
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of SAPO-18/34 intergrowth zeolites and their enhanced stability for dimethyl ether to olefins
Dongpu Zhaoa,
Yan Zhanga,
Zhuo Lia,
Yan Wangb and
Jianqiang Yu*ab
aSchool of Chemistry and Chemical Engineering, Qingdao University, Collaborative Innovation Center for Marine Biomass Fiber Materials and Textiles of Shandong Province, 308 Ningxia Road, Qingdao 266071, China. E-mail: jianqyu@qdu.edu.cn; Fax: +86-532-85955529; Tel: +86-532-85955529
bQingdao Center of Resource Chemistry & New Materials, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Qingdao 266000, China
First published on 4th January 2017
Abstract
In order to investigate the effect of phase composition in the silicoaluminophosphate (SAPO) zeolites on their catalytic performance for the dimethyl ether (DME) to olefin (DTO) reaction, four well crystalline SAPO zeolites with different SAPO-18/34 ratios were synthesized hydrothermally by using a mixed template of diethylamine (DEA) and N,N′-diisopropylethylamine (DIEA). The effects of the composition of the mixed template on the physicochemical properties of the zeolites were investigated by XRD, SEM, TEM, XRF, FT-IR and NH3-TPD techniques. The results revealed that the phase composition, morphology, particle size, chemical composition and surface acidity of the zeolites were greatly affected by the template composition. Catalytic tests showed that both SAPO-18 and SAPO-34 presented relatively faster coking rates compared with that of SAPO-18/34 intergrowth zeolites, and pure SAPO-18 with the smallest crystallite size and weakest acidity exhibited the highest amount of coke deposition. The DME conversion and product distribution were similar over all catalysts, but the selectivities of light olefins showed a strong correlation to the cage size. That is, the catalysts with a higher content of slightly wider AEI cages favor the formation of propylene and butylene, and catalysts with a higher content of relatively narrower CHA cages benefit the formation of ethylene.
1. Introduction
Light olefins, especially ethylene and propylene are key building-block chemicals, widely used in the synthesis of plastics, fibers, and detergents.1 Currently, these alkenes are produced primarily by steam cracking and fluid catalytic cracking (FCC) of petroleum at high temperature.2 However, with the increasing depletion of oil reserves, the price of petroleum is becoming unacceptable; meanwhile, the thermal cracking route is highly energy intensive and non-environmentally benign. Thus, exploring clean, low-cost and non-petrochemical routes for the production of light olefins from other abundant hydrocarbons become the more attractive and practical solutions. Among which, the conversion of methanol-to-olefins (MTO) or dimethyl-ether-to-olefins (DTO) over microporous solid acids has been proven to be the most promising route for the production of light olefins in the past decades, owing to methanol and DME can be easily produced from synthesis gas obtained from natural gas steam reforming and coal gasification. Thus, the development of MTO or DTO route can be an alternative for the production of light olefins from other nonpetroleum carbon resources, such as natural gas, biomass and coal.3Up to now, a variety of solid acid catalysts with different acidities and pore architectures have been studied in the MTO and DTO processes, such as aluminosilicate zeolite H-ZSM-5,4 SAPO zeolites SAPO-34 and SAPO-18. Among which, SAPO-18 (AEI structure) and SAPO-34 (CHA structure) with 8-ring pore opening, moderate acidity and good thermal/hydrothermal stability, were considered as the most promising catalysts applied to industrial production, due to their high selectivity to C2–C4 olefins (>80%) and almost 100% conversion of methanol and DME under relatively mild conditions.5 In the present time, SAPO-34 is mainly prepared by using organic amines as the template, such as, tetraethylammonium hydroxide (TEAOH),6 triethylamine (TEA),7 diethylamine (DEA),8 and morpholine (MOR)9 and so on, and has realized commercial production and application for the UOP/hydro MTO process. However, the as-synthesized SAPO-34 always suffers rapid deactivation resulting from its large particle size and relatively high surface acid density.10
In comparison with SAPO-34, the other potential alternative SAPO-18 catalyst shows comparable selectivity to light olefins, but improved lifetime due to its smaller particle size and weaker surface acid density.11 As what we have known, SAPO-18 is built up of the same primary (Si–O tetrahedral, Al–O tetrahedral and P–O tetrahedral) and secondary structure units (double six-rings) as SAPO-34, and the micropore size in SAPO-18 is closely similar to that in SAPO-34.1c The main differential between the two zeolites lies in the arrangement mode of the double six-rings.12 In SAPO-18, the neighbouring layers of double six-rings are cross arranged and thus possess different orientations. While, in SAPO-34, they are parallel arranged and thereby have the same orientation. However, SAPO-18 suffers from the following problems: complicated synthesis condition, high cost of template and difficult to be separated and recovered because of its smaller particle size. Therefore, SAPO-18 has still not been successfully applied to industrial production.
The development of a bifunctional SAPO-18/34 intergrowth catalyst sharing the advantages of high catalytic activity and long lifetime will be highly desired. Since SAPO-18 and SAPO-34 are structurally close to each other, the formation of SAPO-18/34 intergrowth material is possible in theory. In addition, a number of recent literatures have been published describing the synthesis and enhanced catalytic performance of CHA/AEI intergrowth materials for use as MTO catalysts.13 However, these reported approaches consume huge amount of relatively expensive template such as TEAOH and DIEA, in addition, the obtained zeolites always possess relatively lower AEI/CHA ratios. Therefore, in the viewpoint of whether academic research or practical applications, it is a great challenge to develop SAPO catalyst with higher AEI/CHA ratio, better resistance to coke formation, higher methanol or DME conversion and selectivity to light olefins under low cost in preparation.
To our knowledge, there are few reports on the synthesis of SAPO-18/34 intergrowth zeolites by using mixed template. Herein, we report a facile and low-cost design for the preparation of SAPO-18/34 intergrowth zeolites by using DIEA and DEA as dual templates, also the influences of the molar ratio of DIEA/(DEA + DIEA) on the physicochemical properties and catalytic performance of the catalysts are investigated. The results show that the catalytic performance are comparable to each other, but the stability of intergrowth SAPO-18/34 is more excellent than that of pure SAPO-18 and SAPO-34 for the DTO reaction.
2. Experiment
2.1 Catalyst synthesis
SAPO-34 catalyst was hydrothermally synthesized by using diethylamine (DEA, 99 wt%) as the template. The starting gel composition was 1.6DEA/1.0Al2O3/0.9P2O5/0.6SiO2/50H2O. In detail, 7.39 g pseudoboehmite (69 wt%) was first mixed with 35.43 g distilled water in a beaker. Then, 10.38 g phosphoric acid (85 wt%), 6.92 g silica sol (25.5 wt%) and 5.84 g DEA were added in sequence into the beaker under vigorous stirring. The mixture was stirred for about 3 h at room temperature and then transferred into a 100 mL Teflon-lined stainless steel autoclave. The crystallization was performed at 180 °C for 96 h under autogenous pressure without agitation. After crystallization, the gel was cooled to room temperature and centrifuged at 4000 rpm for 4 min to separate the solid from the mother liquor. The product was filtered and washed with distilled water several times. Finally, the solid was dried at 100 °C for 10 h and calcined at 550 °C for 5 h in a flow of air with a heating rate of 5 °C min−1 to obtain the template-free samples.SAPO-18/34 intergrowth catalysts templated by mixture of DEA and N,N′-diisopropylethylamine (DIEA, 99 wt%) were synthesized with the initial gel composition of 0.2DIEA/1.4DEA/1.0Al2O3/0.9P2O5/0.6SiO2/50H2O and 0.4DIEA/1.2DEA/1.0Al2O3/0.9P2O5/0.6SiO2/50H2O (180 °C, 96 h). The procedure was similar as above except the mixture of DEA and DIEA was used as the structure directing agents.
SAPO-18 templated by DIEA was synthesized with the starting gel composition of 1.6DIEA/1.0Al2O3/0.9P2O5/0.6SiO2/50H2O (180 °C, 96 h). The detailed synthetic procedure was similar as that of SAPO-34–DEA except DIEA was used as the template.
2.2 Characterization
The phase structure of the sample was characterized by powder X-ray diffraction (XRD) on a Rigaku D/max-3C X-ray diffractometer with Cu Kα radiation under instrumental setting of 40 kV and 30 mA. The crystal size and morphology were observed by scanning electron microscopy (SEM) using a JSM-6390LV electron microscope equipped with an energy dispersive spectrometer (EDS). Transmission electron microscope (TEM) images were recorded on a JEOL-2010 TEM with an accelerating voltage of 200 kV.Chemical composition was determined with an X-ray fluorescence (XRF) spectrometer (ZSX Primus II). Nitrogen adsorption–desorption measurement was carried out on a Quantachrome AUTOSORB-1C instrument at −196 °C after the sample was degassed at 200 °C for 10 h under vacuum. The specific surface area and the micropore volume were calculated according to Brunauer–Emmett–Teller (BET) equation and t-plot method, respectively. Fourier transform infrared (FT-IR) spectra were recorded in the range of 400–4000 cm−1 on a Thermo NICOLET 5700 Fourier transform infrared spectrometer. The surface acidity was measured by temperature-programmed desorption of ammonia (NH3-TPD) on a chemisorption analyzer (FINETEC FINSORB-3010) equipped with a thermal conductivity detector (TCD). Typically, 0.08 g sample was first pretreated at 450 °C for 1 h in He to remove the physically adsorbed water. After cooling to 100 °C, the sample was saturated with NH3 for 40 min and purged with He for 10 min to eliminate the weakly absorbed NH3 on the surface. Desorption of NH3 was carried out from 100 to 800 °C at a heating rate of 20 °C min−1.
2.3 Catalytic test and retained coke analysis
The performance of the catalyst for the DTO reaction was performed in an integral fixed-bed reactor (φ 30 mm × 550 mm) at atmospheric pressure. The catalyst (5 g, 20–40 mesh) loaded in the stainless steel reactor was firstly activated at 450 °C in a N2 flow of 50 mL min−1 for 1 h and then the temperature of reactor was adjusted to 400 °C to start the DTO reaction. The flow velocity was 10 mL min−1 for the feedstock of dimethyl ether. The reaction products were analyzed by an on-line gas chromatograph (SHIMADZU GC-2014C), equipped with a flame ionization (FID) detector and AT PLOT Al2O3/S capillary column.The amount of generated coke in the as-synthesized catalysts was determined after 420 min reactions of DME by thermal analysis (TG-DTA) on a TA Q600 analyzer at the temperature range of 50–800 °C with a heating rate of 10 °C min−1 under an air flow of 100 mL min−1.
3. Results and discussion
To investigate the influence of the mixed template on the physicochemical properties and catalytic performance of the catalysts, four SAPO samples were synthesized at the DIEA/DEA molar ratios of 0/1.6, 0.2/1.4, 0.4/1.2 and 1.6/0 under same other parameters, such as the molar ratio of Al/P/Si and amount of template agent. The phase compositions and morphologies of the samples are summarized in Table 1.| Samples | X (DEA) | Y (DIEA) | Y/(X + Y) (%) | Crystal phase | Morphology |
|---|---|---|---|---|---|
| a | 1.6 | 0 | 0 | CHA | Cube |
| b | 1.4 | 0.2 | 12.5 | AEI/CHA | Cube |
| c | 1.2 | 0.4 | 25.0 | AEI/CHA | Cube |
| d | 0 | 1.6 | 100 | AEI | Square-platelet |
Fig. 1 shows the XRD patterns of the samples synthesized with different template compositions. Obviously, the well-resolved peaks of samples a and d prepared with single template DEA and DIEA, are in good agreement with that of pure SAPO-34 with CHA-structure and pure SAPO-18 with AEI-structure, respectively. Meanwhile, for samples b and c synthesized at the DIEA/DEA molar ratio of 0.2/1.4 and 0.4/1.2, typical characteristic diffraction peaks of both AEI-structure and CHA-structure can be observed. It is indicated that the samples synthesized with DEA, DIEA and DIEA–DEA as the template are SAPO-34, SAPO-18 and SAPO-18/34 intergrowth zeolites, respectively. Moreover, it can be seen that the characteristic diffraction peaks of AEI-structure (at 10.7°, 17.0°, 21.5° and 24.0°) enhance, while that of CHA-structure (at 17.8°) decreases simultaneously with the increasing of DIEA ratio in the DEA–DIEA dual templates. These results suggest that the templates play a critical function on the structure of the final products and SAPO-18/34 intergrowth materials can be selectively synthesized by using mixture of DEA and DIEA as template.
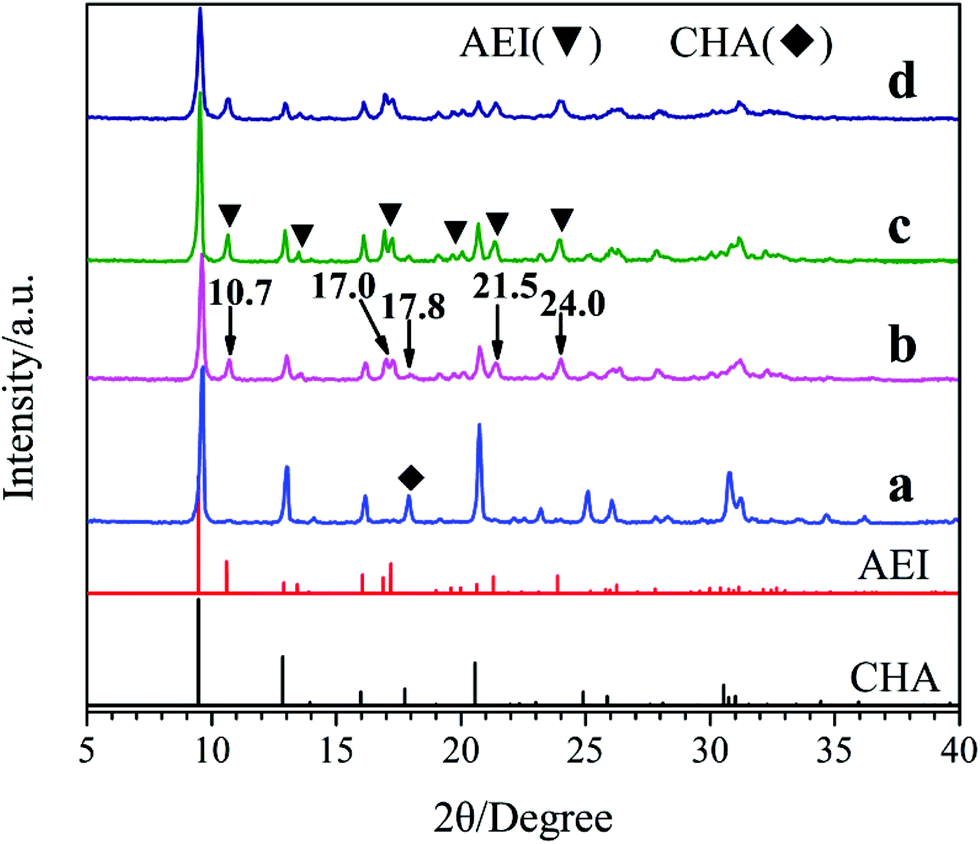 | ||
| Fig. 1 XRD patterns of the samples synthesized at various template compositions. (a) 1.6DEA, (b) 1.4DEA + 0.2DIEA, (c) 1.2DEA + 0.4DIEA, (d) 1.6DIEA. | ||
The XRD patterns in the range of 9–11° of the samples are shown in Fig. 2. Notably, the lines shift toward low-degree with the increasing of DIEA ratio in the DEA–DIEA dual templates. On the basis of Bragg equation, we can conclude that the crystal lattices expand gradually. It is well known, the crystal lattice of a zeolite is mainly affected by its chemical composition and lattice parameter. With respect to the chemical composition, it may be derived from the differentials in the incorporation amount and substitution mechanism of Si atoms. It is known that Si atoms incorporate into the AlPO4 framework through two substitution mechanisms: the first mechanism is one isolated Si substitution for one P (mechanism I: 1Si → 1P), the other is the double substitution of neighboring Al and P by two Si atoms (mechanism II: 2Si → 1Al + 1P).1c,14 In addition, the average bond lengths of Al–O, Si–O and P–O are 1.75 Å, 1.61 Å and 1.54 Å,15 respectively. Thus, when one Si substitutes for one P, the crystal lattice will expand and when two Si substitute for one P and one Al, the crystal lattice will shrink. So it can be inferred that DIEA might be beneficial for the substitution of mechanism I, and TEA favors for the substitution of mechanism II. As for the lattice parameter, it may be derived from the slightly wider cage size of AEI than that of CHA (1.16 nm compared to 0.94 nm).1b However, which factor is dominant can't be obtained only from the XRD pattern, further studies are necessary to build a fully understanding of the crystal lattice.
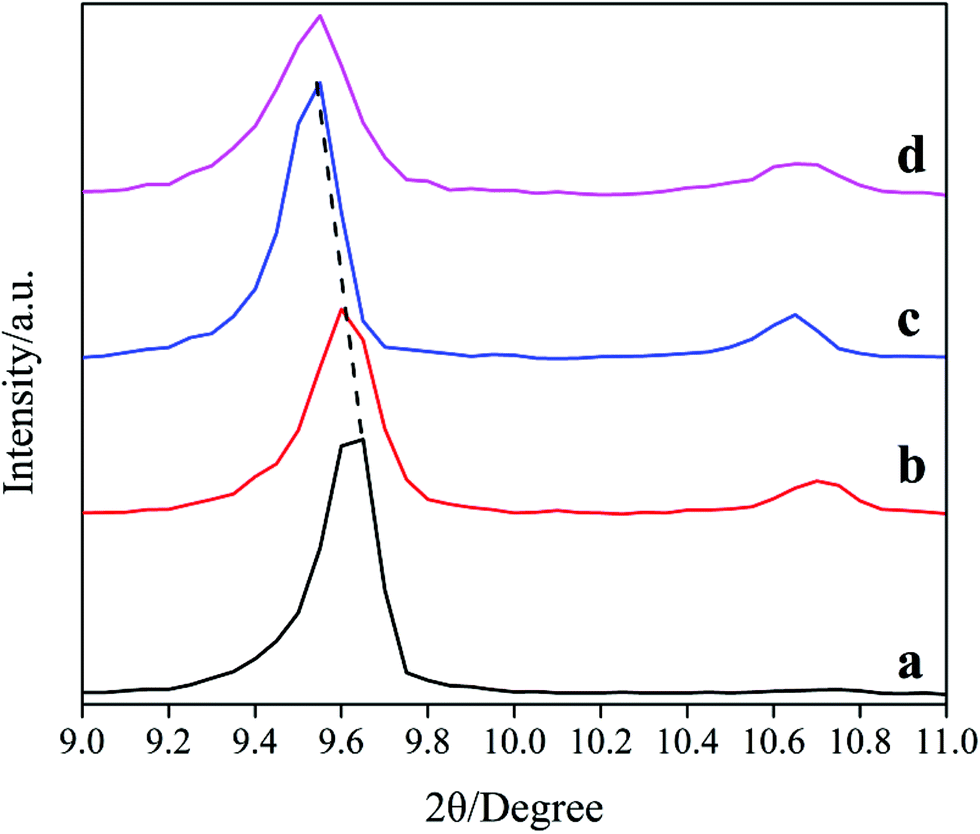 | ||
| Fig. 2 XRD patterns among 9–11° of the samples synthesized at various template compositions. (a) 1.6DEA, (b) 1.4DEA + 0.2DIEA, (c) 1.2DEA + 0.4DIEA, (d) 1.6DIEA. | ||
Fig. 3 shows the SEM images of the SAPO samples. Clearly, the crystal morphology and size are strongly affected by the template composition. The particles become smaller and thinner, and vary from well-distributed cube (typical for SAPO-34 crystal) to square-platelet (typical for SAPO-18 crystal) with the increasing of DIEA ratio in the mixed template,16 which is in good agreement with the XRD analysis. These results may be ascribed to the alkalinity of DEA and the electrophilicity of DEA+, which are all stronger than that of DIEA (as shown in Fig. 4). Since the enhanced alkalinity of the synthesis gel can accelerate the rate of crystal growth, meanwhile, inorganic precursors (such as: silicate, aluminate, phosphate and their oligomers) tend to enrich around DEA+ (electrostatic force) and participate in polymerization, so the more DEA is added, the bigger crystal particle will be produced.
 | ||
| Fig. 3 SEM photographs of samples synthesized at various template compositions. (a) 1.6DEA, (b) 1.4DEA + 0.2DIEA, (c) 1.2DEA + 0.4DIEA, (d) 1.6DIEA. | ||
 | ||
| Fig. 4 The alkalinity and electrophilicity analyses of the two template molecules. | ||
Fig. 5 shows the TEM images of the samples synthesized at different template compositions. As can be seen, all samples are well crystallized and exhibit cubic or sheet-like morphology. The selected area electron diffraction (SAED) patterns of the samples, as shown in the insets of Fig. 5a–d, vary considerably by the usage of template with different compositions. For samples a and d prepared with single template DEA and DIEA, typical diffraction spots of single crystals are obtained; for samples b and c synthesized with mixed template, typical diffraction rings of intergrowth crystals are obtained. These results indicate that samples a and d are pure-phase SAPO-34 and SAPO-18 crystals, respectively, and samples b and c are intergrowth AEI/CHA crystals.
 | ||
| Fig. 5 TEM images of the samples synthesized at various template compositions. (a) 1.6DEA, (b) 1.4DEA + 0.2DIEA, (c) 1.2DEA + 0.4DIEA, (d) 1.6DIEA. | ||
Fig. 6 depicts the FT-IR spectra of the synthesized samples, recorded in the range of 4000–400 cm−1. It is shown that all samples exhibit a broad and strong band (at ∼3450 cm−1) in the region of H-bonds associated hydroxyls, stemming from –OH stretching vibration for physical adsorbed water in SAPO cages.17 Meanwhile, all characteristic absorption peaks of the samples are similar to those reported in the literatures,18 according to which, vibrations at 490 cm−1 and 620 cm−1 are ascribed to Si–O bending of SiO4 tetrahedral and T–O (Si–O, Al–O, and P–O) bending in double-6 rings, respectively. Meanwhile, the absorption peaks at 720 cm−1, 900 cm−1 and 1095 cm−1 are assigned to the symmetric stretching vibration of T–O tetrahedral, the protonated template occluded in the zeolites and the asymmetric stretching vibration of T–O–T (Al–O–Si, Si–O–Si, and P–O–Al), respectively. Furthermore, the bands at 1400 cm−1 and 1640 cm−1 are correspond to the CH3 or CH2 bending vibration from the residual templates and the physically adsorbed water in the SAPO voids, respectively. It can be seen that the peak (490 cm−1) intensities increase and the peaks (at 1090 cm−1) shift toward higher wavelength with the increasing of DEA ratio in the DEA–DIEA dual templates, which can be due to increasing Si content in their structures.19
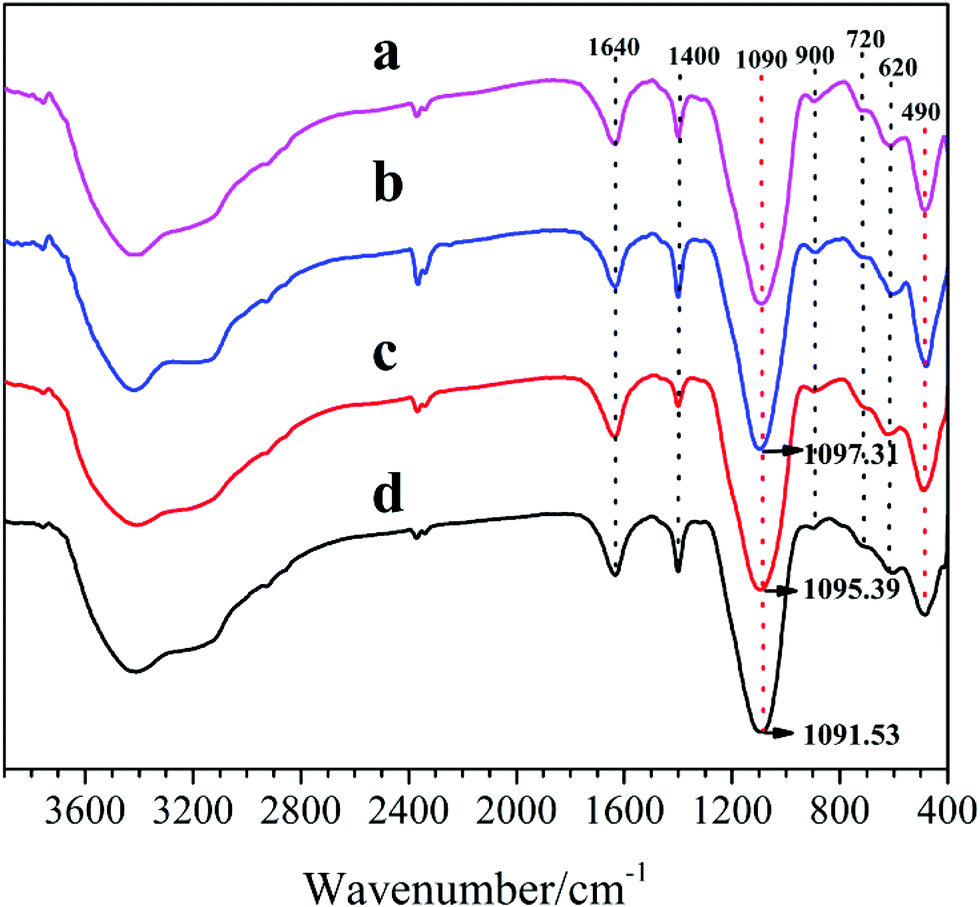 | ||
| Fig. 6 FT-IR spectra of samples synthesized at various template compositions. (a) 1.6DEA, (b) 1.4DEA + 0.2DIEA, (c) 1.2DEA + 0.4DIEA, (d) 1.6DIEA. | ||
The compositions of the prepared samples, analyzed by XRF, are given in Table 2. The results show that samples a and d synthesized with single template DEA and DIEA present the highest and lowest Si content, respectively, and samples b and c synthesized with mixed template exhibit the medium Si content, which are in good agreement with the FT-IR analysis. Moreover, it can be deduced that for SAPO-34 and SAPO-18/34, the Si atoms are inserted only in accordance with mechanism I (1Si → 1P), since their ratios of (Si + P)/Al are equal to 1.20 However, for SAPO-18, the Si atoms are incorporated in accordance with both mechanism I (1Si → 1P) and mechanism II (2Si → 1Al + 1P) because its ratio of (Si + P)/Al is more than 1.21 These results imply that DEA tends to promote more Si atoms incorporation into the AlPO4 framework by means of mechanism I. These observations can be explained by the differences in molecular size and the ability of charge-compensation between the two template molecules.5c The molecular kinetic diameter of DEA+ is smaller than that of DIEA+, so more amount of DEA+ will be filled into the pore channel of zeolite. Furthermore, the electrophilicity of DEA+ is stronger than that of DIEA+, so the higher addition of DEA, the more Si atoms will be incorporated into the AlPO4 framework by means of mechanism I (the main source of negatively charged framework for SAPO molecular sieves) to realize the charge balance.8 Meanwhile, according to the XRF analysis, it can be inferred that it is the lattice parameter differential between AEI and CHA rather than DIEA prefers the substitution of mechanism I (1Si → 1P) that leads to the expansion of crystal lattice.
| Samples | Chemical composition | (Si + P)/Al in solid | MI(1Si → 1P) | MII(2Si → 1Al + 1P) | ||
|---|---|---|---|---|---|---|
| Contenta | Ratio | Contentb | Ratio | |||
| a The content of Si incorporation by means of MI(1Si → 1P) was calculated by subtracting the amount of substance of Al (nAl) to that of P (nP) in the final product.b The content of Si incorporation by means of MII(2Si → 1Al + 1P) was calculated by subtracting the total amount of substance of Si to the Si substitution according to MI(1Si → 1P). | ||||||
| a | Si0.130Al0.500P0.370O2 | 1.000 | 0.130 | (100%) | 0.000 | (0.00%) |
| b | Si0.123Al0.500P0.377O2 | 1.000 | 0.123 | (100%) | 0.000 | (0.00%) |
| c | Si0.100Al0.500P0.400O2 | 1.000 | 0.100 | (100%) | 0.000 | (0.00%) |
| d | Si0.078Al0.489P0.433O2 | 1.045 | 0.056 | (71.8%) | 0.022 | (28.2%) |
The NH3-TPD curves of the samples synthesized with different DIEA/DEA ratios are presented in Fig. 7. Apparently, the two desorption peaks observed in the samples are corresponded to the weak (at 150–200 °C) and strong acid sites (at 450–500 °C), respectively. Meanwhile, with an increase in the ratio of DEA/(DIEA + DEA) from 0 to 1, the amount and strength of both weak and strong acid sites of the samples all increase. In conjunction with the XRF analysis: DEA tends to promote more Si atoms incorporation into AlPO4 framework and substitution for P individually. Meanwhile, it is well known, the Si (4Al) structures generated by each Si substituting for P alone give rise to negatively charged framework and relatively stronger Brønsted acid sites.22 So the acid sites increase with the increasing of DEA ratio in the DEA–DIEA dual templates. With regard to the enhancement of acid strength, it might be attributed to either the increasement of particle size or the higher content of Brønsted acid sites in the catalysts.11
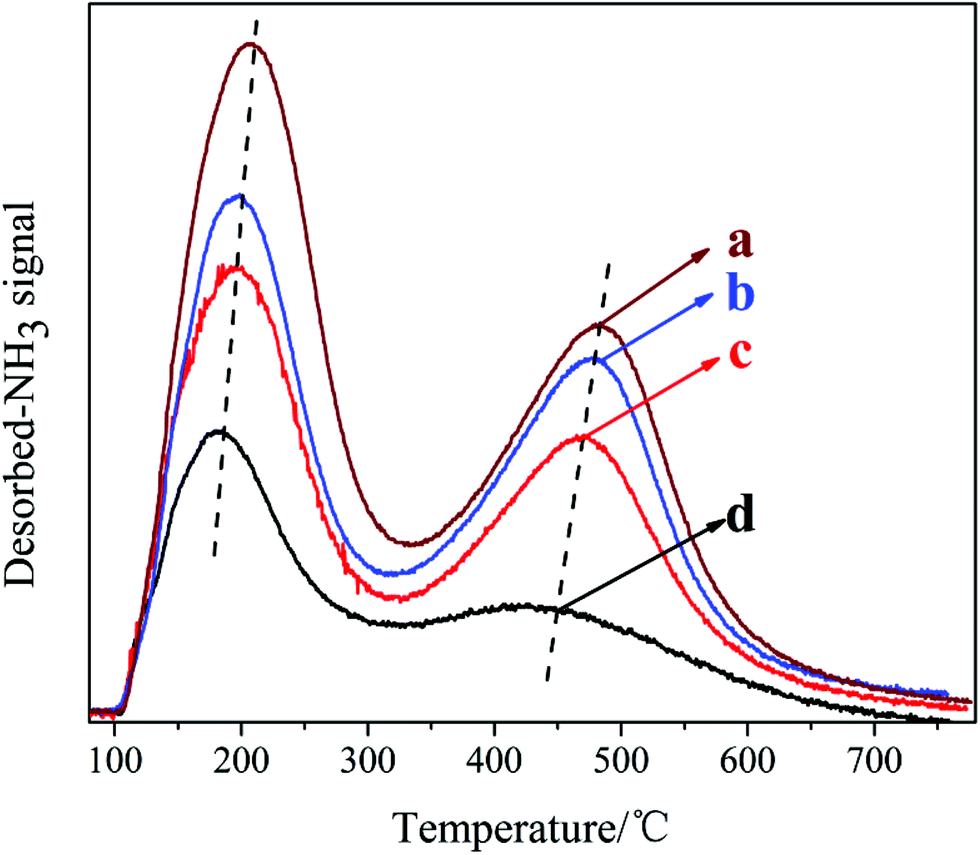 | ||
| Fig. 7 NH3-TPD curves of the samples synthesized at various template compositions. (a) 1.6DEA, (b) 1.4DEA + 0.2DIEA, (c) 1.2DEA + 0.4DIEA, (d) 1.6DIEA. | ||
Catalytic tests of DTO reaction were carried out at 400 °C in a fixed-bed reactor over the catalysts with different AEI/CHA ratios, and the results of DME conversion and coke amount of the discharged SAPO catalysts after 7 h reaction are given in Table 3. It is shown that during the recorded operation period, the conversion of DME is up to 100% for all the catalysts, indicating the high initial activity of the samples, but the rate or amount of coke deposition (an index of catalyst lifetime), varies considerably by the usage of catalysts with different AEI/CHA ratios. In comparison with pure-phase SAPO-18 and SAPO-34, the rate of coke deposition is much slower over the AEI/CHA intergrowth zeolites and it declines with the increasing of AEI/CHA ratio. It is noteworthy that pure-phase SAPO-18 with the smallest crystallite size and lowest acid density presents the most amount of coke deposition, which is even higher than that of pure SAPO-34 with the biggest crystallite size and highest acid density, in good agreement with the previous work showing that SAPO-34 with the smallest crystallites (<150 nm) deactivated rapidly.23 Since the relatively rich external surface of SAPO-18 may negatively influence the long-term operation of DME conversion due to the possible deposition of large organic species on its outer surface.22
| Catalysts | a (1.6DEA) | b (1.4DEA + 0.2DIEA) | c (1.2DEA + 0.4DIEA) | d (1.6DIEA) |
|---|---|---|---|---|
| Conversion (%) | 100 | 100 | 100 | 100 |
| Coke (%, g gcat−1) | 1.97 | 1.84 | 1.58 | 4.20 |
Fig. 8 shows the products distribution of the prepared samples. It can be seen that for all the catalysts, light olefins, especially ethylene and propylene, are the main products (70–80% in total), indicating that even with different AEI/CHA ratios, all the synthesized catalysts are highly selective to light olefins from DME. However, the selectivity of the generated products changes with the variation of phase composition in the catalysts, particularly for ethylene, propylene and butylene. The ethylene selectivity declines, while the propylene and butylene selectivities enhance with the increasing of AEI/CHA ratio in the catalysts, indicating that CHA phase is favorable for the formation of ethylene, and AEI phase favors propylene and butylene. This can be due to the slightly wider size of pear-like AEI cage than that of CHA cage.1b Meanwhile, over SAPO-18, methane, which is regarded as the product stemming from cracking reaction of long-chain alkenes and alkanes over strong acid sites, is more predominantly formed than that over SAPO-34 and SAPO-18/34 intergrowth zeolites. At the same time, over SAPO-18, the selectivity of short-chain ethylene increases but that of long-chain propylene and butylene drop rapidly with time on stream. In combination with the severe coke deposition over SAPO-18, it can be deduced that the accommodation of large amount of coke in SAPO-18 may give rise to increased diffusion resistance of reactant and products, which causes the generation of smaller products (methane and ethylene) and the decline of larger products (propylene and butylene). While slight coke formation over SAPO-34 and SAPO-18/34 intergrowth catalysts has no remarkable influence on the product diffusion, therefore, relatively stable products selectivity is presented.
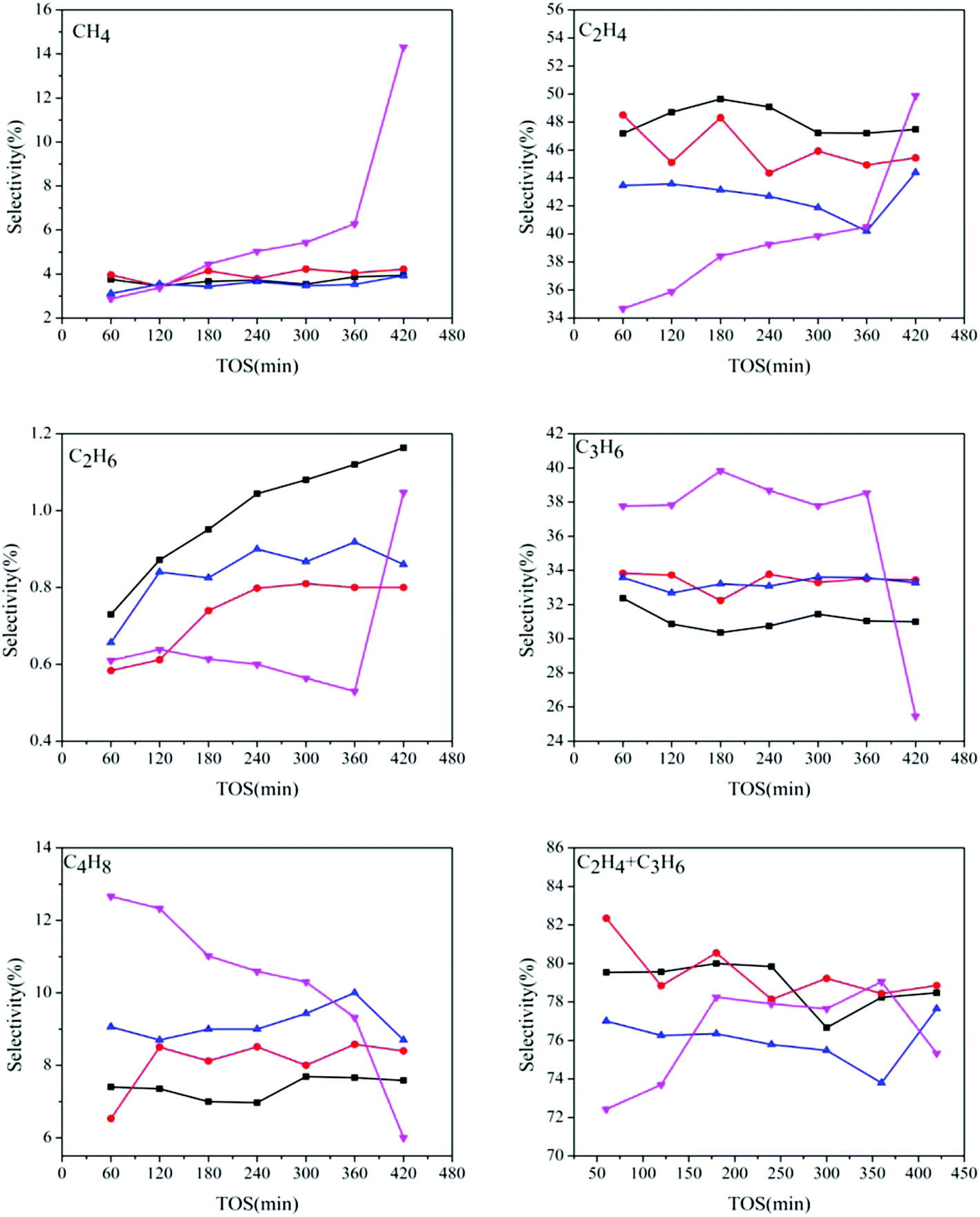 | ||
Fig. 8 Products distribution over the catalysts synthesized at various template compositions. 1.6DEA ( ), 1.4DEA + 0.2DIEA ( ), 1.4DEA + 0.2DIEA ( ), 1.2DEA + 0.4DIEA ( ), 1.2DEA + 0.4DIEA ( ), 1.6DIEA ( ), 1.6DIEA ( ). ). | ||
EDS was further performed to determine the distribution and content of carbon species in the fresh and used catalysts (sample b, as shown in Fig. 9). Obviously, for the used catalyst, the C content was greatly increased and the surface was coated with coke. Meanwhile, the BET surface area and pore volume measurements also confirmed this observation. The BET surface area of fresh catalyst was 532 m2 g−1 and case of the used catalyst the surface area was decreased 63 m2 g−1. The BJH pore volume decreased from 0.35 to 0.24 cm3 g−1, and the pore size also decreased from 4.7 to 3.5 Å. These phenomena can be attributed to the coke formation and accommodation in the catalyst, which decreases the micropore surface area, the pore volume and pore size of the used catalyst.
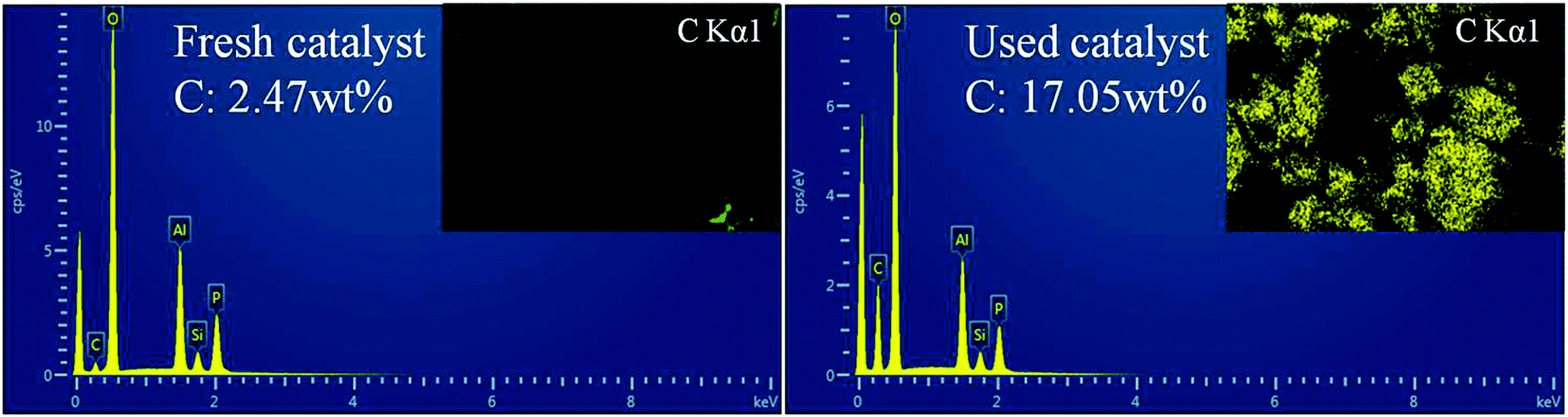 | ||
| Fig. 9 EDS analysis and mapping of C element of the fresh and used catalyst (1.4DEA + 0.2DIEA). | ||
4. Conclusions
Four well crystalline SAPO zeolites with different AEI/CHA ratios were synthesized with a mixed template of DIEA/DEA and investigated for their application as DTO catalysts. Samples a and d synthesized using single template DEA and DIEA were correspond to pure-phase SAPO-34 and SAPO-18, respectively, samples b and c synthesized using dual templates DEA–DIEA were AEI/CHA intergrowth materials. The crystal lattice expanded, the particles became smaller and varied from cube to square-platelet with the increasing of DIEA ratio in the DEA–DIEA dual templates. These could be due to the slightly wider cage size of AEI than that of CHA, meanwhile, the stronger alkalinity and electrophilicity of DEA than that of DIEA. In addition, DEA tended to promote more Si atoms incorporation into the AlPO4 framework by means of mechanism I, while DIEA favored the substitution of both I and II. The acid density and strength decreased with the increasing of AEI/CHA ratio, indicating that the acid sites may reside preferentially in the CHA cages and the acid strength in the CHA cages is stronger than that in the AEI cages.For the DTO reaction, all prepared catalysts exhibited similar DME conversion and products distribution, but the AEI/CHA intergrowth catalysts with medium particle size and acidity showed relatively lower rate of coke deposition than the pure-phase SAPO-18 and SAPO-34. Meanwhile, catalysts with a higher ratio of CHA cages had a higher selectivity to ethylene and catalysts with a higher ratio of AEI cages had a higher selectivity to propylene and butylene, probably due to spatial confinement effects imposed by cage structure of AEI and CHA.
Acknowledgements
The authors are grateful to the financial support from China Postdoctoral Science Foundation (No. 2014M551869), Shandong Excellent Young Scientist Research Award Fund (No. BS2015CL 002), and Qingdao Postdoctoral Application Research Project Fund. The National Natural Science Foundation of China (No. 21503243) also provided the financial support for this manuscript.References
-
(a) K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 4725 CrossRef CAS PubMed
; (b) J. Chen, J. Li, Y. Wei, C. Yuan, B. Li, S. Xu, Y. Zhou, J. Wang, M. Zhang and Z. Liu, Catal. Commun., 2014, 46, 36 CrossRef CAS
- M. Sedighi, M. Ghasemi, M. Sadeqzadeh and M. Hadi, Powder Technol., 2016, 291, 131 CrossRef CAS
-
(a) H. M. Torres Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130 CrossRef CAS
-
(a) D. Lesthaeghe, J. V. D. Mynsbrugge, M. Vandichel, M. Waroquier and V. V. Speybroeck, ChemCatChem, 2011, 3, 208 CrossRef CAS
-
(a) A. G. Gayubo, A. T. Aguayo, A. Alonso, A. Atutxa and J. Bilbao, Catal. Today, 2005, 106, 112 CrossRef CAS
- H. Yang, X. Liu, G. Lu and Y. Wang, Microporous Mesoporous Mater., 2016, 225, 144 CrossRef CAS
- H. Zhou, Y. Wang, F. Wei, D. Wang and Z. Wang, Appl. Catal., A, 2008, 348, 135 CrossRef CAS
- G. Liu, P. Tian and Z. Liu, Chin. J. Catal., 2012, 33, 174 CrossRef CAS
- N. Nishiyama, M. Kawaguchi, Y. Hirota, D. V. Vu, Y. Egashira and K. Ueyama, Appl. Catal., A, 2009, 362, 193 CrossRef CAS
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810 CrossRef CAS PubMed
- J. Chen, P. A. Wright, J. M. Thomas, S. Natarajan, L. Marchese, S. M. Bradley, G. Sankar, C. R. A. Catlow, P. L. Gai-Boyes, R. P. Townsend and C. M. Lok, J. Phys. Chem., 1994, 98, 10216 CrossRef CAS
- K. P. Lillerud and D. Akporiaye, Stud. Surf. Sci. Catal., 1994, 84, 543 CrossRef CAS
-
(a) D. Zhao, Q. Zhao, Y. Zhang, T. Shi, H. Yao and J. Yu, Chem. J. Chin. Univ., 2016, 37, 342 CAS
- J. Chen, J. M. Thomas, P. A. Wright and R. P. Townsend, Catal. Lett., 1994, 28, 241 CrossRef CAS
- R. Xu, W. Pang, J. Yu, Q. Huo and J. Chen, Molecular Sieves and Porous Materials Chemistry, Science Press, Beijing, 2004, p. 39 Search PubMed
- R. Wendelbo, D. Akporiaye, A. Andersen, I. M. Dahl and H. B. Mostad, Appl. Catal., A, 1996, 142, L197 CrossRef CAS
- Z. Li, J. Martínez-Triguero, P. Concepción, J. Yu and A. Corma, Phys. Chem. Chem. Phys., 2013, 15, 14670 RSC
-
(a) L. Marchese, A. Frache, E. Gianotti, G. Martra, M. Causa and S. Coluccia, Microporous Mesoporous Mater., 1999, 30, 145 CrossRef CAS
- S. Abdollahi, M. Ghavipour, M. Nazari, R. M. Behbahani and G. R. Moradi, J. Nat. Gas Sci. Eng., 2015, 22, 245 CrossRef CAS
- P. Wang, A. Lv, J. Hu, J. Xu and G. Lu, Microporous Mesoporous Mater., 2012, 152, 178 CrossRef CAS
- Y. Wei, D. Zhang, Y. He, L. Xu, Y. Yang, B. Su and Z. Liu, Catal. Lett., 2007, 114, 30 CrossRef CAS
- G. Yang, Y. Wei, S. Xu, J. Chen, J. Li, Z. Liu, J. Yu and R. Xu, J. Phys. Chem. C, 2013, 117, 8214 CAS
- B. V. Vora, T. L. Marker, P. T. Barger, H. R. Nilsen, S. Kvisle and T. Fuglerud, Stud. Surf. Sci. Catal., 1997, 107, 87 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2017 |