
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of scalable and versatile nanomaterial libraries for nanosafety studies: polyvinylpyrrolidone (PVP) capped metal oxide nanoparticles†
S. M. Briffaa,
I. Lynch a,
V. Trouilletb,
M. Brunsb,
D. Hapiukc,
J. Liuc,
R. E. Palmerc and
E. Valsami-Jones*a
a,
V. Trouilletb,
M. Brunsb,
D. Hapiukc,
J. Liuc,
R. E. Palmerc and
E. Valsami-Jones*a
aSchool of Geography, Earth and Environmental Sciences, University of Birmingham, B15 2TT Birmingham, UK. E-mail: e.valsamijones@bham.ac.uk
bInstitute for Applied Materials and Karlsruhe Nano Micro Facility, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cNanoscale Physics Research Laboratory, School of Physics and Astronomy, University of Birmingham, UK
First published on 16th January 2017
Abstract
The potential long-term environmental impact of manufactured nanomaterials (NMs) remains poorly understood, and the need to better predict NM fate and transformations and chronic effects is particularly urgent. Compared to their bulk counterparts, manufactured NMs can have distinct physical and chemical characteristics, which influence their behaviour, stability and toxicity. It is therefore essential to develop standard and reference NM libraries for environmental nanoscience and nano(eco)toxicology, and to facilitate a move towards computational prediction of NM fate, through quantitative structure–activity relationships for example. The aim of this work was to develop and fully characterise one such library, which included comparable NMs with a range of core chemistries, but the same capping agent and size range, for use in future studies to test the hypothesis that the core chemistry is a primary factor in controlling toxicity. The library contained the following NMs: 10k, 40k and 360k PVP capped ceria, zinc oxide and copper oxide (9 NMs in total). The work presented here upholds the underpinning hypothesis that the mechanism of NM formation is the same in all cases, suggesting that the protocol is very robust and has the potential to generate a wide range of comparable metal oxide NMs and potentially expand the library further with doped metal oxide and metal NMs. Characterisation by means of DLS (both size and zeta measurements), UV/Vis, XPS, FT-IR, TEM, STEM, EDX and EELS confirms that the tested synthesis protocol can easily and successfully be used to create stable PVP capped metal oxide NMs of reproducible sizes.
1. Introduction
It has become widely accepted that NMs are substances having at least one dimension between 1 and 100 nm.1–4 These materials with nano-scale features have high surface-to-volume ratio and may display distinctive chemical, electronic, optical, magnetic or mechanical properties compared to the equivalent bulk material.2,5,6The nanotechnology field has drastically changed daily life by promising an unlimited range of applications based on the NMs' distinctive properties.5,7 NMs are already extensively used in many industrial and everyday fields such as optics, energy, electronics, structural materials (e.g. composites for aircraft and automotive industries), clothing, food additives, cosmetics, paints and pigments, medical and health care applications.5,8,9 Nanotechnology continues to grow rapidly; since 2010 a 24% increase in consumer products containing NMs has been recorded.10
Three commonly used NMs are ceria (CeO2), zinc oxide (ZnO) and copper oxide (CuO). Ceria was first commercially used in 1999 as a catalyst in diesel particulate filters11 and at present its numerous uses include optical, micro-electronic, solid oxide fuel cells, solar cells, catalysis, bio-medical applications, oxidation resistance, UV absorbents and filters, anti-reflective coatings, abrasives for chemical mechanical planarization and metallurgical and glass/ceramic applications.12–21 Ceria's diversity of uses is due to its properties,19 the most important of which is its redox potential stemming from its ability to cycle between Ce3+ and Ce4+ (which has a characteristic yellow colour).18,22–24
Nano ZnO is used in solar cells, gas sensors, chemical absorbent varistors, electrical and optical devices, electrostatic dissipative coatings, catalysts for liquid phase hydrogenation, and catalysts for photo-catalytic degradation instead of titanium dioxide (TiO2) NMs.25 It is also an approved sunscreen ingredient in many countries around the world due to its UV blocking ability.26 Pinnell et al.27 have found that ZnO absorbs UV-radiation more effectively than TiO2 over a broad range of wavelengths, particularly in the UVA region. This has resulted in ZnO being the sole active ingredient in some broad-spectrum sunscreens.
CuO NMs are being used in catalysts, electronic devices (semiconductors, electronic chips, heat transfer nanofluids, superconductors and electrode materials),28,29 gas sensors, solar cells and lithium batteries.29 CuO has also been used as a biocide against bacteria, fungi and algae, hence its use in face masks, wound dressings and socks.29
The increased use of NMs results in their unintentional and intentional release into the environment.2,9,30 Hence concerns of exposure of humans and other organisms have been increasing.30–32 The impact of NMs on biota is poorly understood,9,28,33 partly due to the lack of data at environmentally realistic NM concentrations and exposure conditions (e.g. in the presence of appropriate macromolecules which can act as dispersants or drive other transformations34). Furthermore, to date, few studies have tried to establish the changes that NMs undergo when incorporated into, and released from, products, and the behaviour and impacts of such modified products have been largely overlooked.2 For instance degradation of cappings and cores may occur at different rates and due to different influencing factors. Many aspects in the study of nanotoxicity remain poorly studied and challenging due to difficulties in identifying, characterising and monitoring NMs particularly after their release.35 The potential exposure routes, NM properties effecting behaviour and possible transformations and interactions need to be determined and understood in a systematic manner. The field has yet to advance sufficiently due to challenges in NM characterisation and in linking systematically physico-chemical properties to toxicity.
In addition, suitable testing and reference NMs, involving the systematic variation of key physico-chemical parameters for (eco)toxicological and environmental studies are still lacking. Where certified reference materials (CRM) do exist, they are generally for size characterisation in water or simple media (e.g. CRM from the IRMM or NIST36), although more recently reference NMs for specific surface area or dimensional standards37 were launched by BAM.38 The other major category of “reference” materials are the OECD sponsored materials39 that are representative of industry NMs; however there is no internal consistency in this materials library in terms of size, surface coating, synthesis route and thus impurities etc., making determination of the key property or combination of properties driving a specific toxicity response near impossible if based on this library. Production of systematically varied NMs and a thorough understanding of their behaviour and degradation in relevant biological milieu are important scientific advances that will support research into nanosafety and in turn contribute to a sustainable nanotechnology industry.40 Indeed, development and validation of systematic sets of test NMs, and their labelled variants, including certification of their behaviour in reference biofluids, was identified as one of the research priorities for the nanosafety research roadmap 2015–2025.41
A key challenge in development of NMs libraries is that the synthesis route must be scalable such that large quantities can be produced easily. Ecotoxicity testing requires g to kg quantities depending on the protocol followed. For example high throughput toxicity screening, mesocosm scale experiments or multi-generation fish tank studies all require far greater quantities than standard protocols can produce. In such cases, it is essential that the protocol is robust enough to allow production of larger quantities whilst maintaining the particle quality of smaller scale synthesis. Furthermore, the protocol should be versatile such that a range of different materials can be produced utilising the same approach and the same chemicals as far as possible, since small changes in, for example, surfactants (used to control NM size or shape42) can result in quite different surface properties/toxicities of the resulting NMs. Additionally, many of the surfactant utilised are themselves toxic to cells, which limits the range of approaches available for development of NMs libraries for nanosafety studies. To date, most of the libraries reported in the literature consist of a simple metal or metal oxide core with a range of surface ligand functionalisations,43,44 thus taking a more pharmaceutical approach, or consist of a panel of metal oxide particles assembled from commercial sources which likely have different synthesis routes as well as the different sizes and properties reported.45 Recent reports of specific property libraries (oxidative stress, dissolution, crystallinity and surface charge46) show progress in the required direction, to allow a move away from extensive toxicity assessment and towards computational or in silico approaches to predict NM toxicity from physicochemical properties, although these materials are not widely available or easily produced utilising a single approach.
The work presented here aimed to develop a reproducible protocol for the synthesis of a broad range of PVP capped metal oxide NMs that would be versatile enough to allow size and core compositional variation whilst keeping the capping agent constant and/or varying the molecular weight of the capping agent (which in principle alters the degree of surface coverage and the accessibility of the core for transformations such as protein binding47). The NM samples have been thoroughly characterised, and the reproducibility and scalability of the method has been assessed. The resulting NM library may thus be used as reference materials for future toxicity and ecotoxicity testing since the robust synthesis results in comparable NMs with systematically varied properties. Toxicity and environmental fate testing using these NMs will help to address the hypothesis that the core chemistry is a primary factor in controlling toxicity and such work is currently under way.
2. Methodology
2.1 Materials and synthesis
Cerium nitrate hexahydrate, zinc nitrate hexahydrate, copper nitrate trihydrate, acetone, (10k, 40k and 360k) polyvinylpyrrolidone (PVP) were all used as received from suppliers. PVP capped CeO2 NMs were prepared by means of a reflux reaction according to Merrifield et al.23 whereby ∼130 mg of cerium nitrate were dissolved in 60 mL of a 3, 0.4 and 0.007 mM solution of either 10k, 40k or 360k PVP respectively and refluxed for 3 hours. Following this the reaction was quenched and the excess PVP was removed using acetone. Centrifuging at 4000 rpm for 10 min resulted in a yellow pellet, which was retained and resuspended in ultra-high purity water (UHP, resistivity 18.2 MΩ cm). The synthesis protocol was modified to produce PVP capped ZnO and PVP capped CuO NMs. This was done by substituting the cerium nitrate hexahydrate reagent with equimolar amounts of zinc nitrate hexahydrate or copper nitrate trihydrate as required. Following synthesis samples were filtered through an 0.1 μm filter. The reaction was scaled-up three and six fold by increasing the amount of starting reagents by a factor of three and six yet keeping the volume of water used in the reaction constant.2.2 Characterisation
Thorough characterisation of NMs, in dry form and in suspension (in Milli-Q water), was carried out by means of DLS, zeta potential, UV/Vis, XPS, FT-IR and TEM. Furthermore, unless stated otherwise, all characterisation is based on the first synthesis, before scaling up, and from a single batch. DLS measurements were performed on a Malvern Zetasizer (nano ZS) at the University of Birmingham. NM samples were analysed dispersed in UHP water. The concentration of these samples is shown in Table 1. Ten consecutive measurements were carried out and averaged to calculate the mean size. The results were obtained at 20 °C with samples equilibrated for 2 minutes before the measurements were started. The same instrument was used to obtain the zeta potential values. The results were obtained at 21 °C with the same samples used for size measurements, equilibrated for 2 minutes before measurements were started. Three consecutive measurements were collected and averaged to calculate the zeta potential.| Sample | X1 (mg L−1) | X3 (mg L−1) | X6 (mg L−1) |
|---|---|---|---|
| Ce 10k PVP | 2.030 ± 0.480 | 11.378 ± 2.123 | 15.631 ± 4.268 |
| Ce 40k PVP | 1.595 ± 0.370 | 8.216 ± 2.121 | 15.561 ± 6.114 |
| Ce 360k PVP | 0.236 ± 0.039 | 3.246 ± 0.929 | 5.178 ± 2.936 |
| Zn 10k PVP | 0.906 ± 0.137 | 3.246 ± 0.728 | 7.353 ± 2.848 |
| Zn 40k PVP | 0.856 ± 0.126 | 2.421 ± 0.718 | 6.563 ± 1.951 |
| Zn 360k PVP | 0.154 ± 0.084 | 0.619 ± 0.139 | 1.818 ± 1.006 |
| Cu 10k PVP | 1.440 ± 0.231 | 3.364 ± 0.980 | 4.273 ± 0.814 |
| Cu 40k PVP | 0.779 ± 0.212 | 1.915 ± 0.266 | 6.261 ± 1.935 |
| Cu 360k PVP | 0.228 ± 0.057 | 0.617 ± 0.347 | 1.972 ± 0.990 |
UV/Vis absorption readings were recorded by means of a Jenway 6800 UV/Vis spectrophotometer at the University of Birmingham. A background spectrum was first obtained using ultrapure water. Following this, three sample readings were obtained. Samples were diluted by adding 50 μL of the NM dispersion to 5000 μL of UHP water prior to UV absorption measurements. The diluted samples were placed in 10 cm long quartz cuvettes. A spectrum scan was taken from 800–220 nm.
XPS characterisation was carried out at Karlsruhe Institute for Technology (KIT) in Germany. Samples were prepared by placing a drop of the NM dispersion on the surface of silicon wafer, which was allowed to air-dry overnight. XPS measurements were performed using a K-Alpha XPS instrument (ThermoFisher Scientific, East Grinstead, UK). All samples were analysed using a microfocused, monochromated Al Kα X-ray source (400 μm spot size). Two random points were analysed for each of the samples. The K-Alpha charge compensation system was employed during analysis, using electrons of 8 eV energy, and low-energy argon ions to prevent any localised charge build-up. The spectra were fitted with one or more Voigt profiles (binding energy uncertainty: ±0.2 eV). The analyser transmission function, Scofield sensitivity factors48 and effective attenuation lengths (EALs) for photoelectrons were applied for quantification. EALs were calculated using the standard TPP-2M formalism.49 All spectra were referenced to the C 1s peak (C–C, C–H) at 285.0 eV binding energy controlled by means of the well-known photoelectron peaks of metallic Cu, Ag, and Au respectively.
FT-IR spectra were obtained using a Varian 660-IR FT-IR Spectrophotometer at the University of Birmingham. Dried samples were placed on the ATR and program ‘Agilent resolutions pro’ was used to run and obtain the spectrometer. Prior to samples being analysed background scans were carried out. TEM samples were prepared on copper grids by means of the drop method. NM imaging was carried out using a JEOL 1200 TEM at an accelerating voltage of 80 kV. STEM (scanning transmission electron microscopy) imaging of the 10k PVP capped NMs was performed at the Nanoscale Physics Research Laboratory at the University of Birmingham. CeO2 and ZnO samples were deposited on Cu grids covered with amorphous carbon film whilst the CuO sample was deposited on a Mo grid. The HAADF (high-angle annular dark field)-STEM imaging was performed in a 200 kV JEM2100F STEM (JEOL) with a spherical aberration corrector (CEOS). HAADF images were acquired with inner and outer angles of 62 and 164 mrad at a probe convergence semi-angle of 19 mrad and camera length of 10 cm. The EELS spectra were recorded with an Enfina detector attached to a JEM2100F STEM (JEOL) operated at 200 kV. The collection semi-angle was 57.8 mrad, the camera length was 2 cm and the aperture size was 5 mm. The FWHM value at the zero-loss peak was 4 eV, with a dispersion of 0.5 eV per channel and a 5 s acquisition time for CeOx and 10 s for CuOx, respectively. Prior to sample analysis reference EELS spectra of commercially available Ce3+ (Ce(NO3)3 salt) and Ce4+ (CeO2 NMs) materials were obtained. The EELS data was processed with Digital Micrograph software (version 2.11). EDX was carried out for 10k PVP capped ZnO at the Nanoscale Physics Research Laboratory at the University of Birmingham. The analysis was carried out on a JEM2100F STEM coupled with a Bruker XFlash 4030 EDX detector with an electron beam energy of 200 keV.
3. Results and discussion
All synthesis experiments produced coloured suspensions, which were stable at room temperature for longer than 6 months. This was confirmed by DLS measurements. An example of these results is shown in Fig. S1 in the ESI.† The NM dispersion colour is dependent on the type and amount of PVP used during the synthesis. The solution colour darkens as the PVP chain length decreases.3.1 NM size
Size measurements were obtained by DLS, TEM and STEM. Results show that the NM size obtained was comparable regardless of the NM core composition. The size obtained was dependent on the type and amount of PVP used during the synthesis. The longer the PVP chain the larger the NM hydrodynamic and core size obtained. Zhou et al.50 controlled the size of as-obtained CeO2 spherical crystallites by varying synthetic parameters such as the molar ratio of PVP (repeating units) to Ce(NO3)3·6H2O. Hence, changing the PVP length (molecular weight), which also constituted a change in molar ratio due to the different quantities used, resulted in a change in NM size. DLS showed the 10k PVP capped CeO2 NM to be c. 5–6 nm, the 40k PVP capped CeO2 NMs to be c. 7–8 nm and the 360k PVP capped CeO2 NMs to be c. 19–20 nm, as seen in Fig. 1. A full table of sizes and polydispersity index (PDI) values can be seen in Table 2. Examples of DLS sizes of different PVP capped metal oxide NM batches are shown in Table S1 in the ESI.†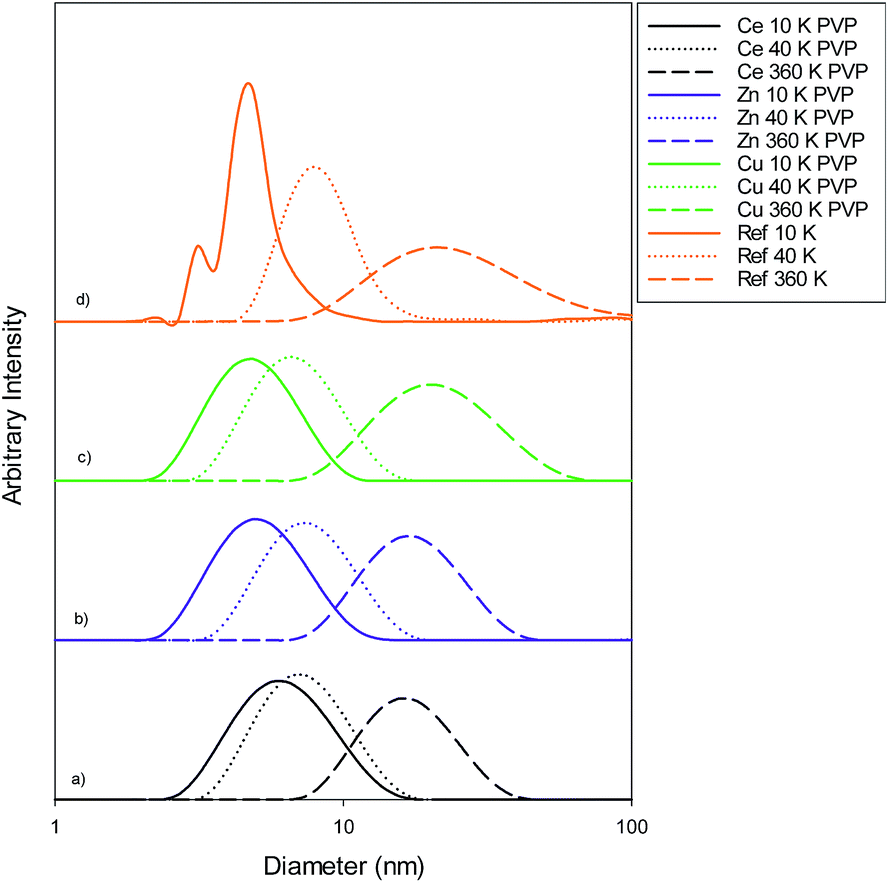 | ||
| Fig. 1 Size distribution graphs for (a) PVP capped ceria, (b) PVP capped zinc oxide, (c) PVP capped copper oxide and (d) PVP reference samples. The data in (a–c) show the 10k PVP capped NM to be c. 5–6 nm, 40k PVP capped NMs to be c. 7–8 nm and 360k PVP capped nanoparticles to be c. 19–20 nm. | ||
| Sample | As prepared | Scaled-up ×3 | Scaled-up ×6 | |||
|---|---|---|---|---|---|---|
| Size (nm) | PDI | Size (nm) | PDI | Size (nm) | PDI | |
| 10k PVP capped ceria | 6.282 ± 0.047 | 0.190 ± 0.006 | 4.816 ± 0.090 | 0.344 ± 0.031 | 4.185 ± 0.212 | 0.213 ± 0.022 |
| 40k PVP capped ceria | 7.347 ± 0.091 | 0.195 ± 0.013 | 7.188 ± 0.175 | 0.195 ± 0.035 | 8.132 ± 0.249 | 0.420 ± 0.065 |
| 360k PVP capped ceria | 19.30 ± 0.136 | 0.385 ± 0.008 | 4.778 ± 0.963 | 0.387 ± 0.110 | 4.183 ± 0.758 | 0.780 ± 0.306 |
| 10k PVP capped zinc oxide | 5.291 ± 0.103 | 0.225 ± 0.016 | 7.040 ± 0.389 | 0.293 ± 0.389 | 4.229 ± 0.805 | 0.362 ± 0.188 |
| 40k PVP capped zinc oxide | 8.075 ± 0.376 | 0.267 ± 0.024 | 8.282 ± 0.272 | 0.229 ± 0.028 | 6.250 ± 0.308 | 0.275 ± 0.085 |
| 360k PVP capped zinc oxide | 19.41 ± 1.272 | 0.354 ± 0.040 | 9.296 ± 0.086 | 0.275 ± 0.018 | 6.364 ± 0.178 | 0.225 ± 0.020 |
| 10k PVP capped copper oxide | 4.999 ± 0.126 | 0.219 ± 0.039 | 3.838 ± 0.033 | 0.791 ± 0.250 | 6.550 ± 0.072 | 0.296 ± 0.014 |
| 40k PVP capped copper oxide | 7.303 ± 0.099 | 0.198 ± 0.020 | 4.765 ± 0.033 | 0.169 ± 0.019 | 4.375 ± 0.138 | 0.476 ± 0.060 |
| 360k PVP capped copper oxide | 19.44 ± 0.183 | 0.222 ± 0.002 | 9.545 ± 0.147 | 0.187 ± 0.030 | 8.119 ± 0.167 | 0.406 ± 0.039 |
TEM results are shown in Table 3. TEM showed comparable values to those obtained by DLS for 10k and 40k PVP capped NMs with the former having a size of c. 5–7 nm and the latter c. 8–9 nm. The size results for the 360k capped metal oxide NMs obtained by means of TEM are smaller than those obtained by DLS and were c. 12–13 nm which is to be expected, since TEM measures primarily the core NM, rather than the electro-transparent PVP coating which makes a larger contribution to the DLS results in the case of the 360k capped NMs.
| Sample | Size (nm) | Histogram | Image |
|---|---|---|---|
| 10k PVP capped ceria | 7.625 ± 2.595 | 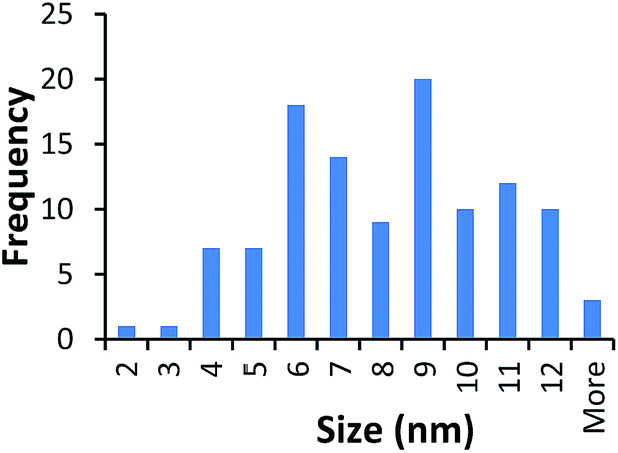 |
 |
| 40k PVP capped ceria | 8.992 ± 2.116 | 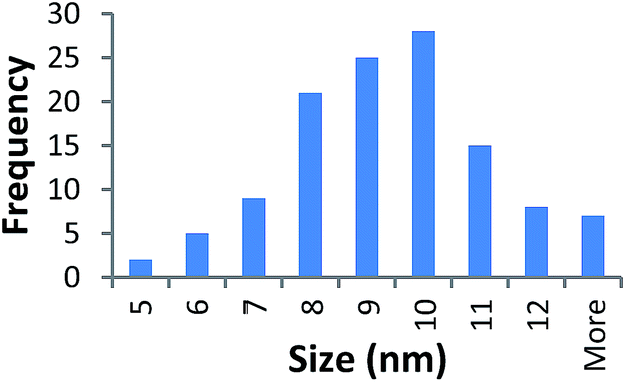 |
 |
| 360k PVP capped ceria | 12.976 ± 5.870 | 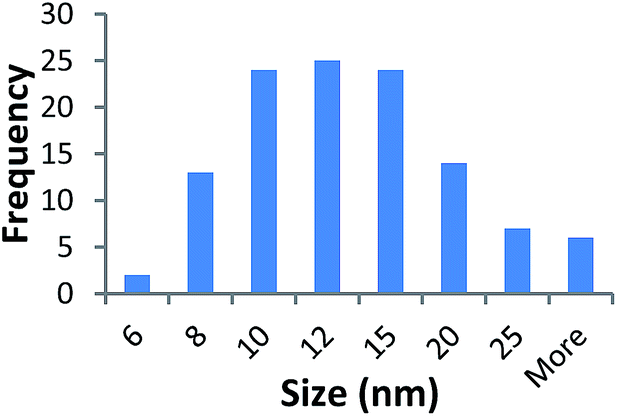 |
 |
| 10k PVP capped zinc oxide | 5.778 ± 2.247 | 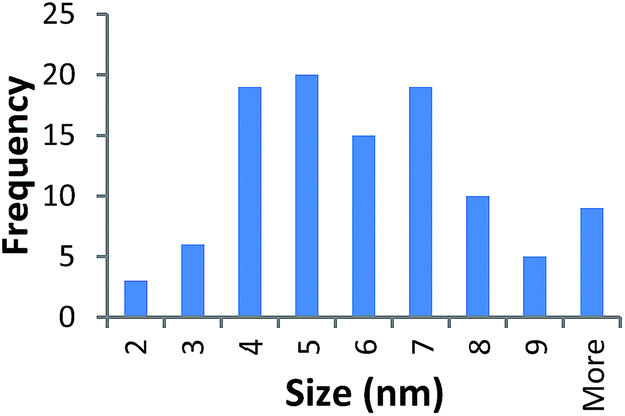 |
 |
| 40k PVP capped zinc oxide | 9.336 ± 3.329 | 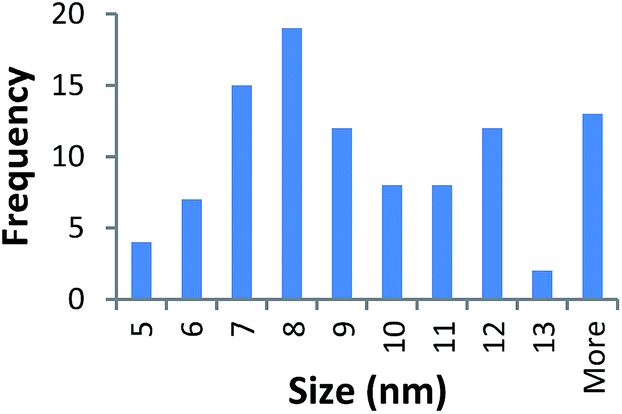 |
 |
| 360k PVP capped zinc oxide | 13.664 ± 5.219 | 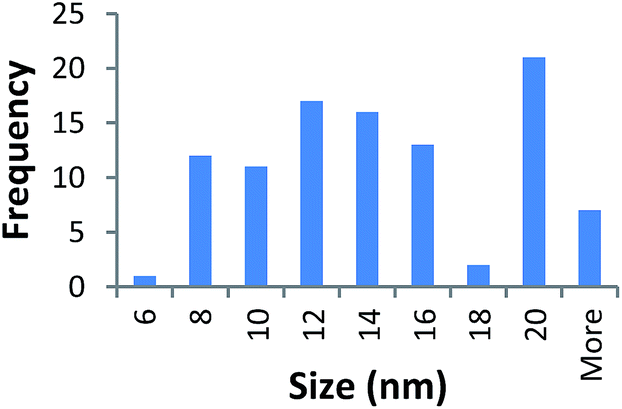 |
 |
| 10k PVP capped copper oxide | 5.950 ± 2.069 | 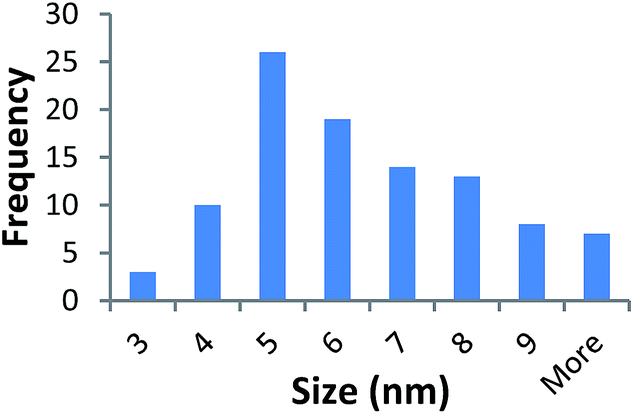 |
 |
| 40k PVP capped copper oxide | 9.320 ± 2.670 | 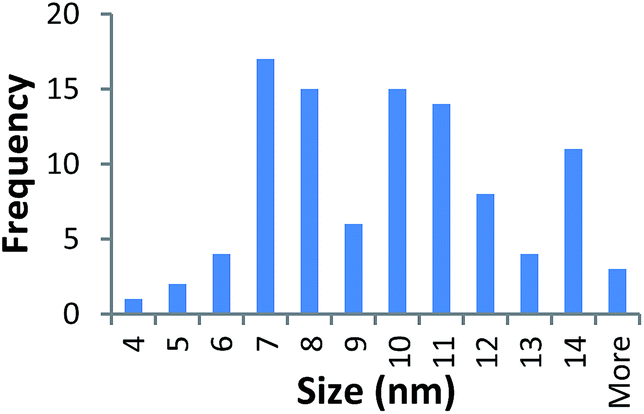 |
 |
| 360k PVP capped copper oxide | 12.144 ± 3.63 | 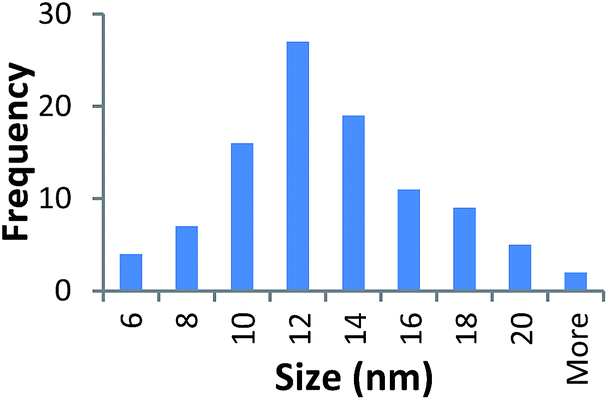 |
 |
It should be noted that differences in the ionic radius of the core metal can influence lattice dimensions and hence size. This can be seen when comparing Ce and the other two smaller ions, Cu and Zn, and is reflected in the particle size measurements, with ceria producing particles nearly 2 nm larger in the case of the smallest 10k PVP capped particles (Table 3). However, no significant effect is observed at larger sizes. Even for the 10k PVP capped particles, the difference is small enough to give overlapping size range for the three different metal oxides.
STEM size values were obtained for 10k PVP capped NMs and showed the core NP size to be slightly smaller than seen by DLS or TEM with a size of c. 3–4 nm since the PVP chains are invisible in HAADF and therefore STEM directly measures the NM core. 10k PVP capped CeO2 NMs had an average diameter of 3.3 ± 0.8 nm, 10k PVP capped ZnO NMs had an average diameter of 4.4 ± 0.9 nm and 10k PVP capped CuO NMs had an average diameter of 3.8 ± 0.9 nm (Table 4).
| Sample | Size (nm) | Histogram | Image |
|---|---|---|---|
| 10k PVP capped ceria | 3.3 ± 0.8 nm | 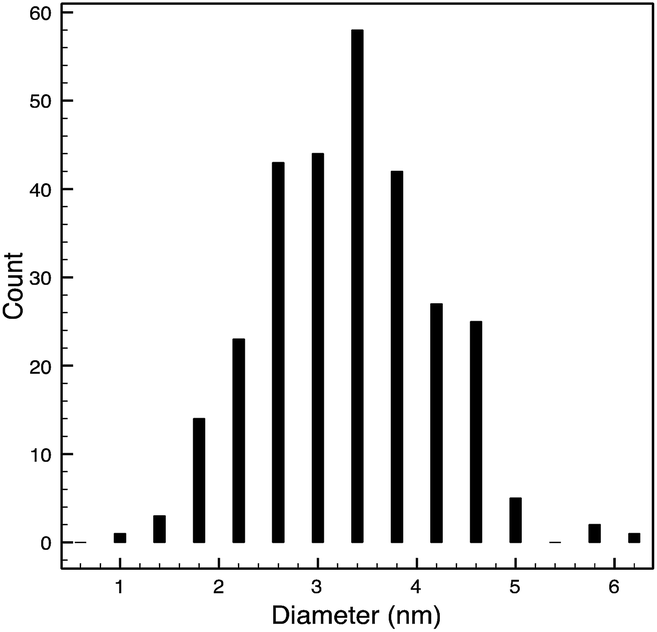 |
 |
| 10k PVP capped zinc oxide | 4.4 ± 0.9 nm | 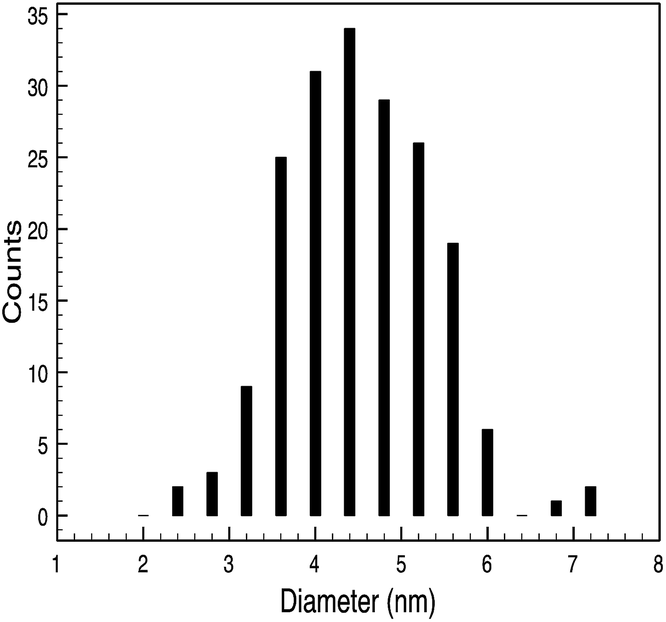 |
 |
| 10k PVP capped copper oxide | 3.8 ± 0.9 nm | 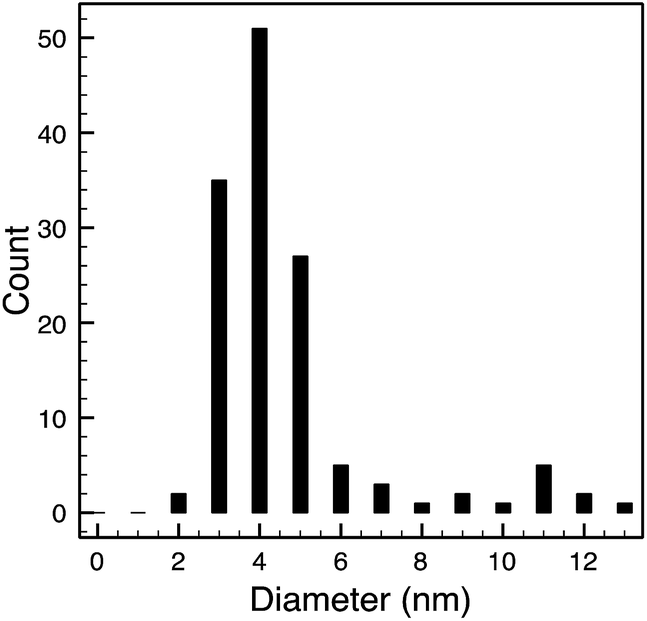 |
 |
3.2 Zeta potential measurements
The average zeta potential values obtained for all PVP capped samples regardless of the NM core, capping agent chain length or scale-up factor were very close to 0, ranging between −4 and +2 mV, the isoelectric point, i.e. the point where the colloidal system is least electrostatically stable. Despite this, the NM dispersions are stable due to steric stabilisation from the PVP chains. This stability remains over a period of at least 28 days for all particles. Fig. S2 in the ESI† shows this for 10k PVP capped ceria.3.3 Chemical characterisation
The chemical composition of the NM library was also characterised by a multi-method approach. FT-IR, UV/Vis, XPS, EELS and EDX were carried out to confirm the presence of the metal oxide NM core.FT-IR spectra were obtained for 10k PVP capped NM samples. The spectra are seen in Fig. 2a–c for CeO2, ZnO and CuO NMs respectively. Ce–O stretching was observed at c. 550 cm−1 as mentioned in the literature.51,52 The Zn–O stretching peaks were found to be the medium intensity peaks at just below 600 cm−1 and that at 517 cm−1. Ismail53 reported the Zn–O stretching peaks at ν = 670 and 555 cm−1. Interaction with PVP could be the reason for the shift in these peaks. The characteristic peaks seen for Cu–O stretching are the very small peak at 538 cm−1, the slightly larger peak above 550 cm−1 at 557 cm−1 and the smaller one at c. 570 cm−1. These are similar to those noted in the literature54 for Cu2O and CuO yet slightly shifted due to the interaction of the NMs with PVP.
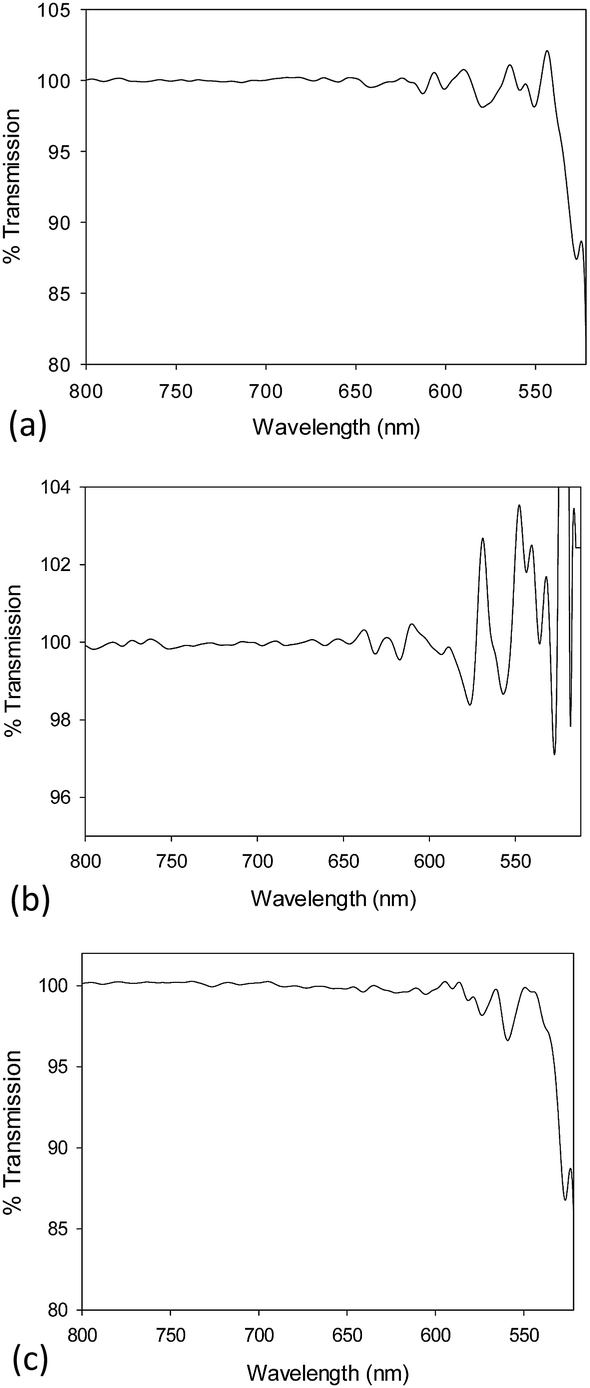 | ||
| Fig. 2 (a) FT-IR spectrum for 10k PVP capped ceria NMs with Ce–O stretching was observed at c. 550 cm−1. (b) FT-IR spectra of 10k PVP capped zinc oxide NMs with Zn–O stretching peaks at just below 600 cm−1 and that at 517 cm−1. (c) FT-IR spectra of 10k PVP capped copper oxide NMs with Cu–O stretching peaks at 538 cm−1, 557 cm−1 and c. 570 cm−1. | ||
Fig. 3a shows the UV/Vis spectra for 10k, 40k and 360k PVP capped metal oxide NMs. The absorption peak of PVP was found to be at 250 nm and is noted in all spectra. The CeO2 NMs spectra show the presence of three peaks found at 254 nm, 300 nm and 250 nm. The first is due Ce3+, the second is due to Ce4+ and the latter is due to the presence of PVP. The ZnO NMs spectra shows a peak at c. 300 nm which is due to the ZnO55 and a peak at c. 250 nm due to PVP. The PVP capped CuO NMs spectra show a small peak located just above 300 nm that could be due to CuO shifted slightly due to the interference of the PVP peak. The peak at 440 nm is characteristic of Cu2O.56 Fig. 3b shows the UV/Vis spectra for PVP capped copper oxide NMs as prepared without further dilution. These spectra show the presence of the peak at 440 nm and a peak at 800 nm which is due to CuO. Therefore the copper oxide NM dispersions are formed of mixed valency oxides of copper. The difference in peak intensities along with the fact that the latter peak was not seen upon sample dilution indicates that the concentration of Cu2O is greater than that of CuO yet this cannot be interpreted quantitatively due to the lack of standards for the sample.
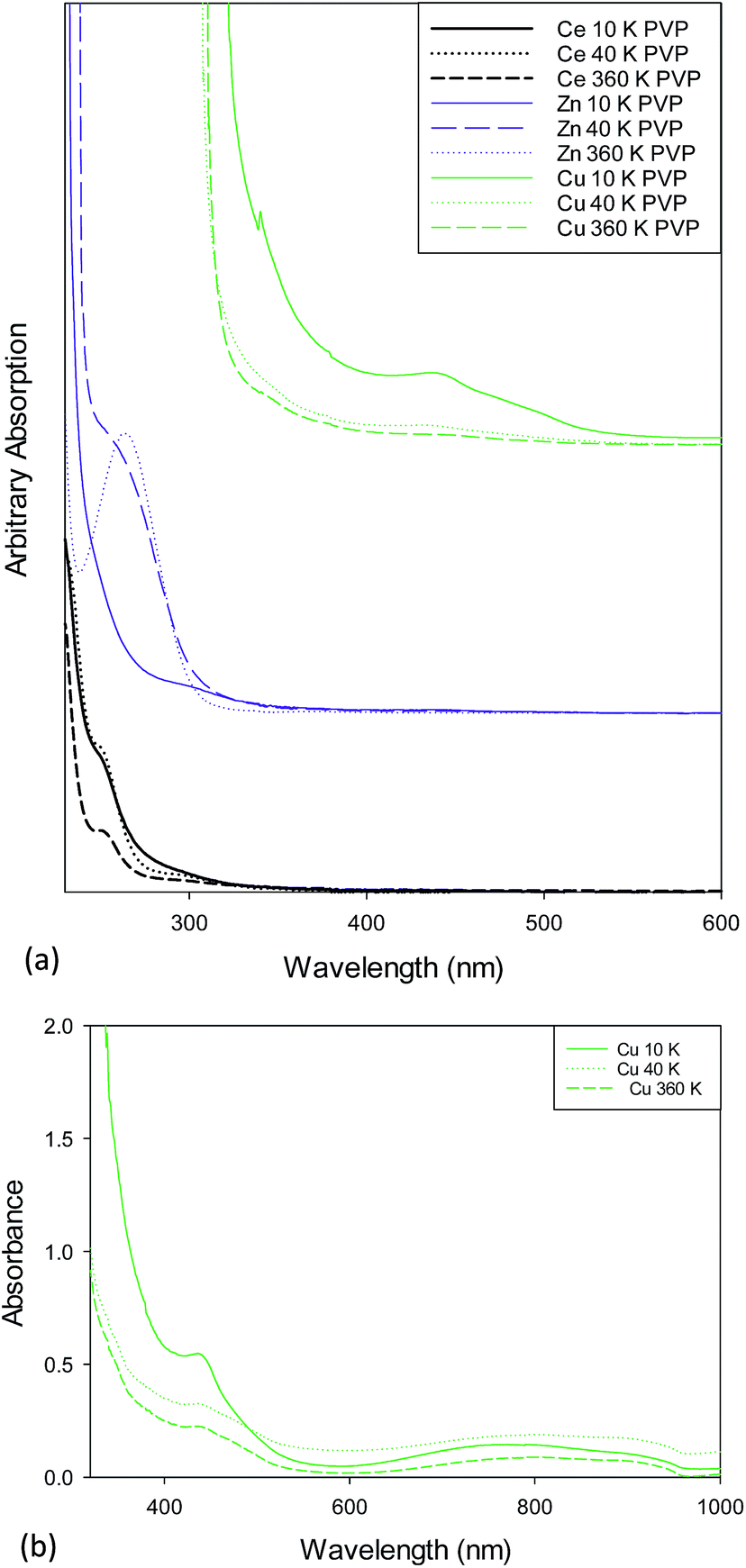 | ||
| Fig. 3 (a) UV/Vis spectra for (a) PVP capped ceria showing a peak at 254 nm due to Ce3+ and at 300 nm due to Ce4+, (b) PVP capped zinc oxide showing the characteristic ZnO peak at just below 300 nm and (c) PVP capped copper oxide NMs showing the characteristic Cu2O peak at 440 nm. The absorption peak at 250 nm is due to PVP. (b) UV/Vis spectra for as prepared 10k, 40k and 360k PVP capped copper oxide NMs showing absorption peaks at 440 nm characteristic of Cu2O and at 800 nm characteristic of CuO. | ||
The XPS spectra were referenced to the carbon 1s peak (C–C, C–H) at 285.0 eV. Kim et al.57 report Ce3+ having XPS peaks at 885.0 and 903.5 eV whilst Ce4+ has peaks at 882.1, 888.1, 898.0, 900.9, 906.4, and 916.4 eV. Peaks between 875 to 895 eV belong to the Ce 3d5/2 levels while peaks between 895–910 eV correspond to the Ce 3d3/2 levels. A peak at 916.7 eV is a characteristic satellite peak indicating the presence of cerium(IV).58 Fig. 4a shows the XPS spectra of the PVP capped CeO2 NMs. 10k and 360k capped ceria show the presence of mixed oxides whilst 40k is seen to be composed of Ce3+. Table 5 shows the Ce3+ and Ce4+ percentages present within the sample, calculated according to the method of DeCaluwe.59 The binding energy of Zn(0) and Zn(II) are very similar with the former having a value of 1021.7 eV and the latter a value of 1022 eV (ref. 60) (Fig. 4b). Therefore to decipher between the two, the kinetic energy needed to be analysed and the Auger peaks allow for this differentiation. The LMM Auger line for the kinetic energy of the ZnO sample was found at about 988 eV implying that the samples were composed of Zn(II).61 The XPS spectra for PVP capped copper oxide samples seen in Fig. 4c are similar in all cases and show the presence of peaks at 940.0 and 944.0 eV that are satellites typical for the presence of Cu(II).62 Besides the satellite peaks the spectra also show two peaks at above and around 935 eV indicating that both copper(II) and copper(I) species are present, further proving the presence of mixed oxide NMs. Since it is well-known that CuO is sensitive to X-rays, care was taken that the copper lines at the beginning and end of the measurement were the same to ensure no X-ray induced changes took place. In addition, since it is not possible to distinguish between Cu(I) and Cu(0) on the basis of the Cu 2p peak only, the Auger line was also measured to allow for this. The Auger line was detected at a kinetic energy of about 916 eV, which proves the presence of Cu(I) and excludes Cu(0).
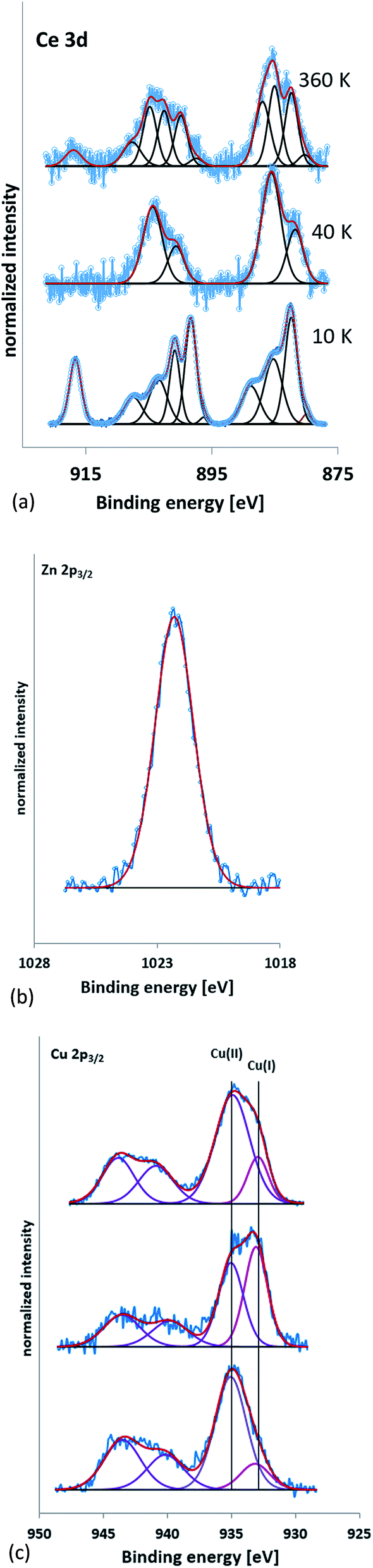 | ||
| Fig. 4 (a) XPS spectra of 10k PVP capped ceria NMs, 40k PVP capped ceria NMs and 360k PVP capped ceria from bottom to top respectively. The peak at 916.7 eV is a characteristic satellite peak indicating the presence of cerium(IV). (b) Zn 2p3/2 XPS spectrum for 10, 40 and 360k PVP capped zinc oxide NM samples. (c) Cu 2p3/2 XPS spectra for 10, 40 and 360k PVP capped copper oxide NMs (bottom to top) indicating the presence of both copper(II) and copper(I) species. | ||
| Sample | Ce3+ (%) | Ce4+ (%) |
|---|---|---|
| 10k PVP capped ceria | 39 | 61 |
| 40k PVP capped ceria | 100 | 0 |
| 360k PVP capped ceria | 81 | 19 |
The 10k PVP capped ceria and 10k PVP capped copper oxide NMs were further characterised by means of EELS. Due to the small size of Cu NMs, the Cu L-edge was very weak in the EELS spectra. Therefore, the O K-edge was used for the analysis of oxidation states. O K-edge reference spectra are taken from CuO (blue line) and Cu2O (red line) NMs. The comparison of the O near edge structures is shown in Fig. 5c. The comparison was performed in thirteen spectra and eight of them showed some similarity to the O K-edge of Cu2O (Fig. 5c). The other spectra showed no similarity to the spectra of either Cu2O or CuO (Fig. 5d). It is difficult to quantitatively determine the oxidation states of the CuOx NMs, since the size is very small and the Cu L-edge is weak. The comparison of the O K-edge suggests that the oxidation state of some CuOx NMs may be at +1 whilst others may indicate a mixture of different type of copper oxides.
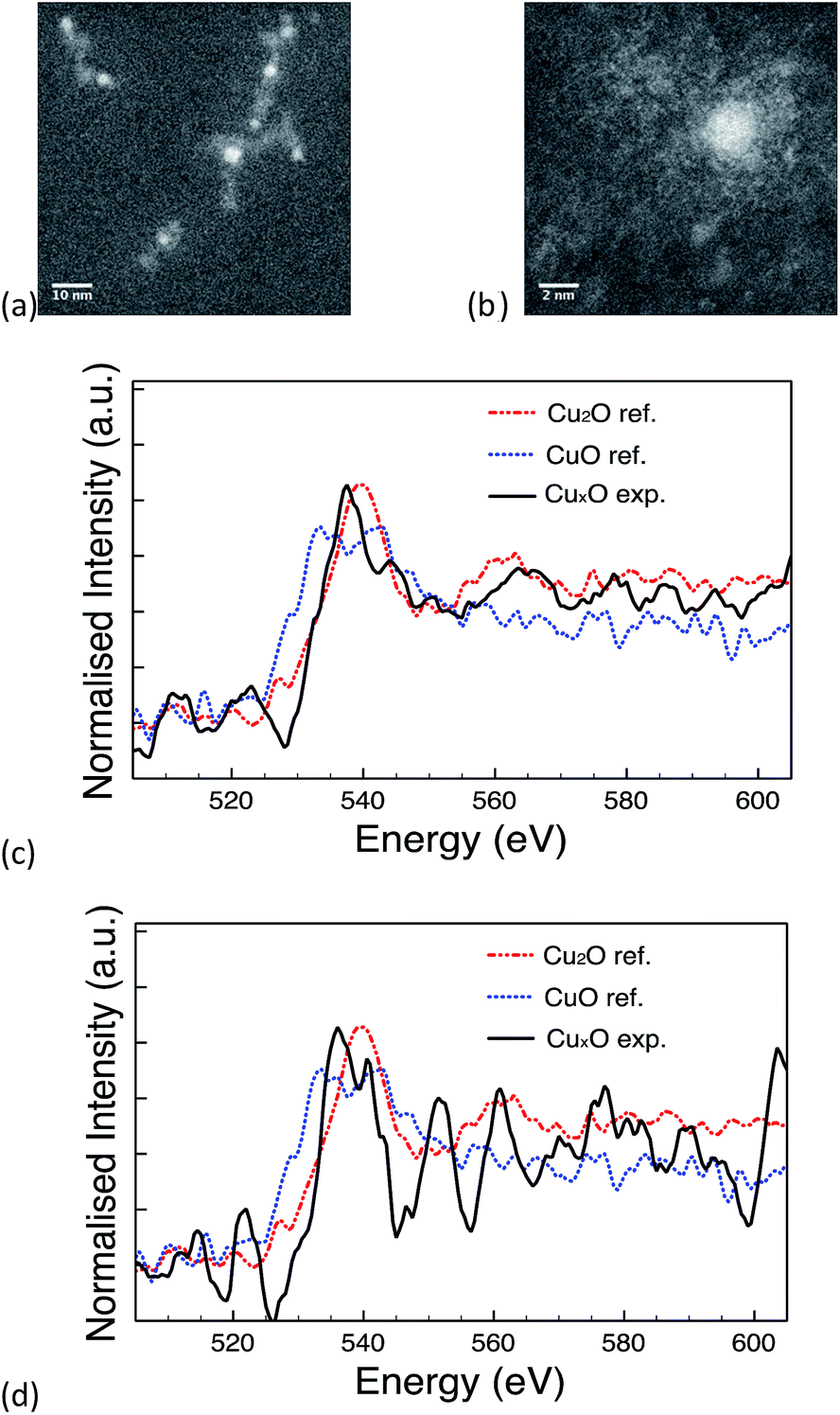 | ||
| Fig. 5 (a) & (b) STEM image of PVP capped copper oxide NMs, (c) spectrum showing similarity to the O K-edge of Cu2O & (d) spectrum showing no similarity to Cu2O or CuO reference NMs. | ||
The EELS spectra of cerium is characterised by two sharp peaks forming due to the transition of a core electron to an unbound state. One peak is due to the 3d3/2 → 4f5/2 transition denoted as M4 and the other is due to 3d5/2 → 4f7/2 transition denoted as M5.23,63,64 These peaks can be used to quantitatively determine the Ce4+/Ce3+ oxidation states ratio by performing multiple linear least-square (MLLS) fitting of the Ce M edge64 and comparing the results obtained to those of reference spectra.
Fig. 6b shows the reference spectra from Ce(NO3)3 and CeO2 standard materials and an example of MLLS fitting result is shown in Fig. 6c. The average oxidation state calculated from 120 individual NPs from a single 10k PVP capped ceria synthesis was 3.38 ± 0.13.
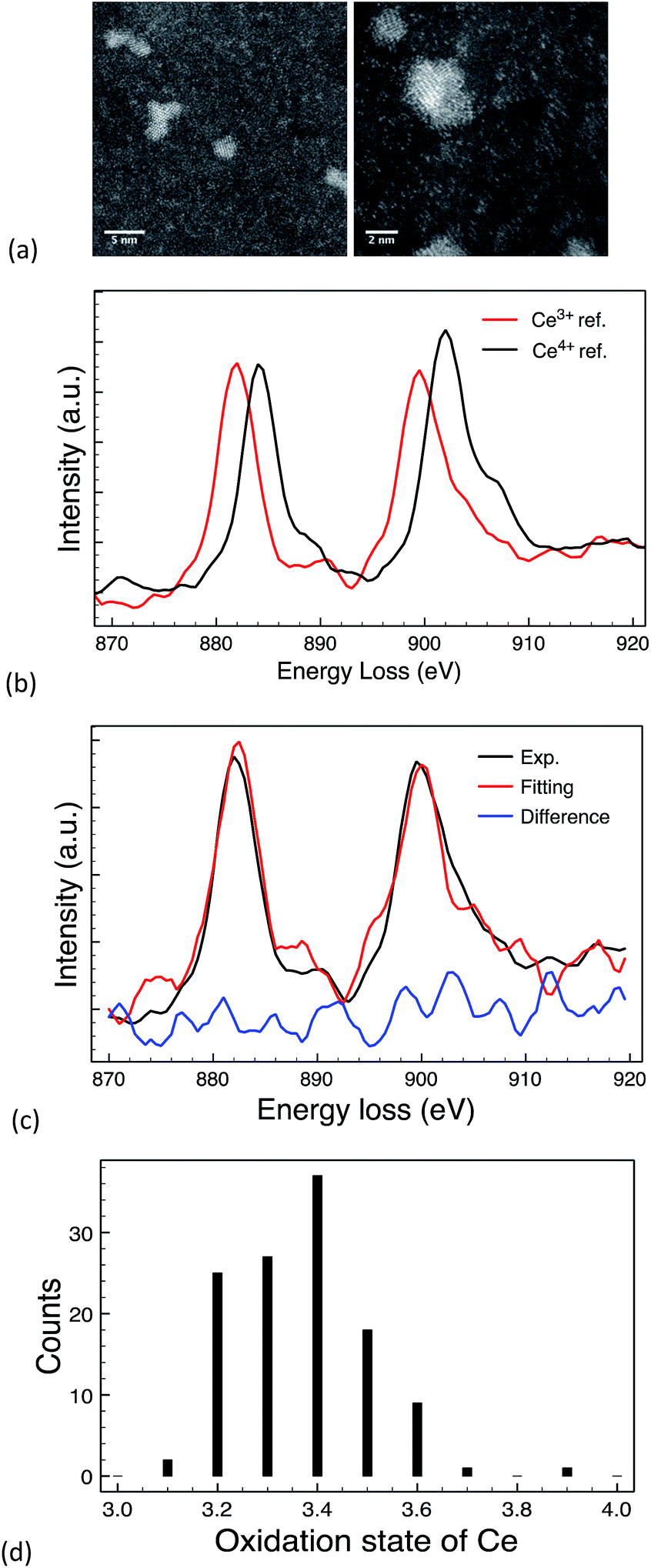 | ||
| Fig. 6 (a) STEM images for 10k PVP capped ceria, (b) reference EELS spectra of commercially available Ce3+ (Ce(NO3)3 salt); and Ce4+ (CeO2 NMs) materials; (c) MLLS fitting of an EELS spectrum from one CeOx NM; & (d) oxidation state distribution of CeOx NPs. | ||
EELS and XPS show different composition percentages, with EELS indicating Ce3+ to be more prominent (Fig. 6d) and XPS showing Ce4+ to be more prominent with 39% Ce3+ and 61% Ce4+ (Table 5). The reason for this is that EELS provides a profile of the ceria oxidation state on a per particle basis and on chosen localised points whereas XPS reveals overall information about the total oxidation state of the sample. This is an important distinction as it indicates that the particles are not completely homogeneous, which can have implications for toxicity also.
The 10k PVP capped ZnO NMs characterised by EDX (Fig. 7) show the presence of both Zn and O in a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.
:3.
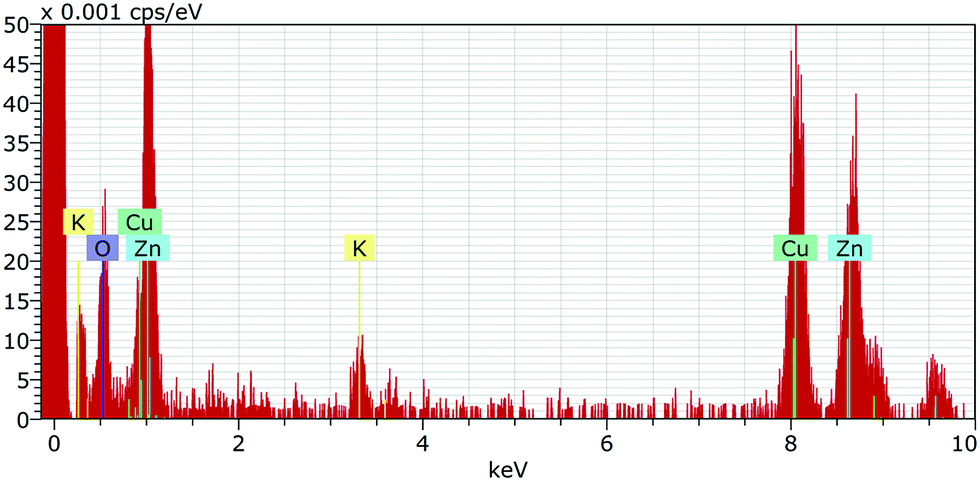 | ||
| Fig. 7 EDX spectrum for 10k PVP capped zinc oxide NMs showing the presence of both Zn and O in a ratio of 2:3. | ||
3.4 Understanding the NM library and its importance
Most synthesis methods for PVP capped NMs are based on the presence of a base or ethylene glycol65–68 or utilise high temperature autoclave reactions.50,69 The method used in this work simplifies the process by eliminating the use of additional precursors and refluxing rather than autoclaving making the synthesis more robust. In this synthesis, the metal nitrate forms ions in solution which behave as Lewis acids, accepting lone pairs of electrons from OH− (Lewis bases). The metal ion's positive charge draws electron density from the O–H bond in the water molecule increasing the bond's polarity and making it easier to break. When the O–H bond breaks, an aqueous proton is released producing an acidic solution. The PVP attaches to the hydroxyls on the growing NMs' surfaces by the carbonyl groups. This results in a strong steric stabilisation of the NMs.The very detailed characterisation of the metal oxide NMs library presented here highlights some important issues for their use as potential reference NMs for nanosafety. Among the key mechanisms identified to date for toxicity is the generation of oxidative stress, often as a result of redox cycling, the presence of surface defects or highly crystalline surfaces.70,71 Thus, different surface oxidation states72 or crystal forms of the same material have quite different toxicities, and certainly have different surface plasmon resonances,73 and likely different bandgaps due to different proportions of the various valence states.74 Indeed, Fernández-García and Rodriguez74 proposed that in the case of Cu2O or CeO2 NMs the deviation of the band-gap from the typical inverse squared dependence on the primary particle size (R2−) appears to be directly related to the presence of Cu2+ (remarkably for very low particle size) and Ce3+ ions at the surface of the nanostructured materials, and suggested that it is not yet clear if the presence of these oxidation states are intrinsic to the nanostructure or result from the specific procedure of preparation.74
Given the fact that several of the NMs in the library described here were mixed or two-phase materials, and the fact that methods that provide particle-by-particle information (such as EELS) indicate different compositions for specific particles compared to the “average” composition given by methods such as XPS (for example), this has important consequences for the prediction of NMs toxicity using QSARs for example, where average properties are typically utilised, which may miss subtle differences, or miss impacts from NMs at the tails of the property distribution function. Thus, we are used to considering size distribution functions, and everyone understands that in a particle size distribution the very small particles at the left hand side of the distribution curve may pose a larger toxicity threat than the larger ones at the right hand tail (for example this is the basis of the EU regulatory definition of NMs where the % of particles on a number basis <100 nm is used to determine whether a material is nano or not). However, nanosafety scientists and modelers are only beginning to consider the distribution functions of other physico-chemical properties such as oxidation state or dissolution. For example, Puzyn et al.75 found that the toxicity of metal oxide NMs decreased in the order Me2+ > Me3+ > Me4+ using a model based on the enthalpy of formation of a gaseous cation which has the same oxidation state as the metal ion in the oxide structure. Whether this model could be extended to NMs with mixed oxidation states remains to be seen, and whether one oxidation state dominates in terms of the toxicity is an important open question in terms of NMs safety-by-design. The NMs library described here, with its very detailed characterisation information will begin to allow questions such as these to be addressed. Basic toxicity screening, and corona fingerprinting of the NMs are underway and will be reported separately, and the combined characterisation and biological data will be provided to the nanosafety modelling community as soon as available.
4. Conclusions
Until now, a simple reflux for the synthesis of a variety of monodisperse metal oxides NMs with controllable sizes using aqueous metal nitrate solution as a precursor, has not been tried for a range of metal salts. One reason for this is that libraries prepared by a single synthesis method, in order to obtain a range of comparable NMs, are not normally needed for commercial applications of NMs, where the main consideration is functionality rather than how a NM is prepared. Yet, for nanotoxicological studies systematically varied NMs where only one condition of the synthesis is varied at a time are vital, in order to allow direct correlation between physico-chemical properties and induction of a biological or toxicological effect. A persistent challenge with NMs synthesis is that, in many cases, to vary a specific physico-chemical parameter requires alteration of several aspects of the synthesis method making these systematic NM structure-correlations impossible. Therefore, this work is the first to use a simple reflux synthesis to prepare a range of differently sized PVP capped metal oxide NMs, and to test and confirm the hypothesis that the mechanism of NM formation would be the same in all cases.The NM library was obtained by successfully modifying a hydrothermal synthesis method using PVP, initially implemented for CeO2 NMs,23 to produce PVP capped zinc oxide and copper oxide NMs. DLS, TEM and STEM analysis showed NM size to be dependent on the MW of the PVP capping molecule and independent of the metal oxide core composition. DLS and TEM showed the NM size to increase as the polymer chain length increased from 10k to 360k. In suspension, the zeta potential of the library NMs was found to be close to zero yet the samples were stable due to PVP steric stabilisation. Physico-chemical characterisation methods revealed the success of the synthesis by confirming the presence of the different metal oxide core. In the case of ceria and copper oxide NMs, where the metal has multiple valency states, mixed metal oxide NMs were found to be present as seen by XPS. PVP was found to play a significant role in the synthesis of the NMs. Additionally PVP influenced both the physical and chemical properties of the NMs.
Characterisation has shown that the modifications were successful and the synthesis is easily adaptable and scalable. More importantly, multi-method characterisation avoided misinterpretation of valency information as results from different techniques providing technique specific information about the samples. UV/Vis, EDX and EELS would have not been sufficient enough and XPS was needed to back up and develop the results interpretation. Specifically, the combination of particle-by-particle characterisation methods with ensemble (average sample) measurements allows the potential to assess distribution curves for a range of properties that may be closely linked to toxicity effects, such as oxidation state.
The NM library provides comparable particles for toxicology or fate and behavioural studies with variables (core composition, shell thickness) systematically altered, thus allowing for correlation of physico-chemical descriptors with toxicological impacts. The scalability of the library makes it particularly suitable for ecotoxicological studies where large quantities of particles are often necessary. Additionally the steric stabilisation of the PVP capping makes the particles robust enough for environmental media where ionic strength and abundance of biomolecules can interfere with stability. It is however important to note that future toxicity studies on this library need to be complemented by stability and characterisation data in the exposure media in order to fully understand and interpret correctly their behaviour.
Finally, the NM library has the potential to be extended further to other NMs such as other rare earth oxides. Furthermore there is the possibility of synthesising doped oxides such as copper doped ceria or zinc doped ceria (work in progress) further widening the scope of the NM library, and allowing for tracking of the engineered NMs at low concentrations in the environment, including against naturally occurring background concentrations.
Abbreviations
| PVP | Polyvinylpyrrolidone |
| DLS | Dynamic light scattering |
| UV/Vis | Ultraviolet/visible light spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| FT-IR | Fourier transform infra-red |
| TEM | Transmission electron microscopy |
| STEM | Scanning transmission electron microscopy |
| EDX | Energy dispersive X-ray spectroscopy |
| EELS | Energy electron loss spectroscopy |
| HAADF | (High-angle annular dark field)-STEM |
| PDI | Polydispersity index |
| MLLS | Multiple linear least-square |
Acknowledgements
The authors acknowledge financial support from FP7 funded projects: QualityNano (Grant Agreement no 262163, via a Transnational Access grant to SMB for KIT visit) and NanoMILE (Grant Agreement no 310451 to EVJ, IL, SMB & RP) and the Endeavour Scholarship Scheme (Group B) (to SMB).References
- E. Valsami-Jones and I. Lynch, Science, 2015, 350, 388–389 CrossRef CAS PubMed.
- B. Nowack and T. D. Bucheli, Environ. Pollut., 2007, 150, 5–22 CrossRef CAS PubMed.
- JRC, Considerations on a Definition of Nanomaterial for Regulatory Purposes, https://ec.europa.eu/jrc/sites/default/files/jrc_reference_report_201007_nanomaterials.pdf, accessed 03/12/2015.
- ISO, DD ISO/TS 80004-1:2010-Nanotechnologies. Vocabulary. Core terms, 2010.
- M. Tejamaya, I. Römer, R. C. Merrifield and J. R. Lead, Environ. Sci. Technol., 2012, 46, 7011–7017 CrossRef CAS PubMed.
- B. S. I. BSI, Nanoparticles Vocabulary, 2011 Search PubMed.
- I. Römer, A. J. Gavin, T. A. White, R. C. Merrifield, J. K. Chipman, M. R. Viant and J. R. Lead, Toxicol. Lett., 2013, 223, 103–108 CrossRef PubMed.
- A. Aydın, H. Sipahi and M. Charehsaz, Recent Adv. Novel Drug Carrier Syst., 2012, 483–500 Search PubMed.
- M. Thwala, N. Musee, L. Sikhwivhilu and V. Wepener, Environ. Sci.: Processes Impacts, 2013, 15, 1830–1843 CAS.
- T. P. o. E. Nanotechnologies, Inventory Finds Increase in Consumer Products Containing Nanoscale Materials, http://www.nanotechproject.org/news/archive/9242/accessed 15th November 2013.
- L. De Marzi, A. Monaco, J. De Lapuente, D. Ramos, M. Borras, M. Di Gioacchino, S. Santucci and A. Poma, Int. J. Mol. Sci., 2013, 14, 3065–3077 CrossRef CAS PubMed.
- D. R. Baer, P. Munusamy, A. Karakoti, S. Kuchibhatla, S. Seal and S. Thevuthasan, Sustainable Nanotechnology Organization Conference, 2012 Search PubMed.
- B. Djuričić and S. Pickering, J. Eur. Ceram. Soc., 1999, 19, 1925–1934 CrossRef.
- W. Lin, Y.-w. Huang, X.-D. Zhou and Y. Ma, Int. J. Toxicol., 2006, 25, 451–457 CrossRef CAS PubMed.
- E. Matijević and W. P. Hsu, J. Colloid Interface Sci., 1987, 118, 506–523 CrossRef.
- S. Logothetidis, P. Patsalas and C. Charitidis, Mater. Sci. Eng., C, 2003, 23, 803–806 CrossRef.
- L. Yin, Y. Wang, G. Pang, Y. Koltypin and A. Gedanken, J. Colloid Interface Sci., 2002, 246, 78–84 CrossRef CAS PubMed.
- S. V. N. T. Kuchibhatla, A. S. Karakoti, S. Thevuthasan, S. Seal and D. R. Baer, 11th IEEE Conference on Nanotechnology, 2011, 646–650 Search PubMed.
- H.-I. Chen and H.-Y. Chang, Ceram. Int., 2005, 31, 795–802 CrossRef CAS.
- G. R. Bamwenda and H. Arakawa, J. Mol. Catal. A: Chem., 2000, 161, 105–113 CrossRef CAS.
- V. Morris, P. G. Fleming, J. D. Holmes and M. A. Morris, Chem. Eng. Sci., 2013, 91, 102–110 CrossRef CAS.
- A. Paulenova, S. E. Creager, J. D. Navratil and Y. Wei, J. Power Sources, 2002, 109, 431–438 CrossRef CAS.
- R. C. Merrifield, Z. W. Wang, R. E. Palmer and J. R. Lead, Environ. Sci. Technol., 2013, 47, 12426–12433 CrossRef CAS PubMed.
- N. V. Skorodumova, S. I. Simak, B. I. Lundqvist, I. A. Abrikosov and B. Johansson, Phys. Rev. Lett., 2002, 89, 166601 CrossRef CAS PubMed.
- R. Y. Hong, J. H. Li, L. L. Chen, D. Q. Liu, H. Z. Li, Y. Zheng and J. Ding, Powder Technol., 2009, 189, 426–432 CrossRef CAS.
- C. Cayrol, J. Sarraute, R. Tarroux, D. Redoules, M. Charveron and Y. Gall, Br. J. Dermatol., 1999, 141, 250–258 CrossRef CAS PubMed.
- S. R. Pinnell, D. Fairhurst, R. Gillies, M. Mitchnick and N. Kollias, Dermatol. Surg., 2000, 26, 309–314 CrossRef CAS PubMed.
- I. A. Mudunkotuwa, J. M. Pettibone and V. H. Grassian, Environ. Sci. Technol., 2012, 46, 7001–7010 CrossRef CAS PubMed.
- O. Bondarenko, K. Juganson, A. Ivask, K. Kasemets, M. Mortimer and A. Kahru, Arch. Toxicol., 2013, 87, 1181–1200 CrossRef CAS PubMed.
- C. Tso, C. Zhung, Y. Shih, Y. Tseng, S. Wu and R. Doong, Water Sci. Technol., 2010, 61, 127–133 CrossRef CAS PubMed.
- K. T. Kim, S. J. Klaine, J. Cho, S.-H. Kim and S. D. Kim, Sci. Total Environ., 2010, 408, 2268–2272 CrossRef CAS PubMed.
- A. S. Karakoti, P. Munusamy, K. Hostetler, V. Kodali, S. Kuchibhatla, G. Orr, J. G. Pounds, J. G. Teeguarden, B. D. Thrall and D. R. Baer, Surf. Interface Anal., 2012, 44, 882–889 CrossRef CAS PubMed.
- D. G. Rickerby and M. Morrison, Sci. Technol. Adv. Mater., 2007, 8, 19–24 CrossRef CAS.
- G. V. Lowry, K. B. Gregory, S. C. Apte and J. R. Lead, Environ. Sci. Technol., 2012, 46, 6893–6899 CrossRef CAS PubMed.
- S. A. Cumberland and J. R. Lead, J. Chromatogr. A, 2009, 1216, 9099–9105 CrossRef CAS PubMed.
- NIST, New NIST Reference Material Could Aid Nanomaterial Toxicity Research, http://www.nist.gov/mml/mmsd/tio2-090512.cfm.
- L. Koenders, T. Dziomba, P. Thomsen-Schmidt and G. Wilkening, in Nanoscale Calibration Standards and Methods, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 243–258, DOI:10.1002/3527606661.ch18.
- BAM, Nanoscaled Reference Materials, http://www.nano-refmat.bam.de/en/category_10_nanoobjects_nanoparticles_nanomaterials.htm.
- OECD, Testing Programme of Manufactured Nanomaterials – Overview, http://www.oecd.org/chemicalsafety/nanosafety/overview-testing-programme-manufactured-nanomaterials.htm, accessed 23/08/2016.
- M. a. R. K. Hassellöv, Analysis and Characterization of Manufactured Nanoparticles in Aquatic Environments, Environmental and Human Health Impacts of Nanotechnology, John Wiley & Sons, Ltd, 2009 Search PubMed.
- K. Savolainen, U. Backman, D. Brouwer, B. Fadeel, T. Fernandes, T. Kuhlbusch, R. Landsiedel, I. Lynch and L. Pylkkänen, Nanosafety in Europe 2015-2025: Towards Safe and Sustainable Nanomaterials and Nanotechnology Innovations, 2013 Search PubMed.
- M. S. Bakshi, Cryst. Growth Des., 2016, 16, 1104–1133 CAS.
- S. L. Harper, J. L. Carriere, J. M. Miller, J. E. Hutchison, B. L. S. Maddux and R. L. Tanguay, ACS Nano, 2011, 5, 4688–4697 CrossRef CAS PubMed.
- A. Albanese, C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, ACS Nano, 2014, 8, 5515–5526 CrossRef CAS PubMed.
- S. Lin, Y. Zhao, Z. Ji, J. Ear, C. H. Chang, H. Zhang, C. Low-Kam, K. Yamada, H. Meng, X. Wang, R. Liu, S. Pokhrel, L. Madler, R. Damoiseaux, T. Xia, H. A. Godwin, S. Lin and A. E. Nel, Small, 2013, 9, 1776–1785 CrossRef CAS PubMed.
- Z. Ji, Sci. Bull., 2016, 61, 755–771 CrossRef CAS.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. De Grossi, A. Riccioli, H. Amenitsch and A. Lagana, Nanoscale, 2014, 6, 2782–2792 RSC.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1994, 21, 165–176 CrossRef CAS.
- F. Zhou, X. Zhao, H. Xu and C. Yuan, J. Phys. Chem. C, 2007, 111, 1651–1657 CAS.
- J. B. B. R. a. U. M. K. Muruganantham Chelliah, J. Appl. Sci., 2012, 12, 1734–1737 CrossRef.
- A. A. Ansari, P. R. Solanki and B. D. Malhotra, J. Biotechnol., 2009, 142, 179–184 CrossRef CAS PubMed.
- H. M. Ismail, J. Anal. Appl. Pyrolysis, 1991, 21, 315–326 CrossRef CAS.
- N. Topnani, S. Kushwaha and T. Athar, Int. J. Green Nanotechnol., 2010, 1, M67–M73 CrossRef.
- P. Kumbhakar, D. Singh, C. S. Tiwary and A. K. Mitra, Chalcogenide Lett., 2008, 5, 387–394 Search PubMed.
- J. Zhang, J. Liu, Q. Peng, X. Wang and Y. Li, Chem. Mater., 2006, 18, 867–871 CrossRef CAS.
- C. K. Kim, T. Kim, I. Y. Choi, M. Soh, D. Kim, Y. J. Kim, H. Jang, H. S. Yang, J. Y. Kim, H. K. Park, S. P. Park, S. Park, T. Yu, B. W. Yoon, S. H. Lee and T. Hyeon, Angew. Chem., Int. Ed., 2012, 51, 11039–11043 CrossRef CAS PubMed.
- E. G. Heckert, A. S. Karakoti, S. Seal and W. T. Self, Biomaterials, 2008, 29, 2705–2709 CrossRef CAS PubMed.
- S. C. DeCaluwe, Quantifying the Role of Ceria as a Catalyst in Solid Oxide Fuel Cell Anodes, University of Maryland, College Park, 2009 Search PubMed.
- Thermo Fisher Scientific Inc., Zinc, http://xpssimplified.com/elements/zinc.php.
- C. Walker and S. Morton, Auger Parameter Energies – Zinc, http://www.uksaf.org/data/zn.html, accessed 03/03/2016.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- T. Manoubi, C. Colliex and P. Rez, J. Electron Spectrosc. Relat. Phenom., 1990, 50, 1–18 CrossRef CAS.
- L. A. J. Garvie and P. R. Buseck, J. Phys. Chem. Solids, 1999, 60, 1943–1947 CrossRef CAS.
- L. Guo, S. Yang, C. Yang, P. Yu, J. Wang, W. Ge and G. K. L. Wong, Chem. Mater., 2000, 12, 2268–2274 CrossRef CAS.
- N. Arsalani, H. Fattahi and M. Nazarpoor, eXPRESS Polym. Lett., 2010, 4, 329–338 CrossRef CAS.
- S. Kittler, C. Greulich, M. Köller and M. Epple, Materialwiss. Werkstofftech., 2009, 40, 258–264 CrossRef CAS.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- H. Zhang, W. Wang, B. Zhang, H. Li and Q. Zhang, RSC Adv., 2016, 6, 30956–30962 RSC.
- T. Xia, M. Kovochich, M. Liong, L. Mädler, B. Gilbert, H. Shi, J. I. Yeh, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 2121–2134 CrossRef CAS PubMed.
- H. Zhang, Z. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. Lin, X. Wang, Y.-P. Liao, M. Wang, L. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Mädler, Y. Cohen, J. I. Zink and A. E. Nel, ACS Nano, 2012, 6, 4349–4368 CrossRef CAS PubMed.
- G. Pulido-Reyes, I. Rodea-Palomares, S. Das, T. S. Sakthivel, F. Leganes, R. Rosal, S. Seal and F. Fernández-Piñas, Sci. Rep., 2015, 5, 15613 CrossRef CAS PubMed.
- G. H. Chan, J. Zhao, E. M. Hicks, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2007, 7, 1947–1952 CrossRef CAS.
- M. Fernández-García and J. A. Rodriguez, in Encyclopedia of Inorganic Chemistry, John Wiley & Sons, Ltd, 2006, DOI:10.1002/0470862106.ia377.
- T. Puzyn, B. Rasulev, A. Gajewicz, X. Hu, T. P. Dasari, A. Michalkova, H.-M. Hwang, A. Toropov, D. Leszczynska and J. Leszczynski, Nat. Nanotechnol., 2011, 6, 175–178 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25064e |
| This journal is © The Royal Society of Chemistry 2017 |