J. Price, CrystEngComm, 2007, 9, 456–459 RSC
- J. E. Beves, E. C. Constable, C. E. Housecroft, M. Neuburger and S. Schaffner, CrystEngComm, 2008, 10, 344–348 RSC
- P. C. Mondal, V. Singh, Y. L. Jeyachandran and M. Zharnikov, ACS Appl. Mater. Interfaces, 2015, 7, 8677–8686 CAS
-
(a) M. García, K. Carfumán, C. Díaz, C. Garrido, I. Osorio-Román, M. J. Aguirre and M. Isaacs, Electrochim. Acta, 2012, 80, 390–398 CrossRef
-
(a) R. A. Bull and J. E. Bulkowski, J. Colloid Interface Sci., 1983, 92, 1–12 CrossRef CAS
-
(a) V. Singh, P. C. Mondal, J. Y. Lakshmanan, M. Zharnikov and T. Gupta, Analyst, 2012, 137, 3216–3219 RSC
- P. C. Mondal, V. Singh and B. Shankar, New J. Chem., 2014, 38, 2679–2685 RSC
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS
- R. Kobayashi and R. D. Amos, Chem. Phys. Lett., 2006, 420, 106 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- P. C. Mondal and A. K. Manna, New J. Chem., 2016, 40, 5775–5781 RSC
- A. K. Manna and B. D. Dunietz, J. Chem. Phys., 2014, 141, 121102–121106 CrossRef PubMed
- S. Zheng, H. Phillips, E. Geva and B. D. Dunietz, J. Am. Chem. Soc., 2012, 134, 6944–6947 CrossRef CAS PubMed
Footnotes |
| † Dedicated to late Prof. Tarkeshwar Gupta, University of Delhi, India. |
| ‡ Electronic supplementary information (ESI) available: Synthesis and characterization data of the complexes, theoretical calculations. See DOI: 10.1039/c6ra25090d |
| § Present address: National Institute for Nanotechnology, University of Alberta, Edmonton, Alberta, T6G 2M9, Canada; E-mail: E-mail: mondalpc@gmail.com |
| This journal is © The Royal Society of Chemistry 2017 |