
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmphiphilic electrospun scaffolds of PLLA–PEO–PPO block copolymers: preparation, characterization and drug-release behaviour†
Lívia M. D. Loiolaa,
Pablo R. Cortez Tornellob,
Gustavo A. Abrahamb and
Maria I. Felisberti*a
aInstitute of Chemistry, University of Campinas – UNICAMP, P. O. Box 6154, 13083-970, Campinas, São Paulo, Brazil. E-mail: misabel@iqm.unicamp.br
bResearch Institute of Materials Science and Technology, INTEMA (UNMdP – CONICET), Av. J. B. Justo 4302, B7608FDQ, Mar del Plata, Argentina
First published on 23rd December 2016
Abstract
Biocompatible amphiphilic copolymers are attractive candidates for the fabrication of electrospun scaffolds to be used for tissue engineering and the delivery of biologically active compounds. The amphiphilic block copolymers poly(L-lactide)-b-poly(ethylene oxide)-b-poly(L-lactide) (PELA) and poly(L-lactide)-b-poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide)-b-poly(L-lactide) (PEPELA), which contain amorphous/crystalline and hydrophilic/hydrophobic blocks, were synthesized by ring-opening polymerization, and then electrospun to obtain non-woven fibrous scaffolds. Acetaminophen (AC) and celecoxib (CL) were used as hydrophilic and hydrophobic model drugs, respectively, to prepare drug-loaded scaffolds. The pure and drug-loaded scaffolds present a morphology characterized by randomly oriented fibres with integrated beads. The drug encapsulation and release profiles were determined by ultraviolet-visible spectroscopy. Due to the amphiphilic nature of PELA and PEPELA, both hydrophilic and hydrophobic drugs could be entrapped within the polymeric scaffolds, allowing the design of drug-delivery systems for specific applications. The combination of confocal Raman spectroscopy mapping and dynamic mechanical analysis revealed that the AC drug is preferentially maintained in the PEO phase, while the CL drug is fractionated between polyether and polyester phases and distributed throughout the fibrous structure due to its hydrophobic character.
Introduction
Electrospun fibrous scaffolds fabricated from biocompatible polymers have recently attracted great interest for biomedical applications, including drug delivery,1–3 tissue engineering,4,5 wound healing6,7 and implantation,8 due to their biomimetic structure that can support cell functions (adhesion, proliferation, differentiation, migration) and nutrient transport.9,10The properties of the electrospun scaffolds, such as hydrophilicity, cell viability, mechanical modulus and strength, are determined by the chemical composition of the polymer and by the scaffold structure.11 Poly(L-lactide) (PLLA) is one of the most used polymers in electrospinning for the biomedical field. However, the high hydrophobic and crystalline character of PLLA limit the bioabsorption and the cell affinity, and consequently its usage for in vivo applications.12 PLLA copolymerization arises as an alternative for combining polymer components to adjust material composition and architecture, hydrophilicity, cell affinity, mechanical properties, morphology and degradation rate. For example, biocompatible PLLA-b-PEO-b-PLLA (PELA) and PLLA-b-PEO-b-PPO-b-PEO-b-PLLA (PEPELA) block copolymers containing poly(ethylene oxide) and poly(ethylene oxide) (PEO)/poly(propylene oxide) (PPO) triblock copolymer (PEO-b-PPO-b-PEO) polyethers as the flexible, amorphous and hydrophilic central blocks, and PLLA as the hard and hydrophobic end blocks are potential materials to drug delivery devices.13
The electrospinning of copolymers based on flexible/hard and hydrophilic/hydrophobic blocks has been described. Thieme et al. prepared electrospun scaffolds from PLA-b-PEO-b-PLA triblock copolymers with central PEO blocks of 35 and 100 kDa and poly(D,L-lactide) (PLA) blocks varying between 10 and 150 kDa. The morphology of the electrospun fibres depended on block lengths, and PLA remained amorphous, while PEO was able to crystallize. The fibres electrospun from the copolymer with PEO 100 kDa were used for inhalation therapy.14 Trinca et al. prepared electrospun scaffolds from segmented and amphiphilic polyurethanes that combine PEO, PLLA and poly(trimethylene carbonate) (PTMC) blocks.15 The scaffolds with randomly oriented fibres presented overall properties dependent on polyurethanes composition, combining the intrinsic properties of each block, such as the hydrophilicity of PEO, stiffness of PLLA and elasticity of PTMC.15,16 Additionally, electrospun scaffolds based on PLLA/PEO/PPO block copolymers were evaluated by Guo et al. regarding their cytotoxicity and cell adhesion, showing to be suitable materials for tissue engineering.17
Electrospinning is an efficient technique to incorporate hydrophobic and/or hydrophilic therapeutic drugs within scaffolds of amphiphilic copolymers by using different strategies, such as blending, coaxial electrospinning, surface adsorption or surface immobilization.1 Electrospun scaffolds from PLA-b-PEO-b-PLA copolymers, whose morphology, water contact angle and in vitro degradation rates were modulated by adjusting the copolymer composition, were used as metronidazole delivery systems against post-surgical infections.12,18 PEO-co-PLA paracetamol-loaded scaffolds presented morphology and water uptake capacity that could be tailored by adjusting the electrospinning parameters, the chemical composition and the molar mass of the copolymers. The hydrophilic drug/polymer interaction and the copolymer degradation rate controlled the sustained paracetamol release.19 Gaharwar et al. reported the encapsulation and the release of dexamethasone, a model hydrophobic drug, within a electrospun beaded scaffold based on the amphiphilic copolymer poly(ethylene oxide terephthalate)–poly(butylene terephthalate) (PEOT/PBT). The amphiphilic beads act as depots integrated to the fibres for the sustained release of dexamethasone by diffusion during 28 days.20
Besides the above-mentioned studies about PLLA and PLLA–PEO copolymers, the comparison between electrospun scaffolds based on the amphiphilic block copolymers PLLA-b-PEO-b-PLLA (PELA) and PLLA-b-PEO-b-PPO-b-PEO-b-PLLA (PEPELA) has not been previously reported. The main difference of these copolymers is the middle blocks, which lead to different hydrophilic/hydrophobic balance, water swelling capacity and surface water contact angle.13 PELA is more hydrophilic compared to PEPELA and this difference is expected to influence the copolymer/drug affinity. Thus, this work aims to study the load and release of hydrophilic and hydrophobic model drugs from electrospun scaffolds as well as the influence of these drugs on their physical–chemical and morphological properties. PELA and PEPELA copolymers were synthesized, characterized and electrospun to amphiphilic fibrous scaffolds, which were produced using the celecoxib (CL) and acetaminophen (AC) drugs. CL is a nonsteroidal anti-inflammatory drug used for the treatment of osteoarthritis, rheumatoid arthritis and acute pain,21 and it was used as hydrophobic model drug due to its very low aqueous solubility (5 μg mL−1).22 AC is widely used as an antipyretic and analgesic, and it was selected as hydrophilic model drug due to its high aqueous solubility (14.3 mg mL−1).23
The chemical structure and molar mass of the copolymers were characterized by proton nuclear magnetic resonance (1H NMR) and gel permeation chromatography (GPC). The morphology of the amphiphilic scaffolds was examined by scanning electron microscopy (SEM). Drugs and neat and drug-loaded scaffolds were characterized by X-ray diffraction (XRD) and differential scanning calorimetry (DSC). The combination of the dynamic mechanical analysis (DMA) and confocal Raman spectroscopy techniques was used to investigate the drug distribution. The drugs release kinetics in phosphate-buffered saline (PBS, pH = 7.4) at 37 °C was also studied.
Experimental
Materials
L-Lactide (LLA, Sigma-Aldrich), poly(ethylene oxide) (PEO, Mn = 8 kg mol−1, Mw/Mn = 1.1, Fluka) and poly(ethylene oxide-block-propylene oxide-block-ethylene oxide) copolymer (PEO-b-PPO-b-PEO, 80 wt% of PEO, Mn = 8 kg mol−1, Mw/Mn = 1.1, Sigma-Aldrich) were freeze-dried before use. Tin(II) 2-ethylhexanoate (Sn(Oct)2, Sigma-Aldrich) was used as a catalyst without previous purification. The solvents N,N-dimethylformamide (DMF, 99.8%, ACS, Sigma-Aldrich), chloroform (CHCl3, 99.9%, Biopack) and diethyl ether (PA ACS, Synth) were used as received, and toluene (PA ACS, Synth) was dried over 5 Å molecular sieves (Sigma-Aldrich). Acetaminophen (AC, N-(4-hydroxyphenyl)ethanamide, C8H9NO2, powder, Sigma-Aldrich) and celecoxib (CL, 4-[5-(4-methylphenyl)-3-(trifluoromethyl)pyrazol-1-yl]benzenesulfonamide, C17H14F3N3O2S, powder, Parafarm) were used as received.Synthesis and characterization of amphiphilic copolymers
Copolymers were synthesized by ring-opening polymerization (ROP) in anhydrous toluene solutions (mcopolymer/Vtoluene = 0.1 g mL−1) under argon atmosphere, as previously described by the authors.13 Briefly, appropriate amounts of PEO or PEO-b-PPO-b-PEO macroinitiators and Sn(Oct)2 catalyst (ncatalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nmacroinitiator = 2:1)24 were solubilized and heated to the reflux (toluene, bp 110 °C) with continuous stirring for 1 h in a round-bottom flask in a pre-activation step. Subsequently, LLA was added, and the system was kept at toluene reflux for 44 h. The resulting PLLA-b-PEO-b-PLLA (PELA) and PLLA-b-PEO-b-PPO-b-PEO-b-PLLA (PEPELA) block copolymers were precipitated in cold diethyl ether (Vsolution:Vether = 1:10), filtered and dried under vacuum at 40 °C for 48 h. Yields of 90%.
:nmacroinitiator = 2:1)24 were solubilized and heated to the reflux (toluene, bp 110 °C) with continuous stirring for 1 h in a round-bottom flask in a pre-activation step. Subsequently, LLA was added, and the system was kept at toluene reflux for 44 h. The resulting PLLA-b-PEO-b-PLLA (PELA) and PLLA-b-PEO-b-PPO-b-PEO-b-PLLA (PEPELA) block copolymers were precipitated in cold diethyl ether (Vsolution:Vether = 1:10), filtered and dried under vacuum at 40 °C for 48 h. Yields of 90%.
1H NMR spectra of the macroinitiators esterified with trifluoroacetic anhydride and the PELA and PEPELA copolymers solutions in deuterated chloroform (chloroform-d 99.8%, containing 0.05% of the internal standard tetramethylsilane, TMS, Cambridge Isotope Laboratories, Inc.) (15 mg in 0.7 mL of chloroform-d) were recorded at room temperature on an Avance AC/P 400 MHz spectrometer, Bruker.
Gel permeation chromatography (GPC) was performed on a Viscotek GPCmax VE2001 instrument equipped with three Shodex KF-806M columns and Viscotek VE3580 refractive index and Viscotek UV2500 detectors. The columns system was kept at 40 °C, and THF was used as the eluent at a flow rate of 1 mL min−1. Molar masses were calculated using polystyrene standards with molar masses from 1050 to 3800000 g mol−1 (Viscotek).
Preparation of electrospun scaffolds
PELA and PEPELA 20 wt% solutions were prepared by dissolving copolymers in a CHCl3:DMF solvent mixture (90:10 by volume) under stirring at room temperature. The drugs were added to the polymeric solution at the copolymer/drug ratio of 2.5, 5.0 and 10.0 wt%. These amounts were selected to ensure the complete drug dissolution. The designed drug contents in the spinning solutions is expressed as the mass fraction xDi = mDi/mPi, where mDi is the mass of the drug and mPi is the copolymer mass in the initial solutions. Fibrous scaffolds were obtained by electrospinning at temperature of 20 °C and relative humidity of 50% in a chamber having a ventilation system. Solutions were loaded into a standard 10 mL plastic syringe connected to a polyamide tube attached to the open end of 18-gauge, stainless steel blunt needle used as a nozzle. The flow rate was controlled by a syringe pump (Activa A22, ADOX S.A.), and a high-voltage power source (ES30P, Gamma High Voltage Research Inc.) was used to charge the solution by attaching the positive electrode to the nozzle and the grounding electrode to an aluminium collector plate. The polymeric solutions were electrospun at a positive high voltage of 17 kV for PELA and of 15 kV for PEPELA copolymer, with a 12 cm distance between the nozzle tip and the collector plate, and with a solution flow rate of 1.0 mL h−1. The electrospun scaffolds were dried under vacuum at room temperature. PELA scaffolds loaded with acetaminophen and celecoxib were denoted as PELA–ACx and PELA–CLy, respectively. PEPELA scaffolds loaded with the same drugs were similarly named as PEPELA–ACx and PEPELA–CLy, where the indexes x and y indicate the initial drug mass fraction xDi of 2.5, 5.0 or 10.0 wt%.
Electrospun scaffolds and drugs characterization
The morphology of the electrospun scaffolds was examined by scanning electron microscopy on a JEOL JSM6460 LV microscope (Peabody, MA, USA) after gold sputtering. Micrographs were analysed using Image-Pro Plus software (Media Cybernetics Inc., USA). Mean beaded fibre diameters and their distribution were obtained by measuring at least 100 fibres by sample.XRD measurements were carried out in a diffractometer Shimadzu XRD-7000. The radiation source (Cu Kα X-ray, λ = 0.1542 nm) was operated at 40.0 kV and 30.0 mA, with the scanning angle ranging from 5 to 50°. The instrument was operated in the fixed time scan mode with 2θ increments of 0.1°, and counts were accumulated for 10 s at each step.
DSC analyses were performed on a TA Instrument MDSC 2910 operating at a 20 °C min−1 heating rate. Samples were heated from −100 to 200 °C (1st heating), kept at 200 °C for 2 min, cooled from 200 °C to −100 °C (cooling), kept at −100 °C for 10 min, and heated again from −100 °C to 200 °C (2nd heating). DSC thermograms were normalized with respect to sample mass.
Rectangular samples with dimensions of approximately 15 × 6 × 0.8 mm3 were analysed by DMA on a Rheometric Scientific DMTA V with a 0.1% strain at a 1 Hz using a film tension fixture with a 9 mm gap. The dynamic mechanical properties of scaffolds were recorded during heating at 2 °C min−1 from −100 °C to 150 °C.
Raman analyses were performed using a Horiba Jobin-Yvon T64000 comprising a micro-Raman spectrometer and an integrated Olympus BX41 confocal microscope. Raman spectra were generated using a 514 nm laser diode as excitation source, focused on the sample with a 50× objective, a slit of 100 μm and an exposure time of 10 s for AC and CL drugs, and 30 s for the PELA and PEPELA pure scaffolds. The exposition times were selected to maximize the Raman signals corresponding to drugs and copolymers. Each spectrum was averaged over 2 scans. Imaging experiments of the drug-loaded PELA–AC10, PELA–CL10, PEPELA–AC10 and PEPELA–CL10 scaffolds were performed by scanning the laser beam over a region of interest and accumulating a full Raman spectrum at each selected point. Raman images were carried out using the LABSPEC5 software (Horiba Jobin-Yvon) and they consist in the integrated intensity plotting of the vibrational bands related to drugs and copolymers as a function of position. For these experiments, the mapping of 42 points equally spaced by the step size of 5 μm within an area of 750 μm2 was acquired with 60 s of exposition time and over 2 scans.
Final encapsulated levels of AC and CL in the scaffolds were determined by ultraviolet-visible spectroscopy using an Agilent 8453 spectrometer (Santa Clara, CA, USA) equipped with a diode-array system. A predetermined mass of sample (m = mDf + mPf, where mDf is the mass of the encapsulated drug and mPf is the copolymer mass in the scaffolds samples) was dissolved in CHCl3:DMF solvent mixture (90:10 by volume), and the absorption band at 260 nm for both drugs was analysed. The encapsulation efficiency (EE) was calculated as:
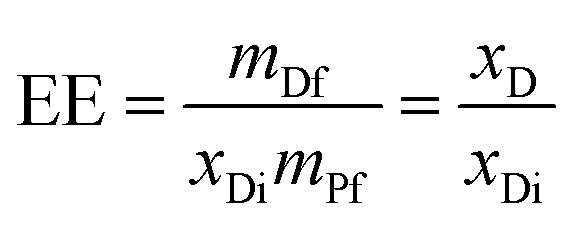 | (1) |
Disc samples for drug release (at least 10 discs of 10 mm of diameter and 0.8 mm of height) were placed in glass flasks containing 100 mL of phosphate-buffered saline (PBS, pH = 7.4) at 37 °C. All experiments were performed in triplicate. Stirring was performed with an orbital shaker at 150 rpm. Samples of 1 mL were taken from the dissolution medium each 1 h during the first 12 h, and one sample per day was taken during 14 days. A total of 26 extractions in each flask were performed, and, after each extraction, 1 mL of fresh PBS was added. The concentration of AC and CL drugs was determined by ultraviolet-visible spectroscopy (UV-Vis 8453, Agilent) measuring the absorption bands at 253 and 251 nm, respectively. The results were plotted as cumulative drug release as a function of time. Cumulative drug release was calculated from ratio of the amount of drug released at each sampling time to the initial loading content.
Results & discussion
The molecular characteristics of PEO and PEO-b-PPO-b-PEO polyether macroinitiators as well as PELA and PEPELA amphiphilic copolymers as obtained by 1H NMR and GPC (Fig. S1 and S2 of the ESI†) are described in Table 1. PELA and PEPELA copolymers present similar molar mass, molar mass distribution and polyether/PLLA mass ratio. The main difference of both copolymers is the insertion of PPO block, which present an intermediate hydrophilicity compared to PLLA and PEO.| Polymer | Reaction medium | Final composition of copolymers | ||||
|---|---|---|---|---|---|---|
| nLLA/nEO+POa | nLLA/nEO+POb | PEO:PPO:PLLA mass ratiob |
Mnb (g mol−1) | Mnc (g mol−1) | Mw/Mnc (—) | |
| a LLA/EO + PO molar ratio.b Determined by 1H NMR.c Determined by GPC. | ||||||
| PEO | — | — | 100:0:0 |
8000 | 11500 |
1.1 |
| PEO-b-PPO-b-PEO | — | — | 80:20:0 |
8000 | 11000 |
1.1 |
| PELA | 1 | 0.9 | 26:0:74 |
31000 |
35000 |
1.4 |
| PEPELA | 1 | 0.9 | 22:5:73 |
31000 |
34000 |
1.5 |
The electrospinning conditions used in this work were selected after a series of screening experiments varying the concentration of the polymer solution (from 5 to 25 wt%), the CHCl3:DMF mixture ratio (from 100:0 to 70:30 volume ratio), the applied voltage (from 10 to 20 kV) and the nozzle tip-to-collector distance (from 10 to 20 cm). Scaffolds with continuous beaded fibres were obtained from neat and drug-loaded copolymer solutions at 12 cm of distance; voltage of 17 kV for PELA and of 15 kV for PEPELA, and therefore, these conditions were adopted to prepare the scaffolds.
Randomly oriented continuous beaded fibres were observed by SEM throughout the structure of all the scaffolds (Fig. 1). Mean fibre diameters (D) of PELA and PEPELA were found in the range of 400–530 nm, as shown in Table 2, being the fibres and beads diameters smaller for PEPELA scaffolds. In general, the viscoelastic properties of the solution, charge density carried by the microjet, and the surface tension of the solution are the key factors that determine the formation of beads, beaded or uniform fibres.25 According to Thieme et al., the PEO/PLA block mass ratio affects the spinnability behaviour of PLA-b-PEO-b-PLA copolymer. Decreasing the PEO/PLA block ratio (i.e., decreasing the hydrophilicity) leads to beads formation and to reduction of fibres diameter, because of the reduction of conductivity and viscosity of the CHCl3 spinning solutions.14
 | ||
| Fig. 1 SEM micrographs showing the effect of AC and CL concentration on fibre diameter and morphology of the scaffolds (a) PELA, (b) PELA–AC2.5, (c) PELA–AC5, (d) PELA–AC10, (e) PELA–CL2.5, (f) PELA–CL5, (g) PELA–CL10, (h) PEPELA, (i) PEPELA–AC2.5, (j) PEPELA–AC5, (k) PEPELA–AC10, (l) PEPELA–CL2.5, (m) PEPELA–CL5 and (n) PEPELA–CL10. The white scale bars in all images correspond to 10 μm. | ||
| Sample | xDi (%) | EE (%) | xD (%) | AC per PEO/PPOa (%) | CL per PLLAb (%) | D ± s.d. (nm) | RD (—) |
|---|---|---|---|---|---|---|---|
| a Amount of AC drug per polyether PEO and PPO fraction in the copolymer.b Amount of CL drug per polyester PLLA fraction in the copolymer. | |||||||
| PELA | — | — | — | — | — | 527 ± 191 | — |
| PELA–AC2.5 | 2.500 ± 0.005 | 88.2 ± 0.3 | 2.2 ± 0.1 | 8.5 | — | 1007 ± 344 | 1.91 |
| PELA–AC5 | 5.000 ± 0.005 | 61.6 ± 0.2 | 3.1 ± 0.2 | 11.8 | — | 1171 ± 414 | 2.22 |
| PELA–AC10 | 10.000 ± 0.005 | 31.5 ± 0.1 | 3.2 ± 0.2 | 12.1 | — | 1330 ± 469 | 2.52 |
| PELA–CL2.5 | 2.500 ± 0.005 | 7.1 ± 0.2 | 0.20 ± 0.01 | — | 0.2 | 783 ± 133 | 1.49 |
| PELA–CL5 | 5.000 ± 0.005 | 18.3 ± 0.1 | 0.90 ± 0.05 | — | 1.2 | 857 ± 285 | 1.63 |
| PELA–CL10 | 10.000 ± 0.005 | 14.3 ± 0.1 | 1.40 ± 0.07 | — | 1.9 | 1050 ± 363 | 1.99 |
| PEPELA | — | — | — | — | — | 407 ± 152 | — |
| PEPELA–AC2.5 | 2.500 ± 0.005 | 91.7 ± 0.3 | 2.3 ± 0.2 | 8.5 | — | 510 ± 156 | 1.25 |
| PEPELA–AC5 | 5.000 ± 0.005 | 61.7 ± 0.2 | 3.1 ± 0.2 | 11.4 | — | 634 ± 103 | 1.58 |
| PEPELA–AC10 | 10.000 ± 0.005 | 41.9 ± 0.1 | 4.2 ± 0.2 | 15.5 | — | 758 ± 262 | 1.86 |
| PEPELA–CL2.5 | 2.500 ± 0.005 | 70.2 ± 0.2 | 1.8 ± 0.1 | — | 2.4 | 624 ± 112 | 1.53 |
| PEPELA–CL5 | 5.000 ± 0.005 | 59.4 ± 0.1 | 3.0 ± 0.2 | — | 4.1 | 652 ± 229 | 1.60 |
| PEPELA–CL10 | 10.000 ± 0.005 | 46.3 ± 0.1 | 4.6 ± 0.2 | — | 6.3 | 815 ± 239 | 2.02 |
Since PELA and PEPELA present similar molar mass and molar mass distribution (Mn = 31 kg mol−1, Mw/Mn = 1.4 and 1.5 for PELA and PEPELA, respectively), the morphological differences of the respective scaffolds is also due to the differences of the copolymer composition, and, consequently, of the hydrophilic–hydrophobic balance. Apparently, the higher hydrophilicity of PELA results in higher fibre and beads diameters.
The addition of AC or CL drugs to PELA and PEPELA results in changes in the fibre diameter, and in number, size and shape of the beads of the scaffolds (Table 2 and Fig. S4 of the ESI†). For PELA–ACx series, the increment of AC contents leads to an increase of the fibres diameter (D) and of the relative fibre diameter (RD = Dcopolymer+drug/Dcopolymer) of 191 to 252% in comparison to neat PELA scaffold (Table 2), and the beads shape changes from almost spherical to ellipsoidal, while the amount of beads decreases (Fig. 1a, c and d). CL drug also induces an increase in the diameter of PELA–CLy fibres (RD of 149 to 199% in comparison to neat PELA scaffold – Table 2). However, this effect is less pronounced compared to PELA–ACx series. Moreover, the number, shape and size of beads remain almost unchanged (Fig. 1a, f and g). The increment of the fibres diameters in PEPELA–ACx series with the increment of drug concentration is less pronounced in comparison to PELA–ACx. On the other hand, the relative increment of the fibre diameter of PELA–CLy and PEPELA–CLy series lies in the same range (approximately 1.5 to 2.0). Moreover, the drug nature (AC or CL) influence on the PEPELA scaffolds morphology is much less important compared to PELA scaffolds.
The encapsulation efficiency (EE, eqn (1)) depends on the combination of drug and polymer of each particular system, where the hydrophilic/hydrophobic characteristics of each component play an important role. The final drug mass fraction (xD) and EE values are listed in Table 2. In both PELA and PEPELA scaffolds, the highest EE values of approximately 90% were found for samples containing 2.5% AC.
It is noteworthy that PEPELA is capable to solubilize both AC and CL drugs despite their hydrophilicity differences. Apparently, PEPELA is less selective for drug nature compared with PELA, being observed a rejection of the hydrophobic CL drug for the last copolymer. As previously reported,13 the substitution of approximately 5 wt% of PEO for PPO segments increases the surface water contact angle (from 40° to 46°), indicating an increase in the copolymer hydrophobicity, which improves the solubility of CL.
Fig. 2a shows the drug mass fraction (xD) in the scaffolds as a function of the drug mass fraction in the initial solution (xDi). A strong deviation is observed from the linearity (dotted line) as the amount of the drugs in the solution increases. Moreover, for PELA–ACx and PELA–CLy series, an asymptotic behaviour is observed. On the other hand, for the PEPELA–ACx and PEPELA–CLy series, in the studied drug concentration range, the drug mass fraction in the scaffolds increases without achieving a maximum drug load. This behaviour suggests a lower miscibility of the drugs in the PELA compared to PEPELA. In the electrospinning process, the polymer/drug solution is ejected from a needle, resulting in a polymer chain stretching in the flow direction while the solvent evaporates. During these processes, part of the drug is rejected from the copolymer as consequence of the low miscibility.
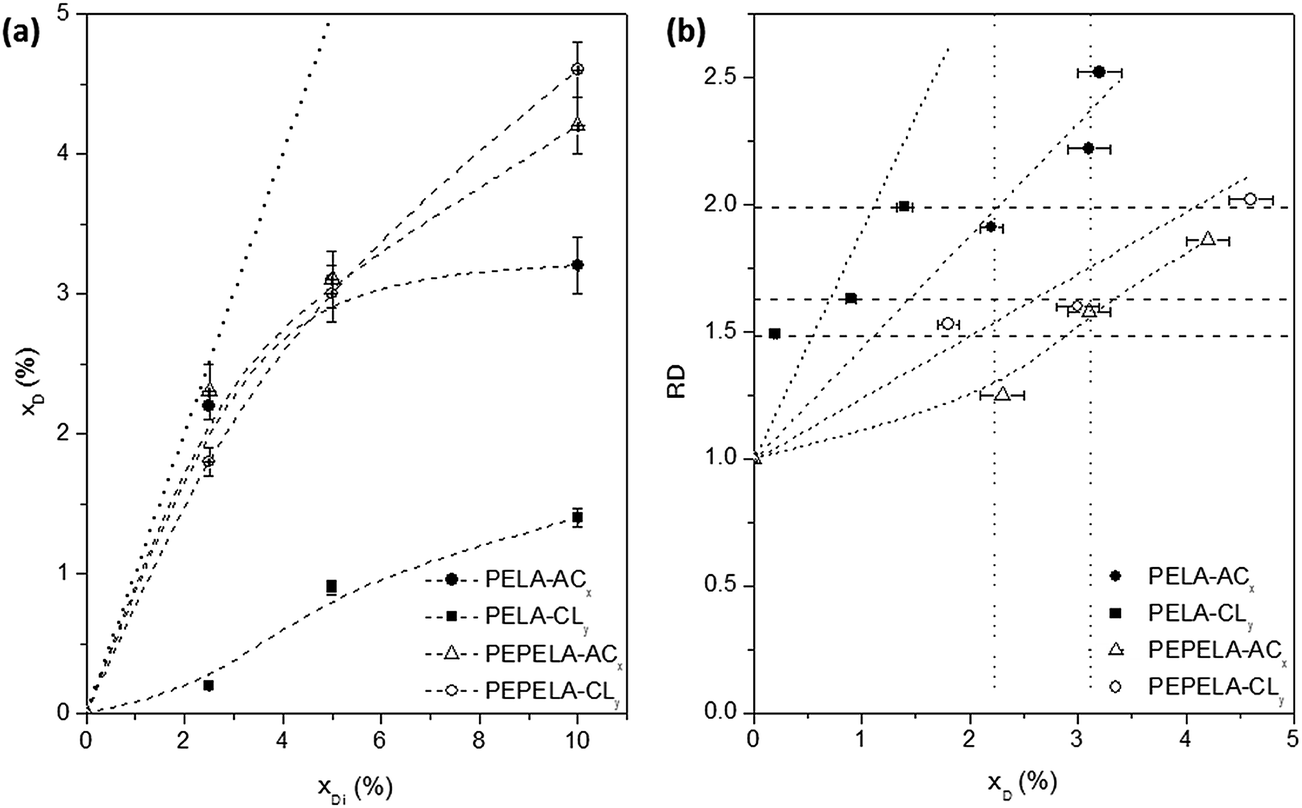 | ||
| Fig. 2 (a) Final drug mass fraction in the scaffolds (xD) as a function of the initial mass fraction of the drug in the spinning solutions (xDi) for (●) PELA–ACx, (■) PELA–CLy, (△) PEPELA–ACx and (○) PEPELA–CLy series. The dashed line depicts the hypothetic linear function (xD = xDi) for 100% of encapsulation efficiency. (b) Relative fibre diameter (RD) as a function of xD for (●) PELA–ACx, (■) PELA–CLy, (△) PEPELA–ACx and (○) PEPELA–CLy series. | ||
The influence of drug loading on the morphology of the electrospun fibres has been described in the literature. Electrospun poly(ε-caprolactone) (PCL) fibres containing ibuprofen and carvedilol, reported by Potrč et al., presented an increase of the average fibre diameters from 465 to 686 nm, when the amount of ibuprofen varied from 10 to 60 wt%, and from 418 to 756 nm for the same amount range of carvedilol.26 The same trend was observed due to the AC and CL addition to PELA and PEPELA scaffolds, as discussed above. On the other hand, Li et al. reported that the addition up to 10 wt% of CL to PLLA-b-PEO diblock copolymer, resulted in uniform fibres, whose diameter decreased with increasing the amount of CL (from 3.52 to 3.21 μm).27
The relative fibre diameter (RD) of PELA and PEPELA presents a complex dependence on the drug content (xD) in the fibres, as showed in Fig. 2b. This parameter allows analysing the effect of the drug on the fibre morphology. For example, fibres containing CL drug present relative increment between 1.5 and 2.0 (horizontal dashed lines in Fig. 2b), however CL mass fraction in the scaffolds is quite different (maximum of 1.4 and 4.6 for PELA and PEPELA, respectively – Table 2). On the other hand, the mass fraction of AC drug in the scaffolds varies in the same range (see vertical dotted lines in Fig. 2b) and the relative increment of the fibre diameter is higher for PELA matrix. These results show that the fibre characteristics clearly depend on polymer, drug and solvent composition and concentration, and they further depend on the processing technique.
Fig. 3 [diffractograms 3a(V) and 3b(V)] shows the XRD patterns corresponding to AC and CL powders. The main XRD peaks for AC were found at 13.8, 15.5, 18.2, 20.2, 23.5, 24.5, 26.5, 27.3, 31.2 and 32.5°, and these signals are in agreement to the diffraction pattern of the monoclinic crystalline acetaminophen, or type I.28 Celecoxib presented the main XRD peaks at 5.35, 10.7, 16.2, 19.7, 21.6, 22.2 and 29.6°, in agreement with the pattern of the thermodynamically stable crystal form of celecoxib at ambient conditions, or type III.22,29
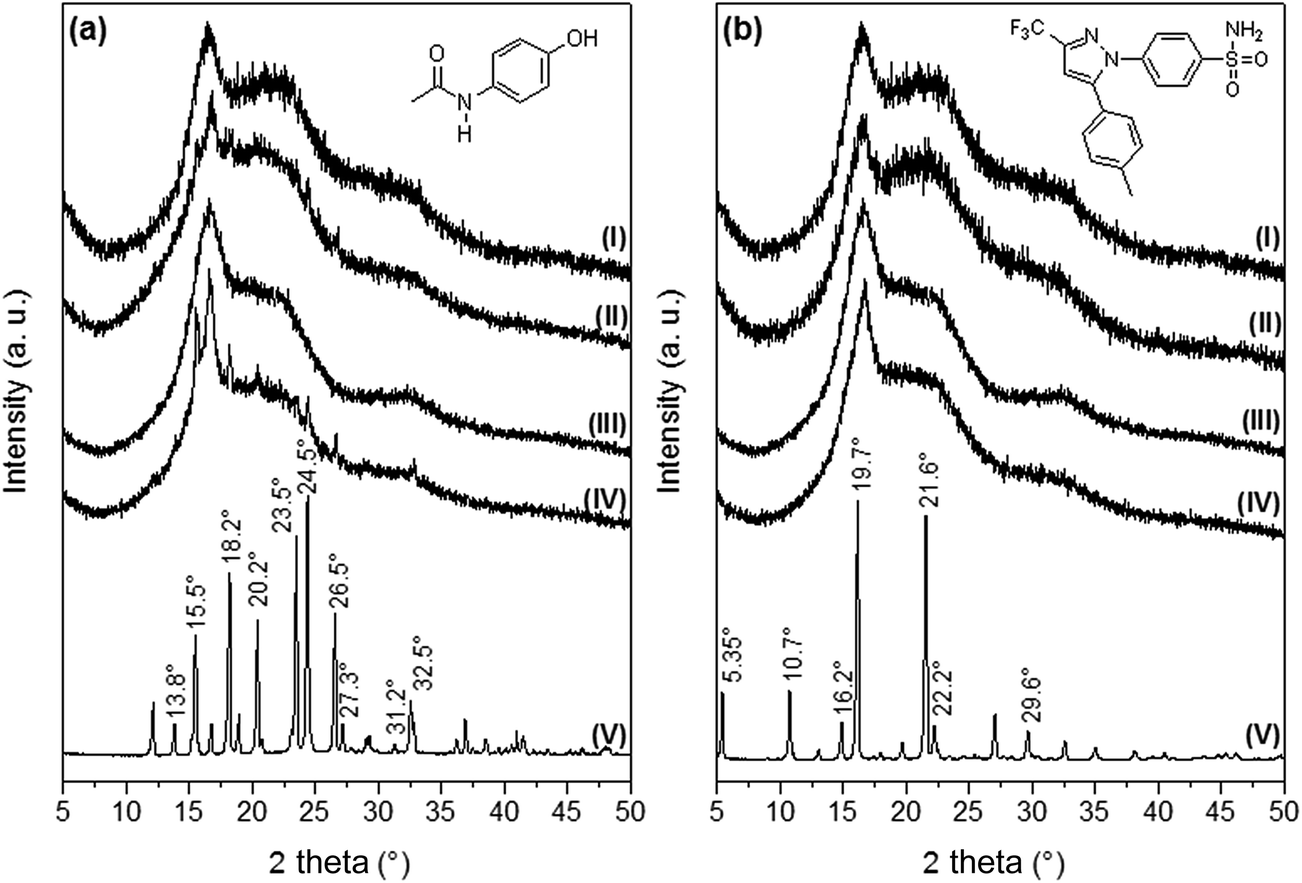 | ||
| Fig. 3 X-ray diffractograms of neat and drug-loaded PELA and PEPELA scaffolds. (a) AC series and (b) CL series: (I) neat PELA; (II) PELA scaffold loaded with 10 wt% of drug; (III) neat PEPELA; (IV) PEPELA scaffold loaded with 10 wt% of drug; (V) neat drug. The chemical structures of AC and CL drugs are also depicted. | ||
XRD patterns of PELA and PEPELA unloaded scaffolds are depicted in Fig. 3 – diffractograms (I) and (III), respectively. The diffractograms of the neat copolymers present peaks at 16.7° and 22°, corresponding to characteristics peaks of PLLA crystalline structure X-ray diffraction.30 The XRD patterns of PELA and PEPELA scaffolds loaded with 10 wt% of AC drug (Fig. 3a – diffractograms (II) and (IV), respectively) and the PELA and PEPELA scaffolds loaded with 10 wt% of CL drug (Fig. 3b – diffractograms (II) and (IV), respectively) show characteristic peaks for the drugs and copolymers. However, the characteristics peaks of AC and CL were absent in the scaffolds with lower drug content (PELA–AC2.5, PELA–AC5, PELA–CL2.5, PELA–CL5, PEPELA–AC2.5, PEPELA–AC5, PEPELA–CL2.5, PEPELA–CL5 – Fig. S3 of the ESI†), suggesting either that the AC and CL were solubilized within the polymer structure as a solid solution or that the low amounts of the drugs could not be detected by XRD technique.
The effect of the addition of AC and CL on the crystallization and relaxations of the copolymers in the electrospun scaffolds was investigated using differential scanning calorimetry (DSC) and dynamic-mechanical analysis (DMA), respectively. The evaluation of the crystallization, melting and glass transitions for drugs and scaffolds was performed on DSC 1st heating, cooling and 2nd heating scans (Fig. 4, S5 and Table S1 of the ESI†). In the 1st heating, AC type I and CL type III showed melting temperatures (Tm,onset) of 171 °C and 162 °C, respectively.
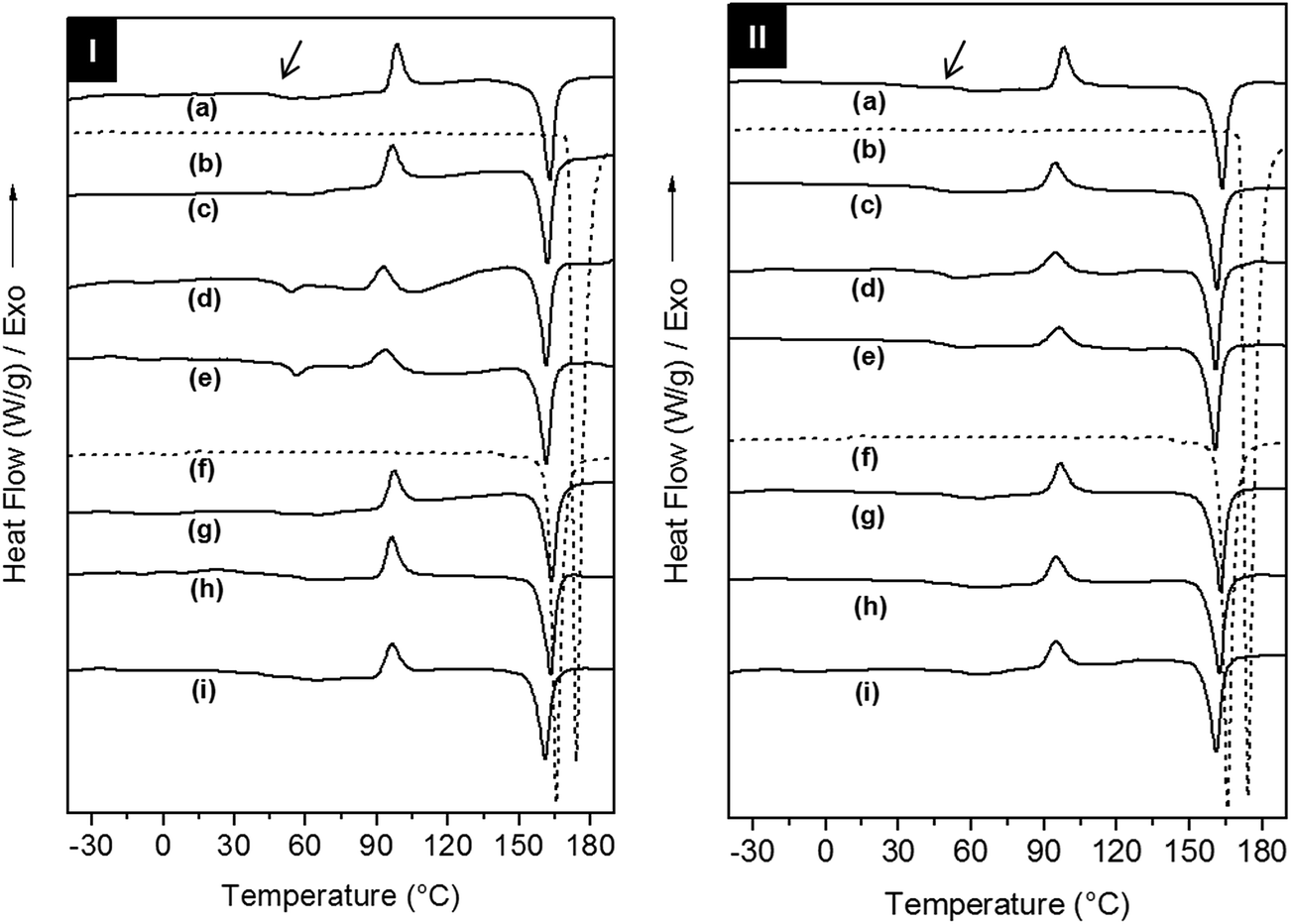 | ||
| Fig. 4 DSC 1st heating curves at 20 °C min−1 of (I) PELA and (II) PEPELA scaffolds: (a) without drug, acetaminophen-loaded scaffolds (c) AC2.5, (d) AC5, (e) AC10 and celecoxib-loaded scaffolds (g) CL2.5, (h) CL5, (i) CL10. The DSC curves of (b) acetaminophen and (f) celecoxib drugs are also depicted. | ||
Drug phase transitions were not observed in the scaffolds DSC 1st heating curves even for the in drug richest formulations. According to Yu and co-workers the quick solvent evaporation and fibres formation in electrospinning process decrease the drug mobility and hinder its crystallization, and thus the amorphous drug is solubilized within the copolymer fibres.31
PELA and PEPELA are heterogeneous and semicrystalline copolymers and both phases of the copolymers, polyether (PEO or PEO-b-PPO-b-PEO) and polyester (PLLA), can crystallize. However, DSC 1st heating curves for PELA and PEPELA scaffolds (Fig. 4) revealed a crystallization peak at 98 °C, due to PLLA cold crystallization, and a melting peak at approximately 163 °C for both copolymers. A glass transition at approximately 52 °C and 60 °C was observed for PELA and PEPELA, respectively, and also assigned to the PLLA phase. No transition could be observed for the polyether phase. Curiously, the addition of AC induced the crystallization of the polyether phase of the copolymers, and a melting peak was observed in the 1st heating scan for PELA–AC5, PELA–AC10 (curves “d” and “e” of the 1st heating scan of PELA-series) and PEPELA–AC5 (curve “d” of the 1st heating scan of PELA-series) at approximately 53 °C. Therefore, the insertion of drugs in the scaffolds has some influence on the crystallization mainly on the polyether phase of the copolymers. The PLLA phase of the loaded scaffolds also presented cold crystallization (T > 80 °C) and melting (T > 150 °C), phenomena at temperatures similar to those observed for the PLLA phase of neat copolymers. These results indicate that during the electrospinning, PLLA end-blocks crystallize first and hinder the PEO and PPO crystallization, resulting in low melting enthalpy ΔHm and melting temperature of PEO phase in comparison to PEO homopolymer (Tm = 70–75 °C). The DSC cooling and 2nd heating scans can be found in the Fig. S5 of the ESI.†
E′ and E′′ moduli of the scaffolds were calculated assuming dense specimens. However, the scaffolds are porous and constituted by fibres with different diameters. Thus, the storage modulus of the scaffolds in the glassy state was lower than 109 Pa and varied with the porosity of each scaffold. In order to minimize the morphological influence in the scaffolds moduli, the relative storage (E′R) and loss (E′′R) moduli were calculated as the E′/E′i and E′′/E′′i ratios, respectively, E′i and E′′i are the moduli at −100 °C. E′R, E′′R and loss factor (tanδ) parameters as a function of temperature curves are depicted in Fig. 5 for PELA and Fig. S6 of the ESI† for PEPELA.
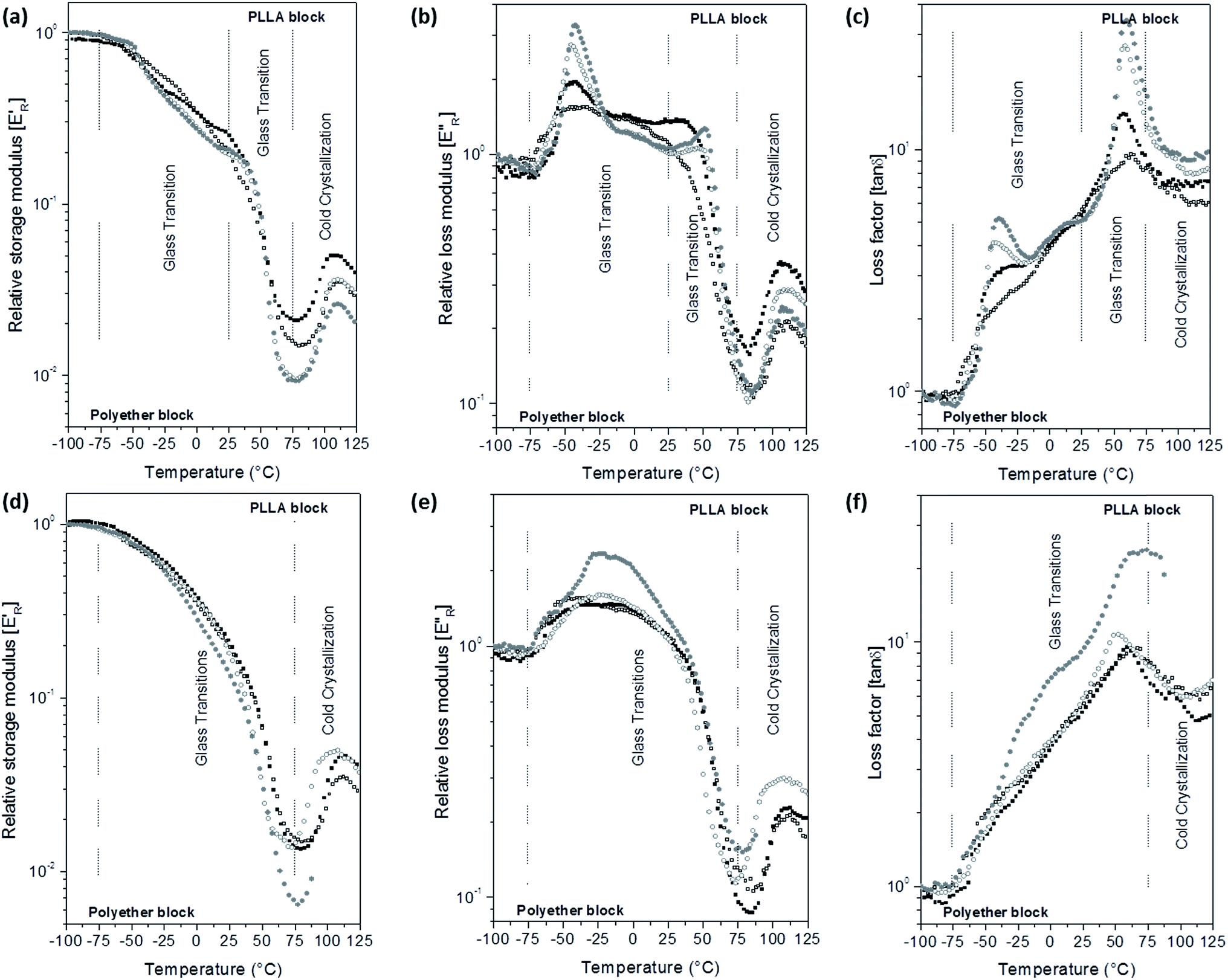 | ||
| Fig. 5 Relative storage (E′R) and loss (E′′R) moduli and loss factor (tanδ) as a function of temperature of PELA scaffolds loaded with acetaminophen (a–c) or celecoxib (d–f) drugs in the concentrations of 0.0 (□), 2.5 (■), 5.0 (○) and 10 wt% (●) of initial drug load. | ||
In a previous study,13 we have demonstrated that PELA and PEPELA copolymers are multiphasic systems in which the minor and amorphous polyether-rich phase is dispersed in the major and semicrystalline PLLA-rich phase. The dynamic-mechanical behaviour of the scaffolds reflects the same phase segregation behaviour. E′R × T curves for unloaded PELA scaffold shows a continuous drop from −100 °C to 75 °C (Fig. 5a), while E′′R × T and tanδ × T curves (Fig. 5b and c) show broad peaks. In this temperature range, both polyether and polyester blocks glass transitions occur. However, the copolymer morphology and interfacial effects did not allow a clear definition of the PLLA block glass transition. According to the DSC 1st heating scan data, the polyether phases of PELA is amorphous; therefore, the broad peaks in the E′′R × T and tanδ × T curves result from the overlap of polyether and polyester amorphous phases relaxation spectra. Above 75 °C, PLLA phase of PELA undergo a cold crystallization characterized by an increase of E′R followed by melting.
Analysis of the E′R × T curves for PELA–ACx scaffolds revealed a two-step decrease in the modulus from −75 °C to 25 °C and from 25 °C to 75 °C, respectively (Fig. 5a). Accordingly, the E′′R × T and tan δ × T curves present two distinguished peaks with maximum temperature values at −45 °C and 60 °C, corresponding to the glass transitions of the polyether and polyester phases, respectively (Fig. 5b and c). The higher the added amount of the hydrophilic AC drug, the more intense and defined are the peaks in the E′′R × T and tan δ × T curves. However, the increase of the relaxations magnitude associated with the glass transition of the polyether and polyester phases is due to different causes: preferential AC drug solvation by the polyether phase and decrease of the crystallinity degree of PLLA phase of the PELA copolymer. The hydrophilic AC drug encapsulated in PELA varies from 8.5 to 12.1 wt%, with respect to the hydrophilic polyether phase corresponding to 26 wt% in the overall mass composition of PELA copolymer. The high amounts of AC drug preferentially dissolved in the polyether phase amplify the magnitude of polymer chain relaxations related to the polyether phase as observed in the DMA curves profiles, as similar as a plasticizer effect. In the temperature range from 50 °C to 100 °C, E′R × T curve presents a valley whose depth is proportional to the PLLA crystallinity degree. The analysis of the E′R in this temperature range suggests the following crystallinity degree order for PLLA phase: 2.5 > 0.0 > 5.0 ≈ 10.0, for PELA loaded scaffolds. Above 75 °C, the cold crystallization and melting of the PLLA phase were observed.
The hydrophobic CL drug is likely to be dissolved in the also hydrophobic PLLA phase that corresponds to 74 wt% in the overall mass composition of PELA copolymer – CL concentration in PELA scaffolds varies from 0.2 to 1.9 wt% with respect to the PLLA mass (Table 2). Therefore, the presence of CL drug to PELA caused only a slight decrease of E′R values in temperatures below the PLLA block cold crystallization (Fig. 5d), and also slight changes in the profile of the peak in the E′′R × T curves for PELA–CL10. The increase of the intensity of the peak in the E′′R × T curve for PELA–CL10 (Fig. 5e) in temperature range between the glass transition of block phases (from −50 °C to 50 °C) suggest that CL drug dissolution in the copolymer amplifies the polymer segments relaxations magnitude of both phases. A similar dynamic mechanical profile is verified for PEPELA scaffolds, as can be observed in the Fig. S6.† The transition temperatures are summarized in Table S1 of the ESI.†
Confocal Raman mapping was used to determine the macroscopic distribution of CL and AC throughout the beaded fibrous structures. The normalized and baseline-corrected Raman spectra of the powders of AC (type I) and CL (type III) drugs and also of the neat PELA and PEPELA scaffolds are depicted in the Fig. S7 of the ESI.†
The PELA–AC10, PELA–CL10, PEPELA–AC10 and PEPELA–CL10 drug-loaded scaffolds were then subjected to confocal Raman spectroscopy mapping of 42 points, spaced 5 μm apart within an area of 750 μm2 in the Raman shift ranges of 1765–1766 cm−1 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) for copolymers (Fig. S7† – green line), 1619–1620 cm−1 for AC and 1618–1619 cm−1 (δN–H) for CL (Fig. S7† – red line).
O) for copolymers (Fig. S7† – green line), 1619–1620 cm−1 for AC and 1618–1619 cm−1 (δN–H) for CL (Fig. S7† – red line).
In observing the beaded fibrous morphology of PELA–AC10 scaffolds under the confocal microscope (Fig. 6), Raman mapping showed that the entrapped AC hydrophilic drug (Fig. 6b – in red) was not uniformly dispersed: the red and green mapping zones are easily distinguishable. Meanwhile, PELA–CL10 scaffolds (Fig. 6) presented a distribution of CL along the overall structure, as it was expected for the hydrophobic CL drug mixed with the major PLLA phase. The red and green mapping zones are overlapped, which means that the CL drug is spread throughout the fibrous scaffolds.
 | ||
| Fig. 6 Confocal Raman spectroscopy mapping of drug-loaded scaffolds (a and b) PELA–AC10 and (c and d) PELA-CL10. The images (a) and (c) represent the Raman shift at 1765–1766 cm−1 mapping for PELA copolymer (in green), and the images (b) and (d) represent the Raman shift at 1619–1620 cm−1 for AC or at 1618–1619 cm−1 for CL (in red) for a selected area of each scaffold. The yellow crosses highlight the mapping points. | ||
Scaffolds prepared from 5 wt% drug solutions were selected for in vitro release testing, because they presented a similar drug mass fraction (approximately 3%), with the exception of PELA–CL5 scaffold (Table 2). Fig. 7 shows the in vitro release profiles of PELA–CL5, PEPELA–CL5, PELA–AC5 and PEPELA–AC5 scaffolds in PBS (pH = 7.4) at 37 °C.
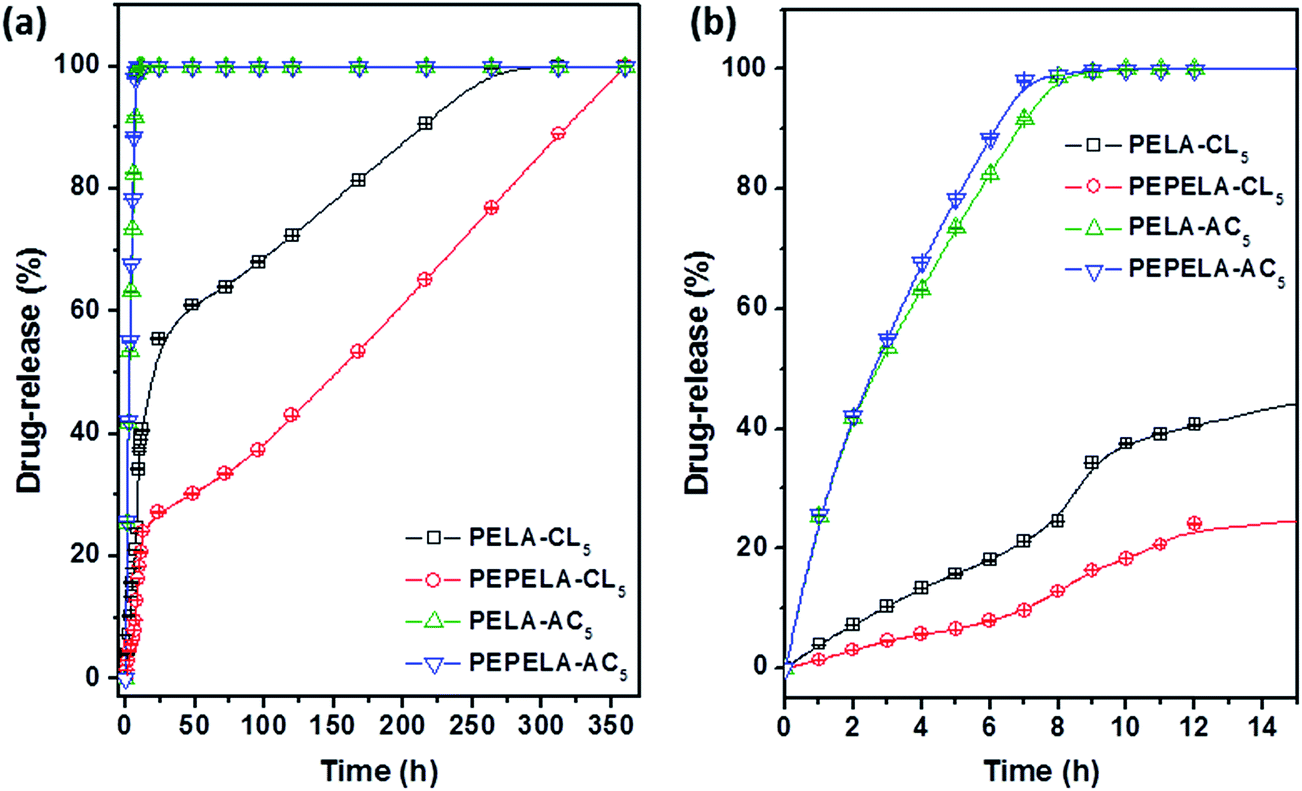 | ||
| Fig. 7 Drug-release profiles of PELA–AC5, PELA–CL5, PEPELA–AC5 and PEPELA–CL5 scaffolds (a) during 350 hours of the assay. (b) The first 15 hours of the drug-releasing assay are highlighted. | ||
The AC drug was quickly released from PELA and PEPELA scaffolds achieving a total drug content release after 10 h. Thus, the predominant mechanism of release was drug dissolution, and it was dependent on drug solubility in the medium. Since hydrophilic AC drug is likely to be dissolved in the polyether blocks, it is possible that the aqueous medium penetrates largely in this phase (water swelling coefficients of PELA and PEPELA at 25 °C after 48 h equal to 36 and 6%, respectively), allowing a fast release by AC dissolution in PBS.
The CL drug-release rate is significantly lower compared to AC drug (Fig. 7), and an almost linear dependence of CL release with time can be observed. CL was released by following a two-stage profile. In a first stage, a high release rate was observed. This burst effect could be explained as a fast release of drug located near the surface of the nanofiber during fibre formation process.32 The heterogeneous drug distribution in the fibrous matrices has been reported and it is due to the migration of drug to the fibre surface during the solvent evaporation that takes place in the electrospinning process.33 Beyond the burst period, the release rate slowed down, following a diffusion-controlled mechanism.
The analysis of release behaviour of PELA–CL5 indicates that burst stage took place during the first 12 h and 41% of drug was released, reaching the complete release after 312 h (13 days). The PEPELA–CL5 scaffold displayed the same burst period, although 24% of drug was released and the release was completed after 360 h (15 days). The second stage also presented an almost linear release profile, which can be considered associated with zero-order diffusion mechanisms.34 This mechanism considers that the diffusion rate of the drug is constant and independent of the amount of encapsulated drug in polymer systems showing neither swelling nor disintegration. As noted above, CL is entrapped in the hydrophobic blocks of PLLA and this would allow a delay in the release of the drug by diffusion through the polymer chains. Results show that the amphiphilic nature of the polymer allows them to act as carrier of both hydrophilic and hydrophobic drugs, and modulate the release profiles for specific applications.
Conclusions
Electrospun matrices of polyester/polyether amphiphilic block copolymers, loaded with acetaminophen and celecoxib as model drugs, were prepared and characterized. Scaffolds consisted of randomly oriented fibres presented a bead-on-string nanofibrous morphology. PELA shows to be more selective for AC hydrophilic drug sorption, while PEPELA could sorb both AC hydrophilic and CL hydrophobic drugs in comparable amounts. Drug-loaded fibres exhibited a mean fibre diameter higher than neat fibres and dependent on the drug amount and nature. XRD patterns indicate that AC and CL were in an amorphous form, as a solid dispersion or forming amorphous molecular aggregates on fibres. While DMA indicates that AC drug is preferentially located into PEO phase and CL drug is fractionated between polyether and polyesters both phases.Confocal Raman spectroscopy mapping techniques revealed that the CL hydrophobic drug was distributed throughout the fibrous structure due to its affinity to the PLLA major phase. Dramatic differences were observed when comparing the release of AC and CL from PELA and PEPELA scaffolds. Data suggested that AC presented a high affinity for the hydrophilic block, allowing the fast release in a swollen matrix. CL exhibited sustained release due to its accommodation in hydrophobic PLLA domains, resulting in a reservoir-like system. Drug-loaded amphiphilic electrospun scaffolds developed in this work presented characteristics, encapsulation efficiency values and release profiles that could be very attractive as potential carriers of both hydrophilic and hydrophobic drugs for specific applications.
Acknowledgements
The authors would like to acknowledge the financial support of the São Paulo Research Foundation – FAPESP (processes 2010/17804-7, 2012/24821-0 and 2015/25406-5), the National Counsel of Technological and Scientific Development – CNPq (process 444392/2014-9), the Argentinean National Agency of Scientific and Technological Promotion (PICT2012-224), and CONICET (PIP2013-089).References
- M. Zamani, M. P. Prabhakaran and S. Ramakrishna, Advances in drug delivery via electrospun and electrosprayed nanomaterials, Int. J. Nanomed., 2013, 8, 2997–3017 Search PubMed.
- T. J. Sill and H. A. von Recum, Electrospinning: applications in drug delivery and tissue engineering, Biomaterials, 2008, 29(13), 1989–2006 CrossRef CAS PubMed.
- K. Kim, Y. K. Luu, C. Chang, D. Fang, B. S. Hsiao and B. Chu, et al., Incorporation and controlled release of a hydrophilic antibiotic using poly(lactide-co-glycolide)-based electrospun nanofibrous scaffolds, J. Controlled Release, 2004, 98(1), 47–56 CrossRef CAS PubMed , available from: http://www.ncbi.nlm.nih.gov/pubmed/15245888.
- S. Agarwal, J. H. Wendorff and A. Greiner, Progress in the field of electrospinning for tissue engineering applications, Adv. Mater., 2009, 21(32–33), 3343–3351 CrossRef CAS PubMed.
- S. H. Saw, K. Wang, T. Yong and S. Ramakrishna, Polymeric Nanofibers in Tissue Engineering, Nanotechnology, 2006, 9(1), 66–134 Search PubMed.
- X. Liu, T. Lin, J. Fang, G. Yao and H. Zhao, M. Dodson and X. Wang, In vivo wound healing and antibacterial performances of electrospun nanofibre membranes, J. Biomed. Mater. Res., Part A, 2010, 94(2), 499–508 Search PubMed.
- R. A. Thakur, C. A. Florek, J. Kohn and B. B. Michniak, Electrospun nanofibrous polymeric scaffold with targeted drug release profiles for potential application as wound dressing, Int. J. Pharm., 2008, 364(1), 87–93 CrossRef CAS PubMed.
- N. Bölgen, İ. Vargel, P. Korkusuz, Y. Z. Menceloğlu and E. Pişkin, In vivo performance of antibiotic embedded electrospun PCL membranes for prevention of abdominal adhesions, J. Biomed. Mater. Res., Part B, 2007, 81(2), 530–543, DOI:10.1002/jbm.b.30694.
- P. C. Caracciolo, P. C. R. Tornello, F. M. Ballarin and G. A. Abraham, Development of Electrospun Nanofibers for Biomedical Applications: State of the Art in Latin America, J. Biomater. Tissue Eng., 2013 Feb 1, 3(1), 39–60 Search PubMed , available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3939572%26tool=pmcentrez%26rendertype=abstract.
- A. Greiner and J. H. Wendorff, Electrospinning: a fascinating method for the preparation of ultrathin fibers, Angew. Chem., Int. Ed., 2007, 46(30), 5670–5703 CrossRef CAS PubMed.
- D. Liang, B. S. Hsiao and B. Chu, Functional electrospun nanofibrous scaffolds for biomedical applications, Adv. Drug Delivery Rev., 2007, 59(14), 1392–1412 CrossRef CAS PubMed , available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2693708%26tool=pmcentrez%26rendertype=abstract.
- D.-J. Yang, C.-D. Xiong, T. Govender and Y.-Z. Wang, Preparation and drug-delivery potential of metronidazole-loaded PELA tri-block co-polymeric electrospun membranes, J. Biomater. Sci., Polym. Ed., 2009, 20(9), 1321–1334 CrossRef CAS PubMed , available from: http://www.ncbi.nlm.nih.gov/pubmed/19520015.
- L. M. D. Loiola, B. A. Más, E. A. R. Duek and M. I. Felisberti, Amphiphilic multiblock copolymers of PLLA, PEO and PPO blocks: synthesis, properties and cell affinity, Eur. Polym. J., 2015, 68, 618–629, DOI:10.1016/j.eurpolymj.2015.03.034.
- M. Thieme, S. Agarwal, J. H. Wendorff and A. Greiner, Electrospinning and cutting of ultrafine bioerodible poly(lactide-co-ethylene oxide) tri- and multiblock copolymer fibers for inhalation applications, Polym. Adv. Technol., 2011, 22, 1335–1344, DOI:10.1002/pat.1617.
- R. B. Trinca, G. A. Abraham and M. I. Felisberti, Electrospun nanofibrous scaffolds of segmented polyurethanes based on PEG, PLLA and PTMC blocks: physico-chemical properties and morphology, Mater. Sci. Eng., C, 2015 Nov, 56, 511–517 Search PubMed , available from: http://linkinghub.elsevier.com/retrieve/pii/S092849311530206X.
- R. B. Trinca and M. I. Felisberti, Segmented polyurethanes based on poly(L-lactide), poly(ethylene glycol) and poly(trimethylene carbonate): physico-chemical properties and morphology, Eur. Polym. J., 2015, 62, 77–86, DOI:10.1016/j.eurpolymj.2014.11.008.
- Q. Guo, X. Li, Q. Ding, D. Li, Q. Zhao and P. Xie, et al., Preparation and characterization of poly(pluronic-co-L-lactide) nanofibers for tissue engineering, Int. J. Biol. Macromol., 2013 Jul, 58, 79–86 Search PubMed , available from: http://linkinghub.elsevier.com/retrieve/pii/S014181301300161X.
- D. Yang, L. Zhang, L. Xu, C. Xiong, J. Ding and Y. Wang, Fabrication and characterization of hydrophilic electrospun membranes made from the block copolymer of poly(ethylene glycol-co-lactide), J. Biomed. Mater. Res., Part A, 2007, 82(3), 680–688, DOI:10.1002/jbm.a.31099.
- H. Peng, S. Zhou, T. Guo, Y. Li, X. Li and J. Wang, et al., In vitro degradation and release profiles for electrospun polymeric fibers containing paracetanol, Colloids Surf., B, 2008, 66(2), 206–212 CrossRef CAS PubMed , available from: http://www.ncbi.nlm.nih.gov/pubmed/18691851.
- A. K. Gaharwar, S. M. Mihaila, A. a Kulkarni, A. Patel, A. Di Luca and R. L. Reis, et al., Amphiphilic beads as depots for sustained drug release integrated into fibrillar scaffolds, J. Controlled Release, 2014, 187, 66–73 CrossRef CAS PubMed , available from: http://www.ncbi.nlm.nih.gov/pubmed/24794894.
- H. Thakkar, R. K. Sharma, A. K. Mishra, K. Chuttani and R. S. R. Murthy, Celecoxib incorporated chitosan microspheres: in vitro and in vivo evaluation, J. Drug Targeting, 2004, 12(9–10), 549–557, DOI:10.1080/10611860400010630.
- G. W. Lu, M. Hawley, M. Smith, B. M. Geiger and W. Pfund, Characterization of a novel polymorphic form of celecoxib, J. Pharm. Sci., 2006, 95(2), 305–317 CrossRef CAS PubMed , available from: http://www.ncbi.nlm.nih.gov/pubmed/16369929.
- S. D. Cohen and E. A. Khairallah, Selective protein arylation and acetaminophen-induced hepatotoxicity, Drug Metab. Rev., 1997 Jan 15, 29(1–2), 59–77, DOI:10.3109/03602539709037573.
- R. B. Trinca and M. I. Felisberti, Influence of the synthesis conditions on the structural and thermal properties of poly(L-lactide)-b-poly(ethylene glycol)-b-poly(L-lactide), J. Appl. Polym. Sci., 2014, 131(13), 40419, DOI:10.1002/app.40419.
- H. Fong, I. Chun and D. Reneker, Beaded nanofibers formed during electrospinning, Polymer, 1999, 40(16), 4585–4592 CrossRef CAS , available from: http://linkinghub.elsevier.com/retrieve/pii/S0032386199000683.
- T. Potrč, S. Baumgartner, R. Roškar, O. Planinšek, Z. Lavrič and J. Kristl, et al., Electrospun polycaprolactone nanofibers as a potential oromucosal delivery system for poorly water-soluble drugs, Eur. J. Pharm. Sci., 2015, 75, 101–113 CrossRef PubMed , available from: http://linkinghub.elsevier.com/retrieve/pii/S0928098715001475.
- L. Li, X. Zheng, D. Fan, S. Yu, D. Wu and C. Fan, et al., Release of celecoxib from a bi-layer biomimetic tendon sheath to prevent tissue adhesion, Mater. Sci. Eng., C, 2016 Apr, 61, 220–226 Search PubMed , available from: http://linkinghub.elsevier.com/retrieve/pii/S0928493115306391.
- M. Rossmann, A. Braeuer, A. Leipertz and E. Schluecker, Manipulating the size, the morphology and the polymorphism of acetaminophen using supercritical antisolvent (SAS) precipitation, J. Supercrit. Fluids, 2013, 82, 230–237 CrossRef CAS , available from: http://linkinghub.elsevier.com/retrieve/pii/S0896844613002660.
- G. P. Andrews, O. Abu-Diak, F. Kusmanto, P. Hornsby, Z. Hui and D. S. Jones, Physicochemical characterization and drug-release properties of celecoxib hot-melt extruded glass solutions, J. Pharm. Pharmacol., 2010, 62(11), 1580–1590, DOI:10.1111/j.2042-7158.2010.01177.x.
- T. d. C. Rufino and M. I. Felisberti, Confined PEO crystallisation in immiscible PEO/PLLA blends, RSC Adv., 2016, 30937–30950 RSC , available from: http://pubs.rsc.org/en/Content/ArticleLanding/2016/RA/C6RA02406H.
- D.-G. Yu, C. Branford-White, K. White, X.-L. Li and L.-M. Zhu, Dissolution Improvement of Electrospun Nanofiber-Based Solid Dispersions for Acetaminophen, AAPS PharmSciTech, 2010, 11(2), 809–817 CrossRef CAS PubMed , available from: http://www.springerlink.com/index/10.1208/s12249-010-9438-4.
- X. Huang and C. S. Brazel, On the importance and mechanisms of burst release in matrix-controlled drug delivery systems, J. Controlled Release, 2001, 73(2–3), 121–136 CrossRef CAS PubMed.
- P. R. Cortez Tornello, G. E. Feresin, A. Tapia, I. G. Veiga, Â. M. Moraes and G. a Abraham, et al., Dispersion and release of embelin from electrospun, biodegradable, polymeric membranes, Polym. J., 2012, 44(11), 1105–1111 CrossRef CAS , available from: http://www.nature.com/doifinder/10.1038/pj.2012.80.
- D. Brooke and R. J. Washkuhn, Zero-order drug delivery system: theory and preliminary testing, J. Pharm. Sci., 1977, 66(2), 159–162 CrossRef CAS PubMed , available from: http://www.ncbi.nlm.nih.gov/pubmed/839408.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25023h |
| This journal is © The Royal Society of Chemistry 2017 |