
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCp2TiCl2-catalyzed hydrocarboxylation of alkynes with CO2: formation of α,β-unsaturated carboxylic acids†
Peng Shaoa,
Sheng Wanga,
Gaixia Du*a and
Chanjuan Xi*ab
aKey Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology, Ministry of Education, Department of Chemistry, Tsinghua University, Beijing 100084, China
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China. E-mail: cjxi@tsinghua.edu.cn
First published on 13th January 2017
Abstract
Cp2TiCl2-catalyzed hydrocarboxylation of alkynes with CO2 (atmospheric pressure) has been reported. A range of alkynes were transformed to the corresponding α,β-unsaturated carboxylic acids in high yields with high regioselectivity. The reaction proceeded with hydrotitanation, transmetalation, and subsequently carboxylation with CO2.
Introduction
α,β-Unsaturated carboxylic acids are important chemicals,1 and can be used as basic materials in the production of plastics, superabsorbent, polymers, and rubbers. Various synthetic methodologies for their synthesis have been developed, for example, oxidation of propene over heterogeneous catalysts at high temperature yielded the α,β-unsaturated carboxylic acids2 and hydrocarboxylation of alkynes utilization of metal carbonyls complexes also formed the α,β-unsaturated carboxylic acids.3 From the viewpoint of sustainable chemistry, the development of efficient routes with high atom and step-economy under mild conditions could be highly desirable. Carbon dioxide (CO2) is an environmental friendly, low-toxic and abundant carbon source. Many reaction have been developed utilization of CO2 as a C1 feedstock in organic synthesis.4 The addition of CO2 to alkynes could serve as a most powerful synthetic route for construction of the α,β-unsaturated carboxylic acids. Recently, Ma and co-workers reported a nickel-catalyzed hydrocarboxylation of alkynes with diethyl zinc and CO2 to form the α,β-unsaturated carboxylic acids.5 In the meantime, Tsuji and co-workers developed a copper-catalyzed hydrocarboxylation of alkynes with CO2 by using hydrosilane as hydrogen source to afford the α,β-unsaturated carboxylic acids.6 On the other hand, direct reaction of vinylmagnesium halides with CO2 could also afford the α,β-unsaturated carboxylic acids, however, the vinylmagnesium reagents are required preparation in advanced and thus limitation their use.7 It was reported that alkynes reacted with isobutylmagnesium bromide (iBuMgBr) in the presence of a catalytic amount of Cp2TiCl2 to afford alkenylmagnesium halides,8 in which isobutylmagnesium bromide is not only as a hydrogen source but also as a transmetallation reagent. Encouraged by this work and as part of our ongoing project on group IV metal complex in organic synthesis,9 Herein we reported a Cp2TiCl2-catalyzed hydrocarboxylation of alkynes with CO2 in the presence of isobutylmagnesium halide to afford the α,β-unsaturated carboxylic acids (Scheme 1). | ||
| Scheme 1 Cp2TiCl2-catalyzed hydrocarboxylation of alkynes with CO2 in the presence of Grignard reagent. | ||
Results and discussion
In initial study, we carried out the first trial in the utilization of diphenylacetylene 1a as the substrate, 5 mol% of Cp2TiCl2 as a catalyst in the presence of isobutylmagnesium bromide in diethyl ether. The reaction mixture was stirred at 30 °C for 6 h. Subsequently, CO2 balloon was connected to the reaction mixture and the mixture was stirred at 30 °C overnight under CO2 atmosphere to obtain an α,β-unsaturated carboxylic acid 2a in 75% yield (eqn (1)).
 | (1) |
Following the hydrocarboxylation of a variety of symmetrical internal alkynes was carried out in the presence of iBuMgBr as hydride source. The representative results are summarized in Table 1. The diaryl alkynes bearing methyl group at any position of the phenyl ring could proceed smoothly and afforded the corresponding diaryl substituted α,β-unsaturated carboxylic acids (entries 2–4). When electron-withdrawing group such as F or Cl was tolerated on the phenyl ring, the hydrocarboxylative acids were obtained with a slightly reduced yield (entries 5 and 6). Notably, for more electron-deficient alkyne, such as di-(4-CF3-phenyl) acetylene was used, no hydrocarboxylative product was obtained. When dinaphthyl or dithienyl substituted alkynes were employed as a substrate, the corresponding α,β-unsaturated carboxylic acids 2h and 2i could also be obtained in acceptable yields, respectively (entries 7 and 8). Moreover, internal alkynes possessing aliphatic substituents could also be applied in this system, giving the corresponding product 2j and 2k in good yield (entries 9 and 10).
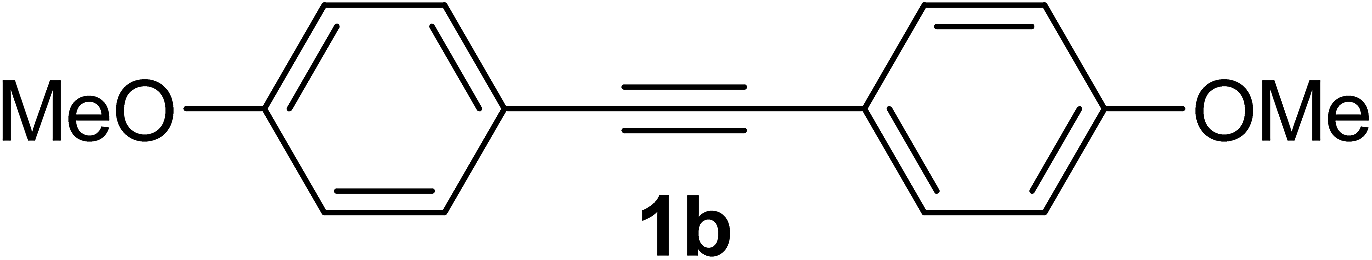


















In further to examine the scope of hydrocarboxylation of alkynes, unsymmetrical internal alkynes were selected as substrates, and the representative results are shown in Table 2. When 1-phenyl-1-propyne 1l was used, the hydrocarboxylation worked well with good regioselectivity, affording desired products with the CO2H group connected to the sp2 carbon atom bearing the phenyl group as a major product in good yield (entry 1). Utilization of unsymmetrical internal alkynes with some other alkyl moieties, such as Et, tBu, cyclopropyl or ether, also proceeded smoothly to afford the corresponding disubstituted acrylic acids with good regioselectivity (entries 2–4 and 6). It is noteworthy that the hydrocarboxylation of the alkyne with hydroxyl group took place regioselectively in moderate yield, albeit one more equivalent of iBuMgBr was necessary (entry 5). The large steric hindrance group TMS was also tolerated in this system with good regioselectivity (entry 7). Alkyne with phenyl and cyclohexenyl groups gave a mixture of two products in 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (entry 8). However, when 1-phenyl-2-thienyl acetylene 1t and 1-(4-F-phenyl)-2-phenyl acetylene 1u were used as substrates, the reaction could proceed, while the regioselectivity of products decreased (entries 9 and 10). When 1-(3,3-dimethylbut-1-yn-1-yl)-4-fluorobenzene 1v was employed in the reaction, less amount of 2v was observed and 3-methylbutanoic acid was obtained as a major product (entry 11). Utilization of 1-(3,3-dimethylbut-1-yn-1-yl)-4-methoxybenzene 1w in this reaction and desired product was obtained in 46% yield (entry 12). When terminal alkynes, for example phenylacetylene and 1-octyne, were applied in this reaction, no hydrocarboxylative products were detected.
:1 ratio (entry 8). However, when 1-phenyl-2-thienyl acetylene 1t and 1-(4-F-phenyl)-2-phenyl acetylene 1u were used as substrates, the reaction could proceed, while the regioselectivity of products decreased (entries 9 and 10). When 1-(3,3-dimethylbut-1-yn-1-yl)-4-fluorobenzene 1v was employed in the reaction, less amount of 2v was observed and 3-methylbutanoic acid was obtained as a major product (entry 11). Utilization of 1-(3,3-dimethylbut-1-yn-1-yl)-4-methoxybenzene 1w in this reaction and desired product was obtained in 46% yield (entry 12). When terminal alkynes, for example phenylacetylene and 1-octyne, were applied in this reaction, no hydrocarboxylative products were detected.
|
|||
|---|---|---|---|
| Entry | Alkyne | Yieldb (%) | Ratio of productc |
| a Reaction conditions: alkyne (1 mmol), Cp2TiCl2 (0.05 mmol), iBuMgBr (1.1 mmol), Et2O (4 mL).b Isolated yield of major product.c NMR ratio.d 2.1 equiv. iBuMgBr was used.e Combined yield.f 3-Methylbutanoic acid was obtained as major product. | |||
| 1 |  |
69 | 2l:2l′ (97:3) |
| 2 |  |
60 | 2m:2m′ (96:4) |
| 3 |  |
51 | 2n:2n′ (96:4) |
| 4 |  |
77 | 2o:2o′ (95:5) |
| 5d |  |
60 | 2p:2p′ (99:1) |
| 6 | 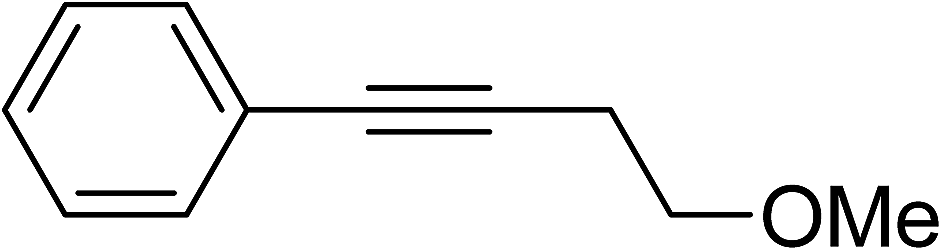 |
41 | 2q:2q′ (99:1) |
| 7 |  |
49 | 2r:2r′ (94:6) |
| 8 |  |
63e | 2s:2s′ (7:1) |
| 9 |  |
51e | 2t:2t′ (2:1) |
| 10 | 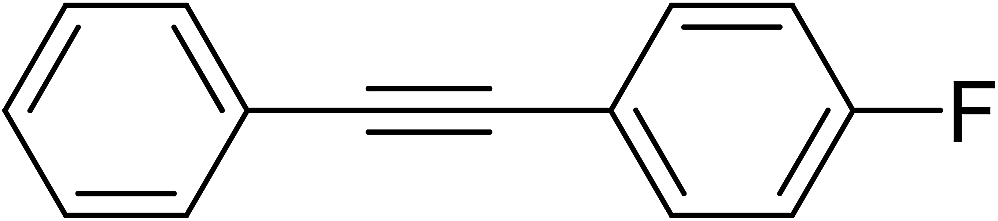 |
54e | 2u:2u′ (2:1) |
| 11f |  |
Trace | 2v:2v′ (100:—) |
| 12 |  |
46 | 2w:2w′ (92:8) |

Based on the above results and literatures about hydromagnesiation of unsaturated hydrocarbons,8 a possible reaction mechanism was proposed as shown in Scheme 2. First, reduction of Cp2TiCl2 by Grignard reagent gives species Cp2TiiBu, which was followed by β-hydride elimination to give Cp2TiH.8 Then addition of alkyne affords an intermediate A, which undergoes transmetallation with the Grignard reagent to produce complex B and release hydromagnesiated product C. The hydromagnesiated C reacts with CO2 and subsequent hydrolysis to afford the α,β-unsaturated carboxylic acids 2. The β-hydride elimination of the complex B to regenerate species of Cp2TiH.
 | ||
| Scheme 2 Possible reaction mechanism. | ||
Conclusion
We have developed Cp2TiCl2-catalyzed an efficient hydrocarboxylation of alkynes with CO2, affording the corresponding carboxylic acids in good yields with high regioselectivity. This protocol provides a convenient pathway for the synthesis of the α,β-unsaturated carboxylic acids by use of CO2 as a renewable source of carbon in organic synthesis.Experimental section
General information
All the reactions were carried out in oven-dried Schleck tubes under N2 atmosphere. Unless indicated, all materials were obtained from commercial sources and used as received. THF and diethyl ether were fresh distilled. Column chromatography was performed on silica gel (particle size 200–300 mesh). 1H NMR and 13C NMR spectra were recorded on 400 MHz at ambient temperature with CDCl3 or DMSO-d6 as the solvent. Chemical shifts (δ) were given in ppm, referenced to the residual proton resonance of CDCl3 (7.26), to the carbon resonance of CDCl3 (77.16). Coupling constants (J) were given in hertz (Hz). The term m, d, s referred to multiplet, doublet, and singlet.General procedure for synthesis of 2a–w
To an oven-dried Schleck tube was added Cp2TiCl2 (5 mol%), which was degassed and refilled with N2 for 3 times. Dry diethyl ether (4 mL) was added via syringe, followed by the dropwise addition of iBuMgBr (1.1 mmol, 2 M in diethyl ether) at room temperature. Then, the alkynes 1a–t (1 mmol) was added. The resulting mixture was stirred at 30 °C for 6 h under N2 atmosphere. Then the solution was stirred overnight under CO2 balloon at room temperature. After that, the reaction was quenched with HCl solution (2 M) till pH = 3–4. Then the mixture was extracted with ethyl acetate (5 mL × 3) and the combined organic phase was dried over anhydrous Na2SO4, followed by filtration and concentration by rotary evaporator. The residue was purified by silica gel (petroleum ether/ethyl acetate = 5/1) to give the corresponding products 2a–w.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21472106 and 21272132) and the National Key Basic Research Program of China (973 program) (2012CB933402). We also thank Dr Chao Chen in Tsinghua University for his kind discussion.References
-
(a) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863 CrossRef CAS
; (b) S. E. Kelly, in Comprehensive Organic Synthesis, Pergamon, Oxford, 1991, vol. 1, pp. 729–817 Search PubMed
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed
- M. Beesu and M. Periasamy, J. Organomet. Chem., 2012, 705, 30 CrossRef CAS
- For selected reviews, see:
(a) P. Braunstein, D. Matt and D. Nobel, Chem. Rev., 1988, 88, 747 CrossRef CAS
- S. Li, W. Yuan and S. Ma, Angew. Chem., Int. Ed., 2011, 50, 2578 CrossRef CAS PubMed
- T. Fujihara, T. Xu, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2011, 50, 523 CrossRef CAS PubMed
-
(a) I. Mutule and E. Suna, Tetrahedron, 2005, 61, 11168 CrossRef CAS
-
(a) H. L. Finkbeiner and G. D. Cooper, J. Org. Chem., 1961, 26, 4779 CrossRef CAS
- For selected reviews, see:
(a) X. Yan and C. Xi, Acc. Chem. Res., 2015, 48, 935 CrossRef CAS PubMed
- X. Wang, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2015, 137, 8924 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25003c |
| This journal is © The Royal Society of Chemistry 2017 |