
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical sensing of aqueous boron based on polymeric hydroxytriphenylene derivatives
L. R. P. Areias ,
A. P. da Costa,
S. P. C. Alves,
C. Baleizão* and
J. P. S. Farinha*
,
A. P. da Costa,
S. P. C. Alves,
C. Baleizão* and
J. P. S. Farinha*
CQFM – Centro de Química-Física Molecular, IN – Institute of Nanoscience and Nanotechnology, Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisboa, Portugal. E-mail: carlos.baleizao@tecnico.ulisboa.pt; farinha@tecnico.ulisboa.pt
First published on 16th January 2017
Abstract
The detection of boron in natural water and wastewaters is still limited to a few methods, requiring a compromise between sensitivity, reliability and accessibility. Here we present a novel polymeric fluorescent boron sensor with excellent water solubility, boron sensitivity, ease of handling, which can be easily recovered and reused. The new off–on fluorescent boron sensor is able to detect and quantify ppb amounts of boron in water, with a limit of detection of 10 ppb of boron, both for boric acid and phenylboronic acid. The sensor was prepared by copolymerizing methacrylic acid with a polymerizable asymmetric hexa-substituted triphenylene, obtained by oxidative cyclization of biphenyl and catechol precursors.
Introduction
Boron is an important micronutrient for plants, animals and humans and its presence is crucial for several body organs in humans and animals (such as the brain, the skeleton and the immune system).1 However, high concentrations of boron can limit plant growth or even lead to its death, and in humans may result in reproductive and developmental toxicity, nausea, vomiting, diarrhoea, dermatitis and lethargy.2–4 Therefore, the consumption of food and water with high boron contents is potentially hazardous. In drinking water, boron is usually present in concentrations below 0.5 mg L−1 (0.5 ppm), with the World Health Organization recommending boron concentrations in water for human consumption below 2.4 ppm,4 and the European Union allowing only 1.0 ppm.5The most sensitive methods for determining boron content in water are Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-AES)6 and Inductively Coupled Plasma-Mass Spectrometry (ICP-MS),7 but these require non-portable and complex equipment, with high operation costs. Spectrofluorimetric and spectrophotometric methods are a good compromise for boron detection, featuring good boron sensitivity, low acquisition and operational costs, simplicity of operation, and the possibility of use in field work. In recent years there has been a growing need to develop chemical sensors for fast monitoring of environmental samples at low cost. Boron-chelating compounds have gained increased attention for use in new boron sensors.8–10
The family of compounds based on 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) have been widely used in the synthesis of discotic liquid crystals (DLC's)11–13 and more recently in covalent organic frameworks (COF's),14,15 due to their unique properties such as one-dimensional charge migration, electroluminescence, and self-assembling behaviour. The chelating properties of HHTP have been also used in optical fluorescent sensing systems for boron10 and picric acid.16 The boron sensor based in unmodified HHTP relies on its large increase in fluorescence emission intensity when in the presence of boron. However, the solubility of HHTP is extremely reduced in water (10−6 M), and sensor aggregation somehow limits the sensitivity and reproducibility of the method. To overcome the low water solubility, a possible strategy is the incorporation of water soluble groups in the HHTP core, or alternatively, the incorporation of HHTP in a water soluble vector (e.g. a polymer chain).
Triphenylene derivatives can be prepared using different strategies.17–20 The first published approach was the trimerization of three phenyl units (precursors of the peripheral rings of triphenylene), which is more indicated for symmetrical derivatives. The oxidative trimerization of catechol derivatives itself would be an attractive and very reasonable method for the direct synthesis, however, the reported oxidative trimerization of catechol with iron(III) chloride is not effective and forms impure HHTP derivatives (because of the formation of triphenylenes with two, four or six positioned OH groups).21,22 In fact, preparation of asymmetrical derivatives through this single step strategy gives low yields and is very laborious due to the statistical formation of several derivatives. Alternative strategies to prepare asymmetrical HHTP derivatives23 rely on: (i) terphenyl intermediates, prepared by oxidative coupling to form biaryl bonds from arenes, by palladium-catalyzed coupling with arylzinc halides, followed by oxidative cyclization with FeCl3/H2SO4,24,25 or by photocyclization of the terphenyl units (although the later presents a problem of regioselectivity for asymmetric substitution, usually decreasing the yield);26 (ii) biphenyl intermediates, obtained mainly by palladium-catalyzed coupling reactions,27 inter or intramolecular Diels–Alder reactions,28 or oxidative cyclization with FeCl3; (iii) naphthyl intermediates, using Diels–Alder cicloadditions to obtain triphenylenes,29 (which can also be obtained by classical Friedel–Crafts chemistry from functionalized naphthalenes); or (iv) phenanthryl cores, where the last peripheral ring of the triphenylene is formed on an isolated phenanthrene derivative by a Diels–Alder reaction, in which the diene is frequently a phenanthrene with exocyclic double bonds.30
Here we describe the design of an optical boron sensor based on a polymerizable hexaalkoxy triphenylene derivative for the detection of boron in wastewaters and natural water. To improve solubility issues previously found for the unmodified HHTP sensor, we use a common strategy for increasing the water solubility of probes and sensors, consisting in coupling these to water soluble moieties or dispersible systems.31,32 We introduce water soluble ethylene glycol groups in the HHTP core, and couple this with a water soluble poly(methacrylic acid) chain (PMAA).
Results and discussion
The synthetic strategy designed to prepare the polymerizable water soluble HHTP derivative take into account that: (i) at least one group of vicinal diols must be available in the final compound for boron chelation; (ii) a spacer between the polymerizable group and the triphenylene moiety is necessary to minimize interaction of the boron-chelating groups with the polymer backbone; and (iii) a hydrophilic group should be added to increase the water solubility of the final sensor. The asymmetric functionalization of the HHTP sensor strongly limits the strategy to obtain the triphenylene ring. The most explored route to prepare symmetrically substituted alkoxytriphenylenes is the oxidative trimerization of catechol derivatives. However, if applied to asymmetrical triphenylenes using different catechol derivatives as starting material, a mixture of products is obtained, which are usually very hard to separate and purify. To overcome this extra difficulty, we selected the oxidative cyclization of biphenyl compounds with catechol derivatives in the presence of FeCl3 as oxidizing agent.The synthetic route starts with the synthesis of an asymmetric catechol, with an ethyleneglycol chain and a methacrylate unit for subsequent polymerization (4, Scheme 1). The mono-pegylated catechol 2 was obtained in a mixture with the di-pegylated derivative, but there are easily separated by successive acid/basic extractions and recovered in moderate yields. A spacer based in an alkyl chain was added to the mono-pegylated catechol 2 and the product (3) was obtained in quantitative yield. The asymmetric catechol 4 was prepared by reacting the terminal hydroxyl group with methacryloyl chloride, in anhydrous conditions and low temperature. The biphenyl intermediate was prepared through a typical Suzuki cross-coupling reaction (Scheme 2).
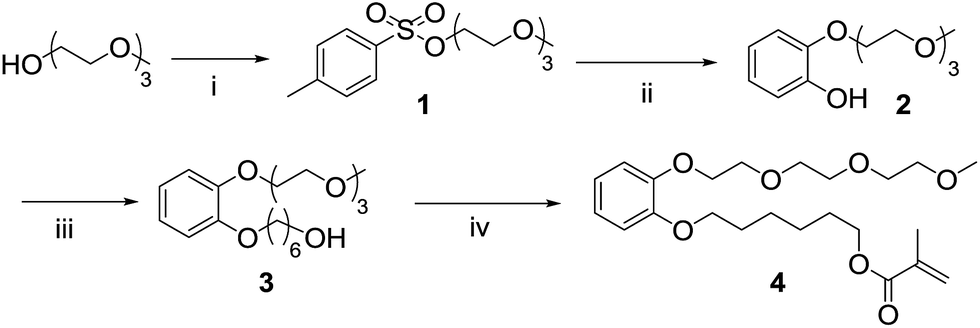 | ||
| Scheme 1 Synthetic route to obtain the asymmetric catechol: (i) TsCl, NaOH, THF, 0 °C to rt, 1 h, 85%; (ii) catechol, NaH, 1,4-dioxane, 16 h, 105 °C, 28%; (iii) 6-bromo-1-hexanol, K2CO3, acetone, 16 h, 65 °C, quantitative yield; (iv) methacryloyl chloride, triethylamine, CH2Cl2, 16 h, −78 °C to rt, 70%. | ||
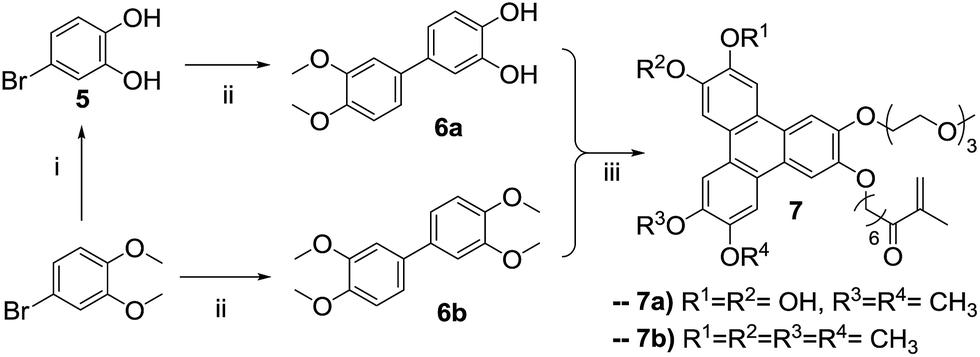 | ||
| Scheme 2 Synthetic route to obtain the asymmetric triphenylene: (i) BBr3, CH2Cl2, −78 °C to rt, 16 h, quantitative yield; (ii) 3,4-dimethoxyphenyl boronic acid, Pd(PPh3)4, K2CO3, toluene, 125 °C, 16 h (6a, 41%; 6b 69%); (iii) 4, FeCl3, CH2Cl2 or 1,4-dioxane, 1 h 30 min, rt (7a, 25%; 7b, 23%). | ||
The demethylation of 4-bromoveratrol with boron tribromide produces the bromo-cathecol 5 in quantitative yield, and the condensation with 3,4-dimethoxyphenyl boronic acid catalyzed by tetrakis(triphenylphosphine)palladium(0) in the presence of potassium carbonate yields the asymmetric biphenyl 6a. The target asymmetric triphenylene 7a was obtained from the reaction of the asymmetric catechol 4 and the asymmetric biphenyl 6a, catalyzed by FeCl3. As intended, the target triphenylene 7a has a polymerizable group, a hydrophilic moiety and vicinal diols for boron complexation. This asymmetric triphenylene is soluble in water and slightly soluble in DMF, and for an accurate characterization (especially photophysical), we prepare its tetra-methylated analogue 7b, following the same strategy employed for the preparation of 7a (Scheme 2). The NMR profile are similar for 7a and 7b, and the photophysical properties (absorption and emission spectra) are identical to those of HHTP. The derivative 7a, was copolymerized with methacrylic acid by a free radical process in DMF, with AIBN as initiator (Scheme 3).
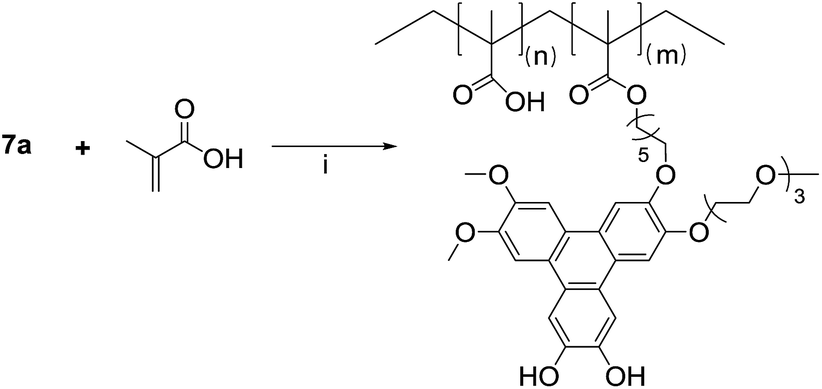 | ||
| Scheme 3 Copolymerization of 7a with methacrylic acid yielding the polymeric boron sensor 7a-PMAA: 7a (1 equivalent), methacrylic acid (20 equivalents), AIBN, DMF, 80 °C, 16 h, quantitative mass yield. | ||
The polymerization reaction was carried out at 80 °C overnight, and the final polymer (7a-PMAA) was obtained as a dark powder after precipitation in cold diethyl ether. The polymer was characterized by GPC-MALS, in DMF with LiBr (0.05 M). The chains have a molecular weight of Mn = 5700 g mol−1, with a size dispersity of 1.2.
The functional polymer 7a-PMAA has UV-vis and fluorescence (excitation and emission) spectra similar to those of the free monomer (7a). In Fig. 1 the UV-vis absorption spectra and the fluorescence (excitation and emission) spectra in DMF of the triphenylene derivatives (7a and 7b) and polymer (7a-PMAA) are compared to those of the unmodified HHTP sensor. The UV-vis absorption spectra and the fluorescence excitation spectra are similar for HHTP, 7a and 7b, presenting two bands (a sharp peak at 280 nm and a shoulder around 310 nm). In the case of 7a-PMAA, the excitation spectrum is identical to the other compounds but the absorption spectra have a broader band, and the shoulder at 310 nm is not visible. This can indicate interaction between the triphenylene derivatives along the polymer chain leading to absorption by non-emissive aggregates (not detected in the excitation spectrum). The fluorescence emission spectra (with excitation at 310 nm) of 7a, 7b and HHTP are also very similar below 460 nm, with a maximum at 382 nm, and three relative maxima or shoulders at 370 nm, 402 nm and 422 nm, characteristic of the local excited state emission of the hydroxytriphenylene chromophore. The emission maximum in DMF is independent of the substituents, indicating no significant intramolecular charge transfer in the excited state.33,34 However, in the case of 7a a small broad peak is visible around 550 nm, possibly due to interactions between the PEG chains and the free hydroxyl groups of the triphenylene moiety, leading to aggregation or excimer formation.35–37
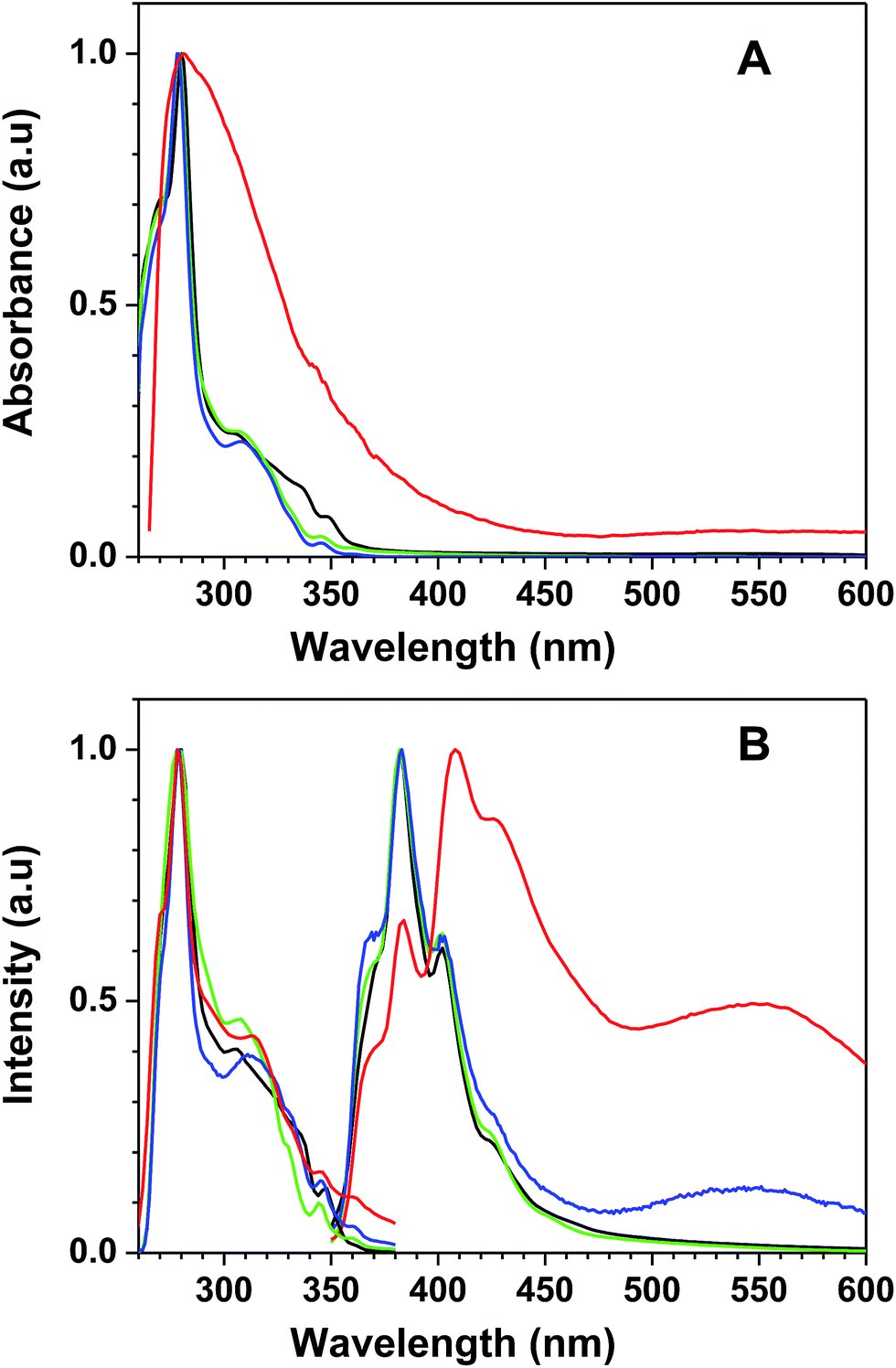 | ||
| Fig. 1 Normalized (A) absorption spectra, and (B) fluorescence emission (λexc = 310 nm) and excitation (λemi = 380 nm) spectra of HHTP (black), 7a (blue), 7b (green), and 7a-PMAA (red), in DMF. | ||
In the case of 7a-PMAA, the emission spectrum is red shifted, with a maximum at 408 nm and three relative maxima or shoulders at 370 nm, 384 nm and 426 nm. As in 7a, an intense broad band appears around 550 nm, probably due to interaction of the PEG chains with the hydroxyl groups of triphenylene core and of the methacrylic acid units of the polymer.
The efficiency of HHTP to chelate and detect boron compounds in aqueous solution at pH 9 was previously studied by our group.10 We found that the fluorescence emission of HHTP is strongly quenched at basic pH, and that a strong emission enhancement is observed at ca. pH ≈ 8–10, proportional to the concentration of boron.
Although the HHTP sensor gave good results when tested with either boric acid or phenylboronic acid, its very low water solubility required very careful preparation procedures. The incorporation of the sensor in a water soluble polymer has two main advantages: (i) to increase the sensor concentration (the HHTP solubility in water is only 10−6 M); and (ii) to allow recovering the sensor after use, by precipitation at acid pH. Here, we performed boron detection experiments with our new boron sensing polymer at pH 9 in aqueous solution (with 20 mM ammonia buffer), with phenylboronic acid (Fig. 2) and boric acid (Fig. 3), in boron concentrations from 0 to 38 ppm.
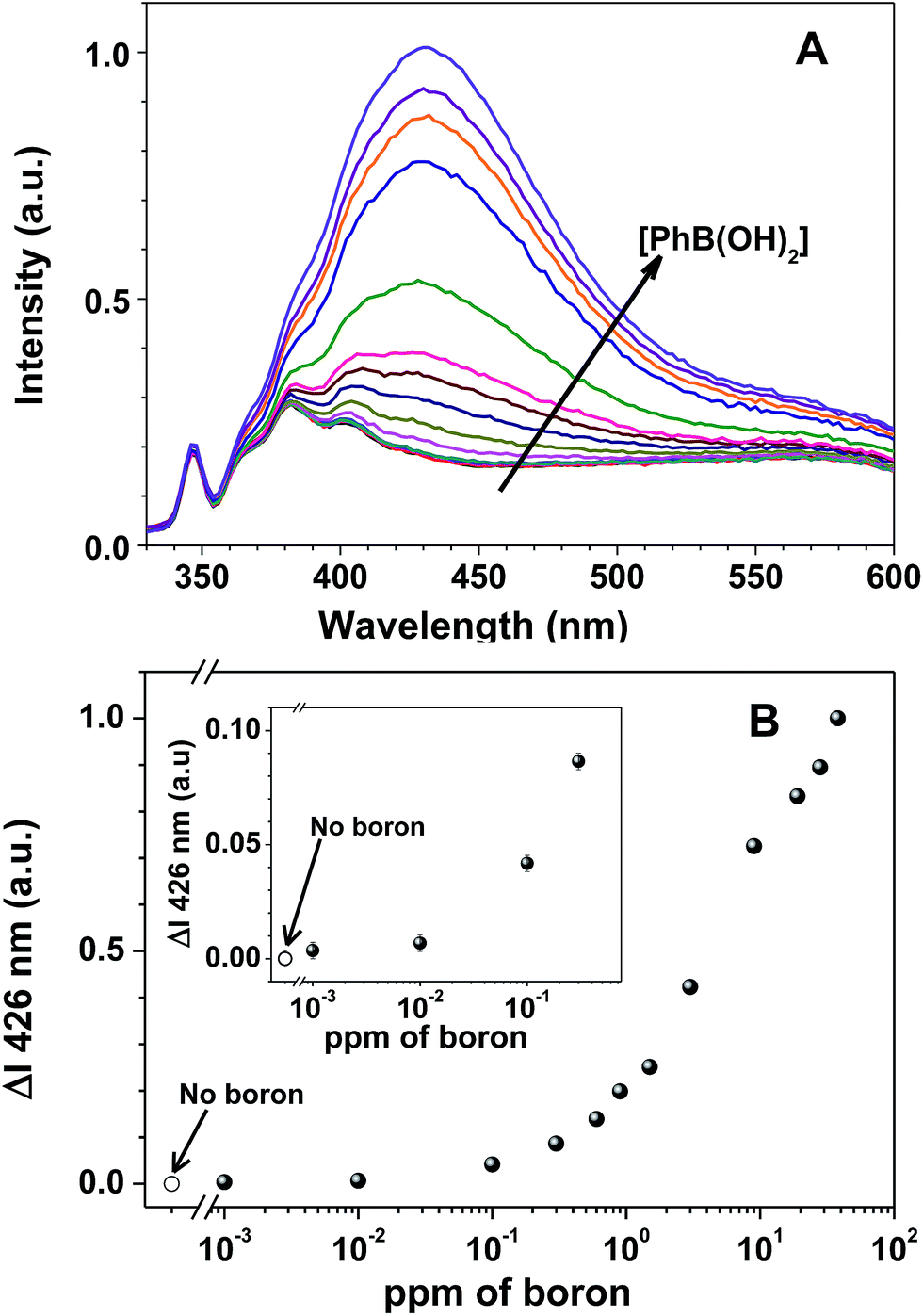 | ||
| Fig. 2 (A) Fluorescence emission spectra of 7a-PMAA (6 × 10−5 M in 7a and 0.02 M of NH3 buffer), with increasing concentrations of phenylboronic acid (0–38 ppm of boron) at pH 9 (the arrow denotes increasing boron concentration); and (B) the corresponding normalized differential fluorescence emission titration curves of 7a-PMAA for increasing phenylboronic acid concentration: 0; 0.001; 0.01; 0.05; 0.1; 0.3; 0.6; 0.9; 1.5; 3; 9; 19; 28 and 38 ppm of boron. The spectra were recorded at λexc = 310 nm after two hours equilibrating in the dark. | ||
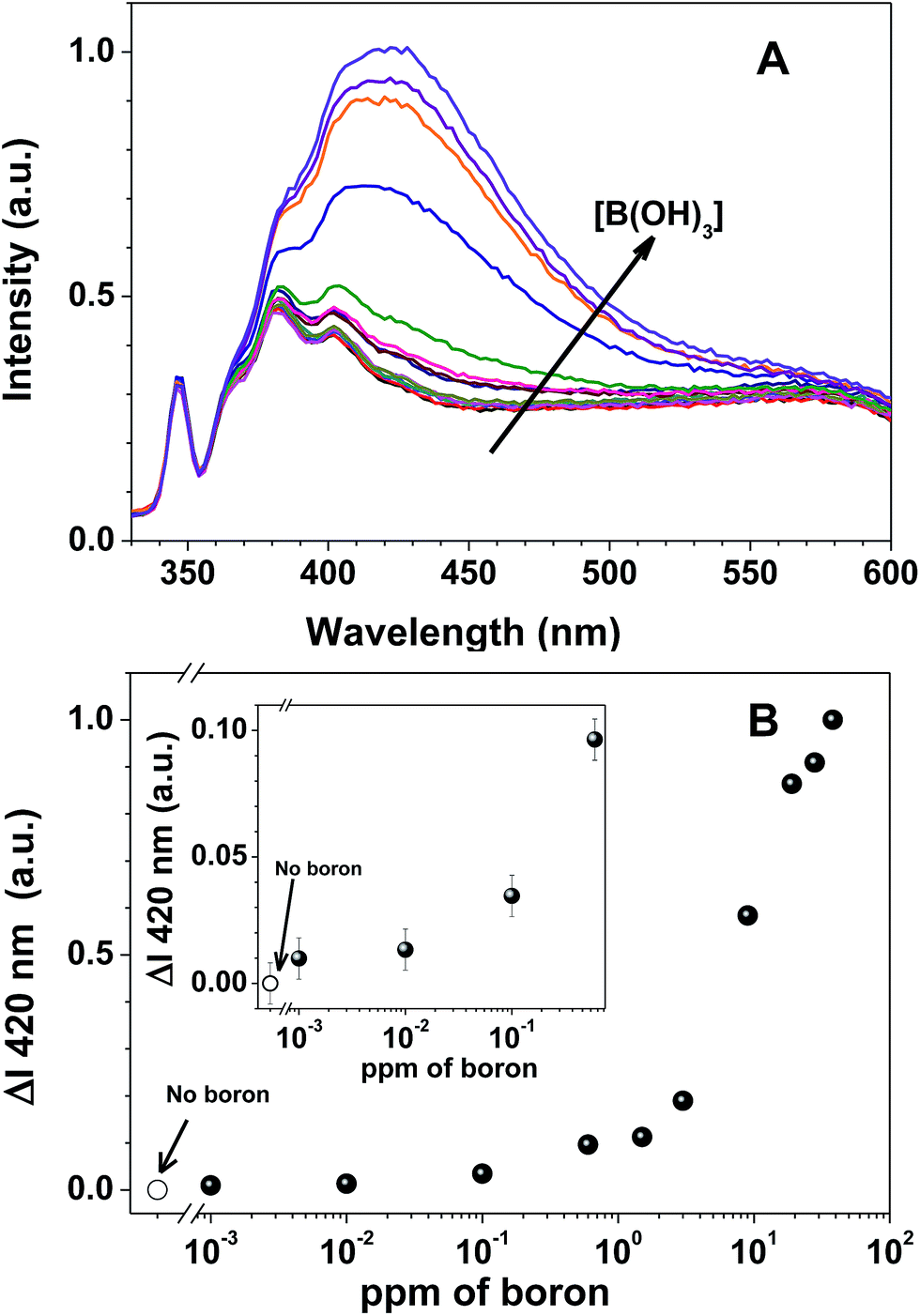 | ||
| Fig. 3 (A) Fluorescence emission spectra of 7a-PMAA (6 × 10−5 M in 7a and 0.02 M of NH3 buffer), with increasing concentrations of boric acid (0–38 ppm of boron) at pH 9 (the arrow denotes increasing boron concentration); and (B) the corresponding normalized differential fluorescence emission titration curves of 7a-PMAA for increasing boric acid concentration: 0; 0.001; 0.01; 0.05; 0.1; 0.3; 0.6; 0.9; 1.5; 3; 9; 19; 28 and 38 ppm of boron. The spectra were recorded at λexc = 310 nm after two hours equilibrating in the dark. | ||
The fluorescence emission spectra of 7a-PMAA strongly depends on the amount of boron in solution, either as boric acid or phenylboronic acid, with an increase in the fluorescence emission intensity with boron concentration being accompanied by a shift in the emission maximum wavelength. The limits of detection, defined as the lowest boron concentration that can be reliably detected, were calculated as the concentration for which the difference in the spectra of 7a-PMAA in the presence of boron relative to that of free 7a-PMAA equals three times the standard deviation of the blank.38
In our case, the limit of detection is LOD = 10 ppb both for boric acid and phenylboronic acid, two orders of magnitude below the lowest established boron concentration limit in water, 1 ppm,5 and comparable with the LOD of the best optical methods, such as azomethine-H (LOD = 10 ppb).4 The limits of quantification (LOQ), defined as the lowest boron concentration that can be reliably quantified, were calculated as the concentration for which the difference in the spectra of 7a-PMAA in the presence of boron relative to that of free 7a-PMAA equals ten times the standard deviation of the blank.38 In our case, the LOQ is 0.1 ppm for phenylboronic acid and 0.6 ppm for boric acid. We estimate the linearity range to be between 0.1–10 ppm for phenylboronic acid and 0.6–20 ppm for boric acid.
We have conducted recovery and reuse experiments with the 7a-PMMA sensor. The recovery was performed at acid pH, with regeneration of the sensor (release of the boron), and the reutilization test was performed using the conditions previously reported. The sensing performance of 7a-PMMA towards boric acid and phenylboronic acid is preserved after recovery and the response is similar to that of fresh sensor within experimental error (Fig. 4).
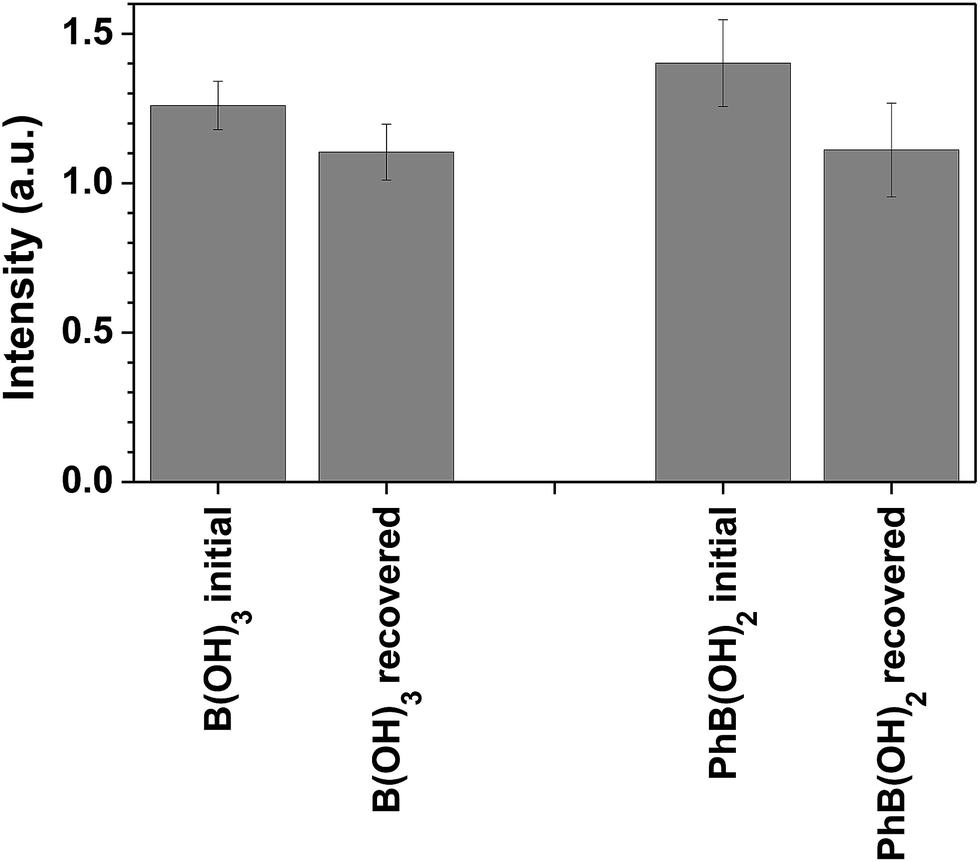 | ||
| Fig. 4 Relative fluorescence intensity of 7a-PMAA (6 × 10−5 M of 7a) in the presence of 38 ppm of boron after initial contact with boron and after one recovery cycle, in NH3 buffer (0.02 M) at pH = 9. The spectra were recorded at λexc = 310 nm after two hours equilibrating in the dark. The emission wavelength was 420 nm when in the presence of B(OH)3 or 426 nm when in the presence of PhB(OH)2. The recovery of the sensor was performed at pH ≈ 1. | ||
The selectivity of the stand-alone triphenylene sensor towards boron in the presence of other metal ions is relatively low, but it can be circumvented by using an anionic chelating agent such as EDTA, as previously reported.10 In the present case, the sensor polymer backbone, based on methacrylic acid (deprotonated under the experimental conditions), acts as a chelating agent for several metal ions. We have performed interference studies on the sensing of boric acid and phenylboronic acid in the presence of Ca2+, Mg2+ and Zn2+ (without EDTA, Fig. 5A). In the case of Ca2+ and Mg2+, the sensor performance was not affected by the presence of the metal ions (the polymer backbone acts as a chelating agent). For Zn2+ we observed a reduction in the sensor performance, which was reverted by adding EDTA (Fig. 5B).
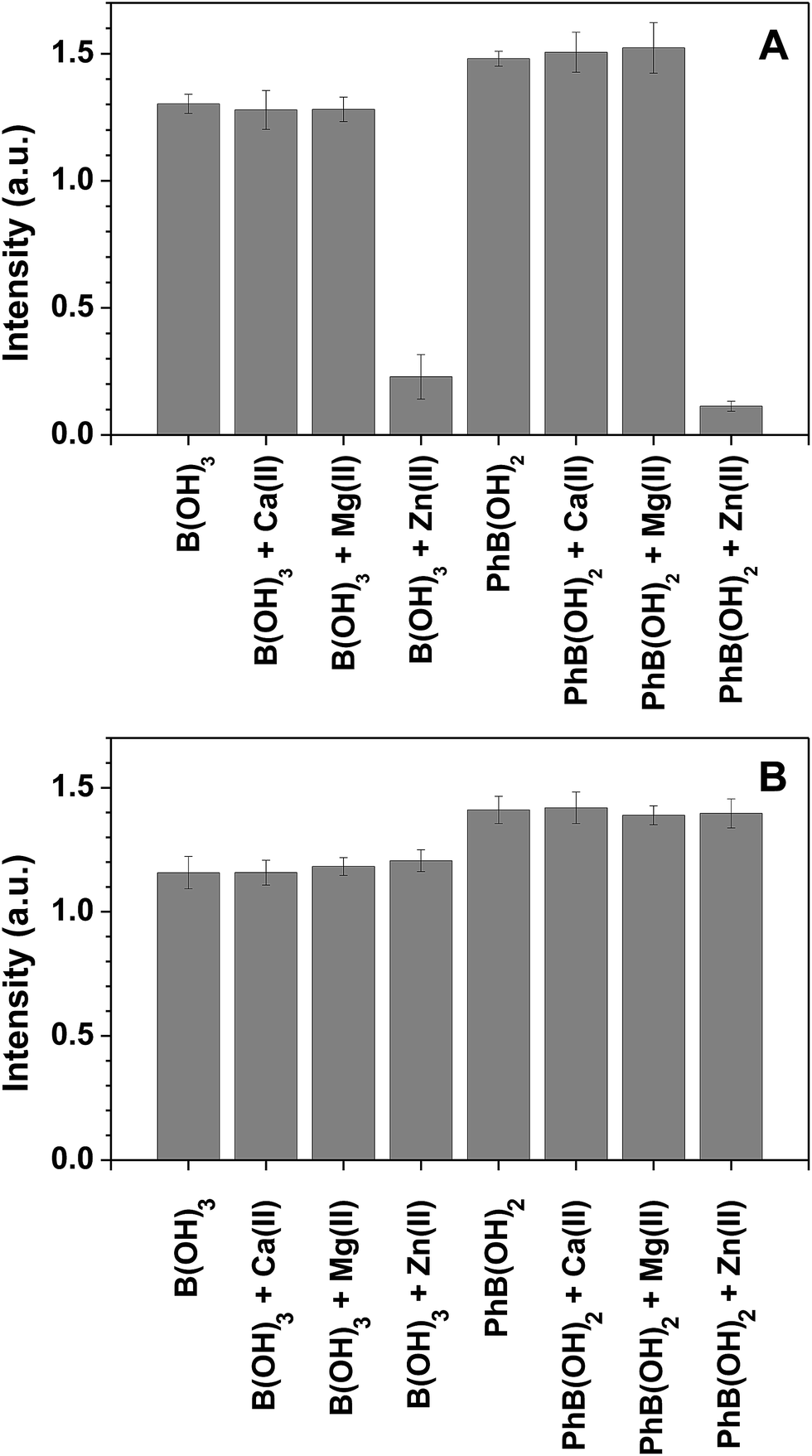 | ||
| Fig. 5 Relative fluorescence intensity of 7a-PMAA (6 × 10−5 M of 7a) in the presence of 38 ppm of boron and several common metal ions 1 mM of Ca(II), Mg(II) and Zn(II), corresponding to an excess of 17 eq. in NH3 buffer (0.02 M) in the absence (A) or presence (B) of 0.01 M EDTA. The spectra were recorded at λexc = 310 nm after two hours equilibrating in the dark. The emission wavelength was 420 nm when in the presence of B(OH)3 or 426 nm when in the presence of PhB(OH)2. | ||
Conclusions
We present a new and very sensitive water soluble polymeric boron sensor, containing an asymmetric substituted triphenylene in a poly(methacrylic acid) backbone. The polymerizable asymmetric substituted triphenylene sensor unit was successfully synthesized by oxidative cyclization of biphenyl and catechol precursors. This new off–on fluorescent boron sensor, able to detect and quantify ppb amounts of boron in water, was tested for boric acid and phenylboronic acid. The limit of detection of the new sensor is 10 ppb for both boric acid and phenylboronic acid. Our novel polymeric fluorescent boron sensor features large water solubility, excellent boron sensitivity and ease of handling, and can be recovered after use by precipitation in acidic conditions, exhibiting the same performance as the fresh sensor.Experimental section
Materials and methods
Tetrahydrofuran (THF, analytical reagent grade, stabilized with 250 ppm of BHT, Scharlau) was distilled prior to use. 1,4-Dioxane (Scharlau) and N,N-dimethylformamide (DMF, Scharlau) were used as received unless stated otherwise. Triethylene glycol monomethyl ether (≥97.0% Fluka), NaOH (pellets, p.a., EKA), p-toluenesulfonyl chloride (>99%, ACROS), NaH (95%, Aldrich), 1,2-dihydroxybenzene (≥99%, Sigma-Aldrich), K2CO3 (anhydrous, 99%, Fluka), 6-bromo-1-hexanol (97%, Aldrich), triethylamine (≥99%, Sigma-Aldrich), methacryloyl chloride (97%, Aldrich), 4-bromoveratrol (98%, Aldrich), BBr3 (1 M in hexane, Aldrich), 3,4-dimethoxyphenylboronic acid (≥95%, Aldrich), tetrakis(triphenylphosphine)palladium(0) (Pd(P(Ph)3)4, 99%, Aldrich), FeCl3 (97%, Sigma-Aldrich), AIBN (98%, Aldrich), boric acid (≥99.8%, Merck), phenylboronic acid (≥97%, Fluka), ammonium hydroxide aqueous solution (28–30%, Fluka), hydrochloric acid aqueous solution (37%, Riedel-de Haën) and methacrylic acid (with 250 ppm MEHQ as inhibitor, 99%, Aldrich) were used as received.NMR spectra were collected on a Bruker 300 or 400 Ultra-Shield (300 or 400 MHz); chemical shifts (δ) are expressed in ppm, and coupling constants (J) are expressed in Hz. Deuterated chloroform (D, 99.8%) and methanol-d4 (D, 99.8%) from Cambridge Isotope Laboratories, Inc; DMF-d7 (D, 99.5%) from Aldrich. Mass spectra were collected on a Micromass Quattro Micro API using electrospray ionization. Column chromatography was performed using Merck silica gel 60 (0.040–0.0063 mm). Thin layer chromatography was performed using Merck silica gel 60 F254 aluminium plates and visualized by UV light.
Spectroscopic N,N-dimethylformamide (≥99.9%, Aldrich) was used as received for the characterization of hydroxytriphenylene compounds. The aqueous solutions were prepared in demineralized water from a Millipore system Milli-Q 18 MΩ cm.
Synthesis of 2-(2-(2-methoxyethoxy)ethoxy)ethyl benzenesulfonate (1). To a solution of triethyleneglycol monomethyl ether (4.00 g, 0.0243 mol) in 15 mL of dry THF in nitrogen atmosphere, was added under vigorous stirring a solution of NaOH (1.61 g, 0.0414 mol) dissolved in 15 mL of water. This mixture was cooled to 0 °C, and a solution of tosyl chloride (5.57 g, 0.0292 mol) in 15 mL of THF was slowly added dropwise. After the addition, the temperature was raised to room temperature and stirred for 1 hour. The reaction mixture was extracted with diethyl ether (2 × 50 mL) and the organic layers were washed with an aqueous solution of NaOH (1 M). The organic phase was dried with anhydrous MgSO4 and filtered; the solvent was removed under reduced pressure, dried under vacuum, to yield a colourless liquid (6.5 g, 85%) 1H NMR (300 MHz, CDCl3) δ (ppm): 2.37 (s, 3H), 3.28 (s, 3H), 3.43–3.46 (m, 2H), 3.51 (m, 6H), 3.60 (t, J = 4.8 Hz, 2H), 4.08 (t, J = 4.8 Hz, 2H), 7.26 (d, J = 8.07 Hz, 2H), 7.71 (d, J = 8.31 Hz, 2H). 13C (300 MHz, CDCl3) δ (ppm): 21.5, 58.9, 68.5, 69.2, 70.04, 70.06, 71.8, 127.9, 129.8, 132.9 and 144.8. HRMS (ESI-TOF): m/z for C14H23O6S calcd 319.12150; found 319.12099 (M + H+).
Synthesis of 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)phenol (2). A mixture of NaH (55 mg, 2.25 mmol) in dry dioxane (3.0 mL) was stirred under nitrogen atmosphere for 30 minutes. To this mixture, a solution of catechol (0.5 g, 4.5 mmol) in dry dioxane (3.0 mL) was added dropwise, and after 30 min, a second solution, of compound 1 (0.72 g, 2.25 mmol) in dry dioxane (3.0 mL) was added dropwise. The reaction mixture was left under reflux overnight, cooled down, and treated carefully with MeOH to destroy the remaining NaH. The solvent was removed under reduced pressure and a solution of HCl (10%) was added to the resulting yellow oil. The aqueous phase was extracted with dichloromethane. The organic phase was washed with a solution of NaOH (10%) to remove the di-substituted catechol. The basic aqueous phase was then acidified with HCl (1 M) and extracted with dichloromethane, to isolate the desired mono-substituted catechol 2. The organic phases were dried with anhydrous MgSO4, filtered and the solvent removed under reduced pressure to afford 2 as a yellow oil (0.325 g, 28%). 1H NMR (300 MHz, CDCl3) δ (ppm): 3.40 (s, 3H), 3.57–3.69 (m, 4H), 3.70–3.76 (m, 4H), 3.85 (t, J = 3.86 Hz, 2H), 4.18 (t, J = 4.18 Hz, 2H), 6.78–6.84 (m, 1H), 6.89–6.94 (m, 3H), 7.22 (s, 1H). 13C (300 MHz, CDCl3) δ (ppm): 59.00, 69.50, 69.58, 70.43, 70.46, 70.62, 71.90, 114.81, 115.78, 119.76, 122.82, 146.06 and 147.47. HRMS (ESI-TOF): m/z for C13H21O5 calcd 257.13845; found 257.13835 (M + H+).
Synthesis of 6-(2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy) phenoxy)hexan-1-ol (3). To a solution of 2 (0.23 g, 0.89 mmol) in acetone (20 mL), K2CO3 (0.62 g, 4.45 mmol) was added under nitrogen atmosphere. After 30 min, 6-bromohexan-1-ol (0.13 mL, 0.99 mmol) was added to the initial mixture and refluxed overnight. The solvent was removed under reduced pressure, and the solid residue was dissolved in dichloromethane (25 mL) and the resulting suspension filtered. The filtrate organic solution was washed with water (3 × 20 mL), dried with anhydrous MgSO4 and solvent evaporated under reduced pressure resulting in 3 in quantitative yield as a yellow oil (0.32 g). 1H NMR (300 MHz, CDCl3) δ (ppm): 1.46–1.64 (m, 6H), 1.80–1.89 (quint, J = 1.85 Hz, 2H), 3.40 (s, 3H), 3.55–3.58 (m, 2H), 3.65–3.71 (m, 6H), 3.78–3.81 (m, 2H), 3.90 (t, J = 3.90 Hz, 2H), 4.01 (t, J = 4.00 Hz, 2H), 4.18 (t, J = 4.18 Hz, 2H), 6.91 (s, 4H). 13C (300 MHz, CDCl3) δ (ppm): 25.49, 25.88, 29.23, 32.66, 59.05, 62.78, 68.98, 69.81, 70.53, 70.72, 70.86, 71.94, 113.87, 114.83, 120.99, 121.67, 148.73 and 149.31. HRMS (ESI-TOF): m/z for C19H33O6 calcd 357.22740; found 357.22717 (M + H+).
Synthesis of 6-(2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy) phenoxy)hexyl methacrylate (4). To a solution of 3 (0.36 g, 1.01 mmol) in dry dichloromethane (15 mL), triethylamine (0.21 mL, 1.52 mmol) was added under nitrogen atmosphere. The reaction temperature was dropped to −78 °C, in a liquid nitrogen/acetone bath. To this mixture distilled methacryloyl chloride (0.15 mL, 1.52 mmol) was added dropwise and the reaction temperature was then raised to room temperature and left overnight. The solvent and remaining methacrylate, either in the acid or acyl form, were removed under vacuum. The oil residue was washed with water to remove the triethylamine salts and the aqueous phase extracted with dichloromethane (3 × 15 mL). The combined organic phases were washed with a solution of NaOH (10%) (3 × 40 mL) and brine (3 × 40 mL). The organic phase was dried with anhydrous MgSO4, the solvent evaporated under reduce pressure to afford 4 (0.3 g, 70%) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm): 1.41–1.50 (m, 4H), 1.70–1.75 (m, 4H), 1.96 (s, 3H), 3.39 (s, 3H), 3.55–3.58 (t, J = 3.56 Hz, 2H), 3.65–3.70 (m, 4H), 3.76–3.79 (t, J = 3.58 Hz, 2H), 3.88 (t, J = 3.88 Hz, 2H), 4.00 (t, J = 4.00 Hz, 2H), 4.13–4.20 (m, 4H), 5.56 (s, 1H), 6.11 (s, 1H), 6.91 (s, 4H). 13C (300 MHz, CDCl3) δ (ppm): 18.33, 25.70, 25.80, 28.58, 29.21, 59.03, 64.05, 68.94, 70.55, 70.86, 71.94, 114.02, 114.95, 121.06, 121.66, 125.17, 125.22, 129.00, 136.50, 148.73, 149.30 and 167.52. HRMS (ESI-TOF): m/z for C23H37O7 calcd 425.25333; found 425.25338 (M + H+).
Synthesis of 4-bromobenzene-1,2-diol (5). To a solution of 4-bromoveratrol (0.5 g, 2.3 mmol) in dry dichloromethane (20 mL) at −78 °C under nitrogen atmosphere, a solution of BBr3 in hexane (1 M, 5.8 mL, 5.75 mmol) was slowly added dropwise. The reaction temperature was raised to room temperature and left overnight. The solvent was removed under reduce pressure and the residue washed with water, and the aqueous phase was extracted with diethyl ether (3 × 20 mL). The organic phases were dried with anhydrous MgSO4, the solvent was removed under reduced pressure and dried under vacuum to afford 5 as an off-white powder in quantitative yield (0.47 g). 1H NMR (300 MHz, MeOD) δ (ppm): 6.66–6.68 (m, 1H), 6.67–6.80 (m, 1H), 6.90 (s, 1H). 13C (300 MHz, MeOD) δ (ppm): 110.44, 116.22, 117.89, 121.99, 144.57 and 146.28. HRMS (ESI-TOF): m/z for C6H6BrO2 calcd 188.95461; found 188.95457 (M + H+).
Synthesis of 3′,4′-dimethoxybiphenyl-3,4-diol (6a). To a solution of 4-bromobenzene-1,2-diol (0.4 g, 2.16 mmol), 3,4-dimethoxyphenyl boronic acid (0.43 g, 2.38 mmol) and tetrakis(triphenylphosphine)palladium(0) (0.13 g, 0.11 mmol) in degassed toluene (40 mL), was added an aqueous solution of K2CO3 2 M (0.22 mmol, 0.11 mL). The reaction mixture was refluxed at 125 °C for 4 h under argon atmosphere. The solvent was removed under reduce pressure, the residue washed with water, and the aqueous phase was extracted with ethyl acetate (3 × 30 mL). The organic phases were dried with anhydrous MgSO4 and the solvent removed under reduced pressure. The residue was purified by column chromatography with different eluent polarity (1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 up to 1:1 of ethyl acetate:n-hexane). The biphenyl 6a was obtained as a brown powder (0.22 g, 41%). 1H NMR (300 MHz, CDCl3) δ (ppm): 3.83 (s, 3H), 3.86 (s, 3H), 6.80–6.89 (m, 3H), 6.99–7.02 (m, 3H). 13C (300 MHz, CDCl3) δ (ppm): 55.77, 55.86, 110.21, 111.52, 113.75, 115.35, 118.51, 118.80, 133.45, 134.35, 143.76, 144.65, 147.88 and 148.86. HRMS (ESI-TOF): m/z for C14H15O4 calcd 247.09644; found 247.09649 (M + H+).
:3 up to 1:1 of ethyl acetate:n-hexane). The biphenyl 6a was obtained as a brown powder (0.22 g, 41%). 1H NMR (300 MHz, CDCl3) δ (ppm): 3.83 (s, 3H), 3.86 (s, 3H), 6.80–6.89 (m, 3H), 6.99–7.02 (m, 3H). 13C (300 MHz, CDCl3) δ (ppm): 55.77, 55.86, 110.21, 111.52, 113.75, 115.35, 118.51, 118.80, 133.45, 134.35, 143.76, 144.65, 147.88 and 148.86. HRMS (ESI-TOF): m/z for C14H15O4 calcd 247.09644; found 247.09649 (M + H+).
Synthesis of 3,3′,4,4′-tetrametoxybiphenyl (6b). To a solution of 4-bromoveratrol (0.50 g, 2.30 mmol), 3,4-dimethoxyphenyl boronic acid (0.46 g, 2.53 mmol) and tetrakis(triphenylphosphine)palladium(0) (0.14 g, 0.12 mmol) in degassed toluene (40 mL), was added an aqueous solution of K2CO3 2 M (0.24 mmol, 0.12 mL). The reaction mixture was refluxed at 125 °C for 4 days under argon atmosphere. The solvent was removed under reduced pressure, the residue washed with water, and the aqueous phase was extracted with ethyl acetate (3 × 30 mL). The organic phases were dried with anhydrous MgSO4 and the solvent removed under reduced pressure. The residue was purified by column chromatography with different eluent polarity (1
:3 up to 1:1 of ethyl acetate:n-hexane). The biphenyl 6b was obtained as a light yellow powder (0.44 g, 69%). 1H NMR (300 MHz, CDCl3) δ (ppm): 3.94 (s, 6H), 3.97 (s, 6H), 6.94 and 6.97 (d, 2H), 7.08 (s, 2H), 7.10 and 7.13 (d, 2H). 13C (300 MHz, CDCl3) δ (ppm): 56.00, 56.02, 110.42, 111.50, 119.13, 134.25, 148.34, 149.12. HRMS (ESI-TOF): m/z for C16H19O4 calcd 275.12766; found 275.12779 (M + H+).
Synthesis of 6-(6,7-dihydroxy-10,11-dimethoxy-3-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)triphenylen-2-yloxy)hexyl methacrylate (7a). To a solution of 6a (0.06 g, 0.24 mmol) and 4 (0.36 g, 0.85 mmol) in dry dioxane (8 mL), a solution of FeCl3 (0.51 g, 3.12 mmol) in dry dioxane (7 mL) was added under argon atmosphere. The reaction was kept for 90 min. at room temperature and after that time half of the solvent was evaporated and methanol was added to the reaction crude. This mixture was kept overnight in the freezer, to precipitate the desired asymmetric triphenylene. The resulting powder was isolated under filtration and exhaustively washed with methanol and water to ensure that all iron has been removed. The black power was dried under vacuum to afford 7a (0.04 g, 25%). 1H NMR (300 MHz, DMF-d7) δ (ppm): 1.56 (m, 6H), 1.75 (m, 2H), 1.92 (s, 3H), 3.47 (s, 3H), 3.60 (m, 2H), 3.65 (m, 2H), 3.76 (m, 2H), 3.95 (m, 2H), 4.11 (s, 6H), 4.17 (m, 2H), 4.38 (m, 4H), 4.61 (m, 2H), 5.68 (s, 1H), 6.08 (s, 1H), 7.21–7.26 (double singlet, 6H). ESI (+) [M + H]+ = 668.24 m/z.
Synthesis of 6-(6,7,10,11-tetramethoxy-3-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy) triphenylen-2-yloxy)hexyl methacrylate (7b). To a solution of compounds 6b (0.05 g, 0.18 mmol) and 4 (0.27 g, 0.64 mmol) in dry dichloromethane (7 mL), a suspension of FeCl3 (0.38 g, 2.34 mmol) in dry dichloromethane (6 mL) was added under argon atmosphere. The reaction was kept for 90 min. at room temperature and after that time half of the solvent was evaporated and methanol was added to the reaction crude. This mixture was kept overnight in the freezer, to precipitate the hexa, substituted triphenylene. The resulting powder was isolated under filtration and exhaustively washed with methanol and water to ensure that all iron has been removed. The white greenish power was dried under vacuum to afford 7b (0.029 g, 23%). 1H NMR (300 MHz, CDCl3) δ (ppm): 1.54–1.66 (m, 4H), 1.75–1.80 (m, 4H), 1.95 (s, 3H), 3.38 (s, 3H), 3.56 (m, 2H), 3.68–3.74 (m, 4H), 3.85 (m, 2H), 4.03 (m, 2H), 4.13 (s, 12H), 4.18–4.25 (m, 4H), 4.44 (m, 2H), 5.56 (s, 1H), 6.12 (s, 1H), 7.78–7.93 (m, 6H). 13C (300 MHz, CDCl3) δ (ppm): 18.35, 25.87, 28.64, 29.38, 29.69, 56.11, 59.18, 64.63, 69.45, 70.08, 70.65, 70.81, 71.01, 71.98, 104.26, 104.35, 104.42, 107.03, 107.16, 108.38, 123.12, 123.16, 123.19, 123.25, 123.27, 123.32, 123.52, 123.56, 123.97, 125.28, 125.32, 136.48, 148.50, 148.76, 148.82, 148.91, 148.99, and 167.54. HRMS (ESI-TOF): m/z for C39H51O11 calcd 695.34273; found 695.34259 (M + H+).
Acknowledgements
This work was partially supported by Fundação para a Ciência e a Tecnologia (FCT, Portugal) within projects PTDC/CTM-POL/3698/2014, RECI/QEQ-QIN/0189/2012, and UID/NAN/50024/2013. L. A. acknowledges a doctoral grant from FCT, Portugal (PD/BD/113533/2015). S. P. C. A. acknowledges a postdoctoral grant from FCT, Portugal (SFRH/BPD/74654/2010).References
- P. P. Power and W. G. Woods, Plant Soil, 1997, 193, 1–13 CrossRef CAS.
- F. H. Nielsen, Ultratrace elements of possible importance for human health, an update, in Essential and toxic trace elements in human health and disease: an update, ed. A. S. Prasad, Wiley-Liss, New York, 1993 Search PubMed.
- World Health Organization, Boron in drinking-water. Background document for development of WHO Guidelines for drinking-water quality, 2009 Search PubMed.
- World Helth Organization, Guidelines for drinking-water quality, 4th edn, 2011 Search PubMed.
- Counsil Directive 98/83/EC of 3 November 1998, On the quality of water intended for human consumption. Official Journal of the European Communities, p. 330/32.
- A. Şimşek, D. Korkmaz, Y. S. Velioǧlu and O. Y. Ataman, Food Chem., 2003, 83, 293 CrossRef.
- I. López-García, P. Viñas, R. Romero-Romero and M. Hernández-Córdoba, Spectrochim. Acta, Part B, 2009, 64, 179 CrossRef.
- Y. Hattori, M. Ishimura, Y. Ohta, H. Takenaka, T. Watanabe, H. Tanaka, K. Ono and M. Kirihata, Org. Biomol. Chem., 2015, 13, 6927 CAS.
- D. Frath, S. Azizi, G. Ulrich, P. Retailleau and R. Ziessel, Org. Lett., 2011, 13, 3414 CrossRef CAS PubMed.
- S. P. C. Alves, C. Baleizao and J. P. S. Farinha, Anal. Methods, 2014, 6, 5450 RSC.
- N. Boden, R. C. Borner, R. J. Bushby, A. N. Cammidge and M. V. Jesudason, Liq. Cryst., 1993, 15, 851 CrossRef CAS.
- S. Kumar, Liq. Cryst., 2004, 31, 1037 CrossRef CAS.
- S. K. Pal, S. Setia, B. S. Avinash and S. Kumar, Liq. Cryst., 2013, 40, 1769 CrossRef CAS.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053 CrossRef CAS PubMed.
- H. Arora, V. Bhalla and M. Kumar, RSC Adv., 2015, 5, 32637 RSC.
- H. Wu, C. Zhang, J. Pu and Y. Wang, Liq. Cryst., 2014, 41, 1173 CrossRef CAS.
- N. Boden, R. J. Bushby and A. N. Cammidge, J. Chem. Soc., Chem. Commun., 1994, 4, 465 RSC.
- N. Boden and R. J. A. N. Cammidge, J. Am. Chem. Soc., 1995, 117, 924 CrossRef CAS.
- D. Perez and E. Guitian, Chem. Soc. Rev., 2004, 33, 274 RSC.
- H. Bengs, O. Karthaus, H. Ringsdorf, C. Baehr, M. Ebert and J. H. Wendorff, Liq. Cryst., 1991, 10, 161 CrossRef CAS.
- K. Morimoto, T. Dohi and Y. Kita, Eur. J. Org. Chem., 2013, 9, 1659 CrossRef.
- A. Zelcer, B. Donnio, C. Bourgogne, F. D. Cukiernik and D. Guillon, Chem. Mater., 2007, 19, 1992 CrossRef CAS.
- R. C. Borner and R. F. W. Jackson, J. Chem. Soc., Chem. Commun., 1994, 7, 845 RSC.
- R. Freudenmann, B. Behnisch and M. Hanack, J. Mater. Chem., 2001, 11, 1618 RSC.
- R. J. Bushby and C. Hardy, J. Chem. Soc., Perkin Trans. 1, 1986, 721 RSC.
- J. W. Goodby, M. Hird, K. J. Toyne and T. Watson, J. Chem. Soc., Chem. Commun., 1994, 14, 1701 RSC.
- H. Hart, C.-y. Lai, G. Chukuemeka and S. Shamouilian, Tetrahedron, 1987, 43, 5203 CrossRef CAS.
- G. W. Gribble, R. B. Perni and K. D. Onan, J. Org. Chem., 1985, 50, 2934 CrossRef CAS.
- D. N. Nicolaides, R. W. Awad, G. K. Papageorgiou, J. Stephanidou-Stephanatou, A. Terzis and C. P. Raptopoulou, Tetrahedron Lett., 1996, 37, 1097 CrossRef CAS.
- P. Relogio, M. Bathfield, Z. Haftek-Terreau, M. Beija, A. Favier, M.-J. Giraud-Panis, F. D'Agosto, B. Mandrand, J. P. S. Farinha, M.-T. Charreyre and J. M. G. Martinho, Polym. Chem., 2013, 4, 2968 RSC.
- V. Augusto, C. Baleizao, M. N. Berberan-Santos and J. P. S. Farinha, J. Mater. Chem., 2010, 20, 1192 RSC.
- M. Ikeda, M. Takeuchi and S. Shinkai, Chem. Commun., 2003, 12, 1354 RSC.
- R. Nandy and S. Sankararaman, Beilstein J. Org. Chem., 2010, 6, 992 CrossRef CAS PubMed.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, 1991 Search PubMed.
- J. B. Birks, Excimers, Rep. Prog. Phys., 1975, 38, 903 CrossRef CAS.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, 1970 Search PubMed.
- American Chemical Society, Committee on Environmental Improvement and Subcommittee on Environmental Analytical Chemistry, Anal. Chem., 1980, 52, 2242 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |