
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Post-synthetic ion-exchange process in nanoporous metal–organic frameworks; an effective way for modulating their structures and properties
Yasamin Noori
and
Kamran Akhbari
 *
*
School of Chemistry, College of Science, University of Tehran, P. O. Box 14155-6455, Tehran, Islamic Republic of Iran. E-mail: akhbari.k@khayam.ut.ac.ir; Fax: +98 21 66495291; Tel: +98 21 61113734
First published on 12th January 2017
Abstract
Over the last two decades, metal–organic frameworks (MOFs) have received great attention and the number of compounds reported is still growing, which is mainly due to their amazing structures and various pore topologies, accessible cages and their potential applications in different fields, such as gas storage, separation, ion-exchange and catalysis. Among these, ion-exchange processes are considered as the post-synthetic parturient technique as it can achieve dominant applications. There are two primary types of ion-exchange processes (anionic and cationic) and each type may use naturally occurring and synthetic materials. The cation-exchange process at the SBUs of MOFs and metal-exchange involving metal nodes in MOFs, have been previously reported. In this study, we attempt to give an overview of all types of ion-exchange processes that occur in the pores and study their applications. We studied the aspects relating to the new MOFs that are driven during their use in ion-exchange: for example, why ion-exchange occurs, a comparison between the MOFs before and after the ion-exchange process and their current applications.
 Yasamin Noori | Yasamin Noori was born in Tehran, Iran. She received her Bachelor's Degree in Pure Chemistry from the University of Tehran. Yasamin is currently a second year student at the University of Tehran working towards earning her Master of Science Degree in Nanochemistry. She works under the guidance of Assistant Professor Kamran Akhbari and her research focused on the post-synthetic ion-exchange process in nanoporous metal–organic frameworks and the solid-state structural transformations of lead(II) and cadmium(II) coordination polymers and supramolecular compounds using mechanochemical methods. |
 Kamran Akhbari | Kamran Akhbari joined the Department of Chemistry of the University of Tehran in 2013. He earned a Bachelor's Degree in Chemistry from Shahid Beheshti University in 2005, a Master of Science Degree in 2008 and PhD Degree in 2012 in Inorganic Chemistry from Tarbiat Modares University. He passed a post-doctoral fellow under the supervision of Prof. Ali Morsali at Tarbiat Modares University. He has published more than sixty papers and his current research focuses on the crystal engineering of coordination polymers and supramolecular compounds, metal–organic frameworks (MOFs) and their potential applications in energy storage materials, separation, drug delivery and catalysis. In 2013, he won the second place in the Khwarizmi Young Award for his work in the field of modulating methane storage in nanoporous anionic MOFs via a post-synthetic cation-exchange process. |
1. Introduction
Over the last two decades, supramolecular compounds have monopolized the attention of researchers and the number of their compounds being synthesized is still growing.1–3 A wide branch of supramolecular compounds is “coordination polymers”. CPs are obtained from the coordination bonds formed between metal centers and appropriate ligands.4 CPs offer significant advantages over conventional molecular compounds due to their very low solubility in conventional organic solvents and water and much higher thermal stability.1,5 Recently, CP nanocrystals, with finite repeating units, have aroused a growing interest due to their particular properties that differ from the conventional bulk CP.6–9 As the prototype of polymeric crystal engineering, CPs are an intriguing class of hybrid materials, which exist as infinite crystalline lattices extended from inorganic vertices (metal ions or clusters) and organic ligand struts via coordination interactions.10 The structure and properties of coordination polymers depend on the coordination habits and geometries of both metal ions and connecting ligands.11,12 Metal coordination polymers represent an important interface between synthetic chemistry and materials science.13 Metal–organic coordination polymer is a general term used to indicate an infinite array of metal ions linked by coordinated ligands.13 A coordination compound is any compound that contains a coordination entity. A coordination entity is an ion or neutral molecule that is comprised of a central atom, usually that of a metal, to which is attached a surrounding array of atoms or groups of atoms, each of which is called a ligand.14 This is a general term that incorporates a wide range of architectures including simple one-dimensional chains15 with small ligands to large mesoporous frameworks like metal–organic frameworks.16 The diversity in both the focus and scientific basis of the researchers involved has led to numerous terminology suggestions and practices for this class of compounds and several subgroups within them. While “coordination” seems entirely reasonable to use at this level, a generic term describing both 2D- and 3D-coordination polymers namely “coordination network solids” may be useful.17 Porous metal–organic frameworks are zeolite analogues with constant porosity and adsorption capacity.18 In the formation of MOFs, several parameters are involved, such as the metal and its coordination possibilities, the nature of the counter anion, the metal-to-ligand ratio and flexibility of the organic building blocks.19 MOFs have attracted a substantial amount of attention because of their unique properties as highly tailorable microporous materials20 and their applications in gas sorption,21–23 catalysis,24–27 separation,28,29 drug delivery,30,31 and the synthesis of nanomaterials.32–34 Ion-exchange is one of the interesting properties of anionic or cationic MOFs that do not have neutral frameworks.35–38 To date, two bunches of exchange that occur in the metal nodes and secondary building units (SBUs) of MOFs, have been studied. In the first group, metal-exchange involving metal nodes and metalated struts in MOFs has been reported. Their discussion focused on the system where metal cations are exchanged directly in the node (Fig. 1).39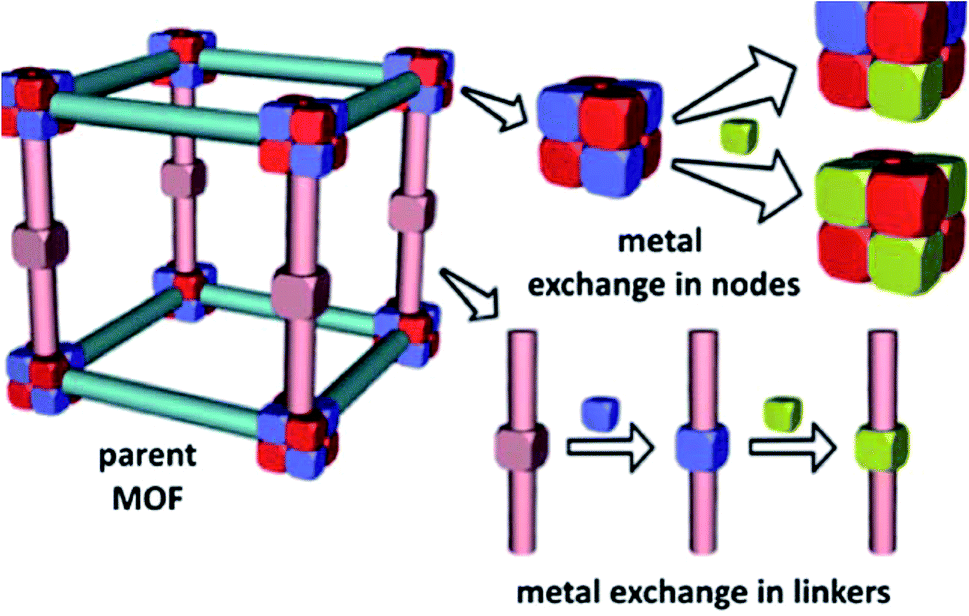 | ||
| Fig. 1 Metal-exchange within MOF nodes and metal-exchange in metal-containing linkers. Reproduced with permission from ref. 39, copyright 2013, Royal Society of Chemistry. | ||
The other group studied the cation-exchange occurring in the SBUs of MOF's. They also limited their discussion to the substitution that occurs at the SBUs and not in the pores.40 In addition, both of them studied cation-exchange, while the ion-exchange that occurs at MOFs is not confined to cation-exchange. Recently, post-synthetic exchange (PSE) of MOFs through the metathesis of organic linkers has been discovered, as well as metal ions in the SBUs of MOFs (Fig. 2) have been revealed.41,42 Similar cation and anion-exchange reactions with nanoparticles and other inorganic materials have been apperceived; however, the observation of a likewise phenomenon in MOFs is relatively novel.43 There are two primary types of ion-exchange, organic and inorganic. Most inorganic ion-exchanges occur in small pore sizes and thus, the kinetics of the ion-exchange is slow, though organic exchangers, such as polymer resins, display rapid ion-exchange.44
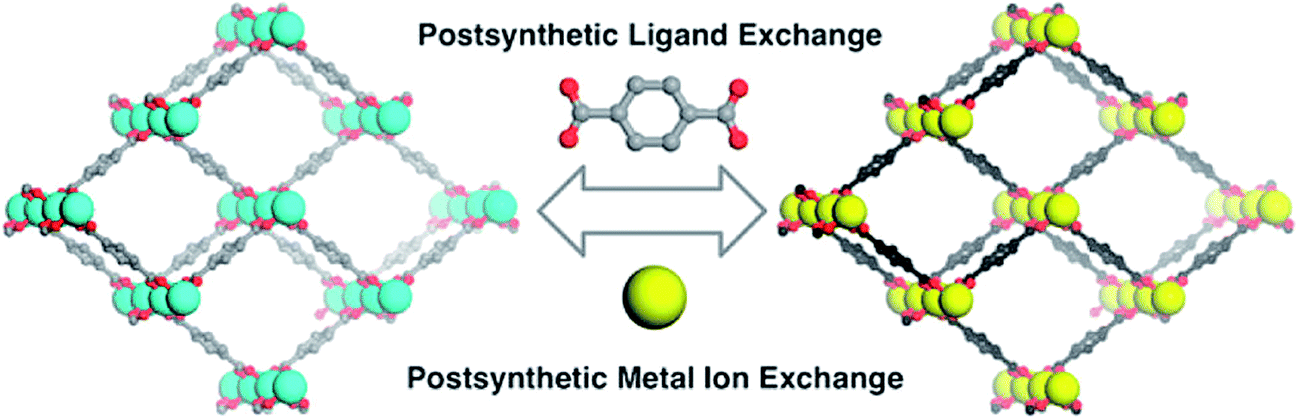 | ||
| Fig. 2 Post-synthetic ligand and metal ion-exchange (PSE) processes. Reproduced with permission from ref. 42, copyright 2012, American Chemical Society. | ||
In the other aspect, ion-exchange can be divided into cation-exchange and anion-exchange. This study outlines the available observations of all types of exchange that occurs in the pores of MOFs and the general trends and future studies can be outlined. We organized the data around cation-exchange and anion-exchange. All known examples of substitution in the MOFs pores and relevant details are reported. We also note that we have limited our discussion to the exchange that occurs in the pores and not at the SBUs or nodes. In addition, we focused on the applications that have been attained through ion-exchange, such as the separation of gases, catalysis, adsorption and luminescence properties. A comparison between MOFs has also been carried out in these cited cases via ion-exchange. Ion-exchange has already yielded some amazing results and new materials that have not been accessible otherwise, but the limit of its use for architecting new MOFs in a systematic and predictive manner depends on understanding its mechanism. This tutorial review is envisaged to provide a blueprint towards this goal.
2. Ion-exchange in metal–organic frameworks
To prepare metal–organic frameworks, a wide range of metal centers and multi-purpose organic ligands, which can be befitted for ion-exchange have been used.45 If we can predict which MOFs are capable to ion-exchange, it will become a logical tool for synthesizing new materials with targeted properties. When the factors that generate exchangeable ions in the pores are elucidated, particular materials may be selected for ion-exchange including cation-exchange and anion-exchange, and their exact compounds may be designed. In this section, we classify two branches of ion-exchange based on their examples, reported cases and their applications.2.1 Anion-exchange
In this content, it has been well known that the counter ions can play an important role in the coordination interactions46 and for MOFs that have cationic coordination frameworks, the anions are generally included for charge balance and/or serve as templates in the empty spaces available. As a result, the anion-exchange properties for such MOF materials have been extensively investigated (Fig. 3).47 In addition, if a neutral open framework can be oxidized, it will contain free counter anions in the channels or pores, which may be applied in anion-exchange materials.35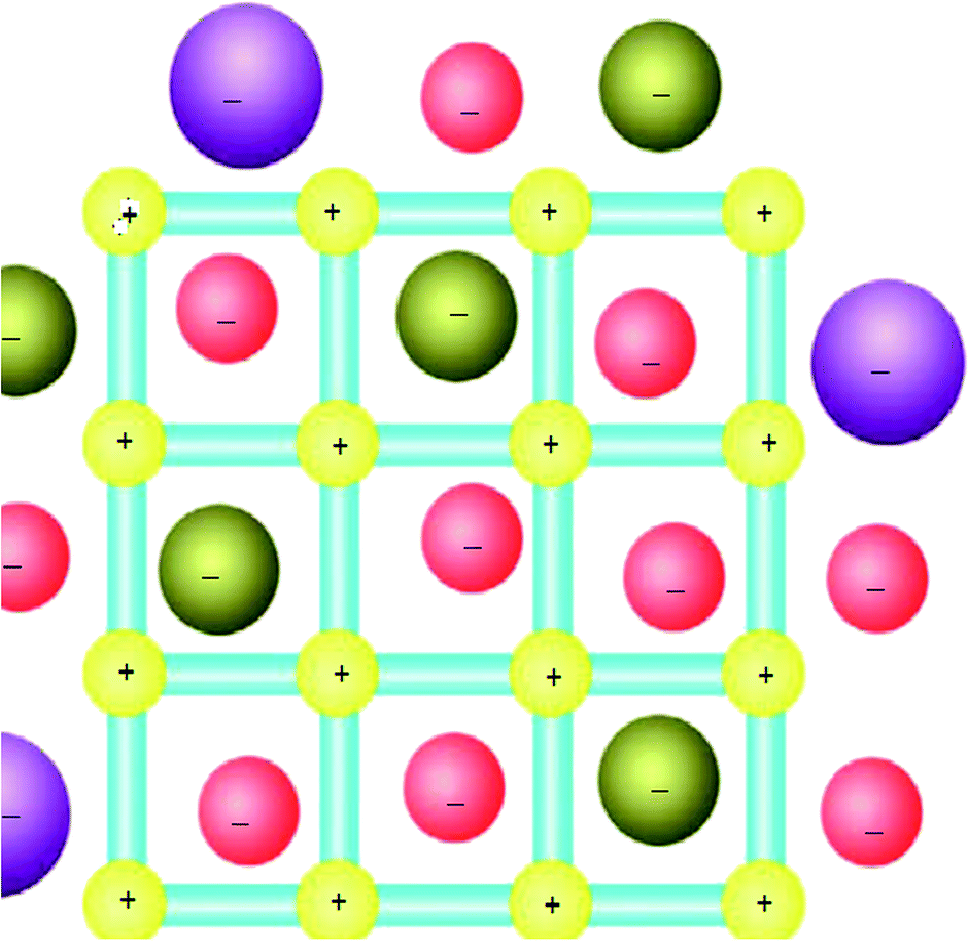 | ||
| Fig. 3 The MOFs-based ion-exchange system (with a cationic framework). Reproduced with permission from ref. 47, copyright 2013, Nature publishing group. | ||
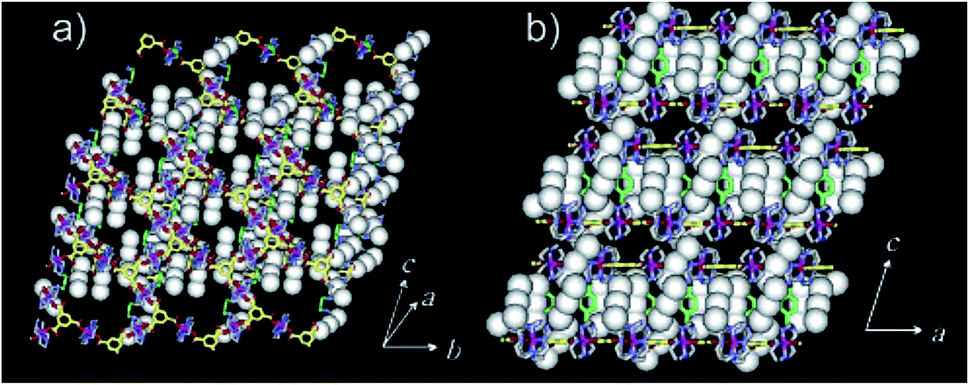 | ||
| Fig. 4 The X-ray structure showing oxidized framework with I3− and I2 included in the channels. (a) Top-view. (b) Side-view. Reproduced with permission from ref. 49, copyright 2004, American Chemical Society. | ||
As another example, a new MOF with zeolite topology, constructed from a tetrahedral building block, has been reported. The novel open-framework metal–organic polymer was [{Cd(H2O)3}34(N4C6H12)17]Cl68·46H2O·68DMF (2). Anion-exchange was carried out for the cationic framework by substituting SCN− for Cl− in the cages.50
Several dipyrromethane (dipyrrinato, diprrin) coordination complexes that are generally used to prepare the structures found in a class of organic ligand MOFs reported before,51 have been described by X. Z. Kiang et al. In addition, they have reported a more executive and systematic investigation of these dipyrin-based metalloligand systems describing 14 new MOF structures. The topology generated upon the formation of the MOFs was found to be robust in certain cases, as demonstrated by anion-exchange. Among the 14 new MOFs, anion-exchange of (MOF-Co/AgBF4-1) (3) and (MOF-Co/AgoTf-1) (4) has been investigated. Crystals of 4 were grown according to the procedures used to prepare 3. Once single crystals were formed, the mother liquid was removed and replaced with a solution of tetrabutylammonium tetrafluoroborate quickly. An identical experiment was performed using MOF 4 and tetrabutylammonium hexafluorophospate. In this case, the PF6− anion was exchanged. The ability of the large PF6− anion to completely replace the triflate anion in 4 strongly suggests that the template effect observed in this MOF was not exclusively due to the size of the anion. In their experiment, during the anion-exchange process, no change in crystal morphology or stability was observed. The PF6− anions occupy the same positions in the crystal lattice as the displaced triflate anions (Fig. 5).52
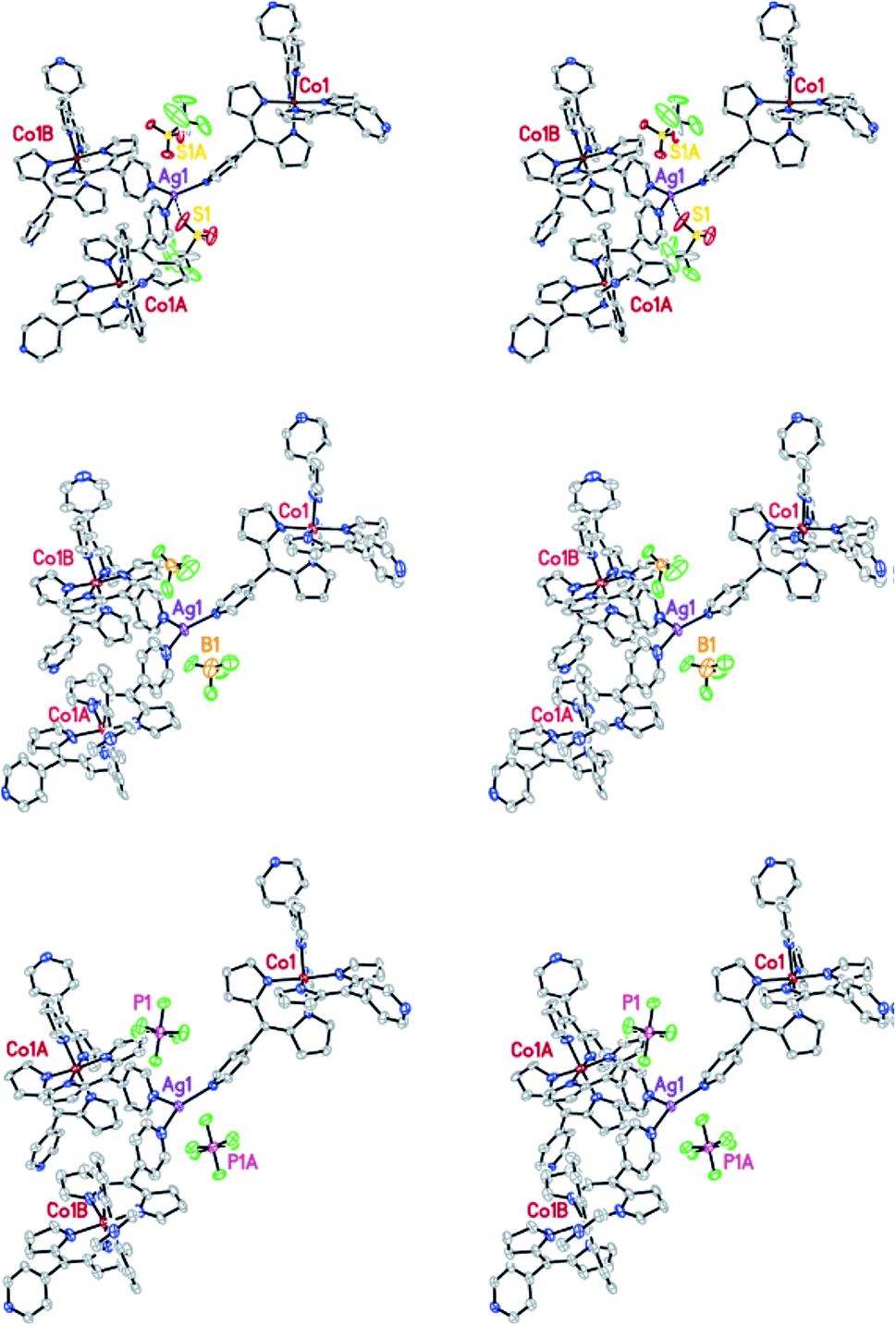 | ||
| Fig. 5 The stereo-view of MOF-Co/AgOTf-1 highlighting the location of the anions before (top) and after anion-exchange. Reproduced with permission from ref. 52, copyright 2006, American Chemical Society. | ||
A novel polymeric coordination complex [Ni(timpt)2](ClO4)2 (5) has been synthesized via a solvothermal reaction using a template ligand with Ni(ClO4)2·6H2O by Sh. Y. Wan and co-workers. The anion-exchange of this MOF is another studied case. Their research was the first example of a self-penetrating MOF that was established from a flexible three-connecting ligand. The powder of this MOF was suspended in an aqueous solution of NaNO3 to allow possible anion-exchange process to occur. Both bands of NO3− and ClO4− anions have similar intensities. Among the anion-exchange, the ClO4− anions in the complex were exchanged with NO3− anions. The anion-exchange was also carried out using NaBF4 and the same partial anion-exchange was observed.53
Reversible anion-exchange of porous MOFs has also been studied. Two novel new rigid tripodal arene core based nitrogen ligands have been synthesized and used in the synthesis of {[Ag3(1,3,5-tris)2X2]X}n (X = ClO4− (6), X = NO3− (7)). The counter-ions located in the cationic frameworks can be exchanged reversibly. This observation indicates that these MOFs can act as zeolite-like porous materials for anion-exchange. The powder of the first MOF was suspended in water containing excess NaNO3. The uncoordinated ClO4− ions exchange with the nitrate ions. To indicate the reversibility of the anion-exchange process in more detail, the exchanged solid was suspended in an aqueous solution of NaClO4. It is very important to confirm that the anion exchange occurred in the solid state. Their experiments display the anion-exchange of this MOF occurs completely through the entire porous structure and was not just a surface phenomenon. The reversible anion-exchange for the second MOF was also carried out in the same way.54
Another study on selective anion-exchange properties has been recognized for a series of 3D microporous Cu coordination polymers. Their research describes a novel 3D MOF {[Cu(2-(2-pyridyl))2](ClO4)(H2O)1/2}n (8) and an unusual anion-exchange was observed for this material. In this case, the crystalline sample of MOF was suspended in an aqueous solution of NaX, where X represents the anion to be used in the exchange process. Notably, the ClO4− in the MOF can be completely replaced by C6H5COO−. They also reported that the inverse anion-exchange reaction cannot be observed upon mixing the exchange product and NaClO4 in water under similar conditions. Further experiments show that the ClO4− in the first MOF cannot be replaced by familiar inorganic anions, such as BF4−, OAc−, NO3− and Cl−, and even the other analogous organic carboxylates, such as O–CH3–C6H5COO−, m-CH3–C6H5COO−, p-CH3C6H5COO−, picolinate, nicotinate and isonicotinate.55
In other study, a research group proposed a rational approach to synthesize MOFs that include OH− ions based on salt inclusion into alkaline-stable MOFs. Thus, they chose ZIF-8 (as the mother framework) and reported a novel MOF containing hydroxide ions (NBU4)m(A)n{Zn(mim)2}6 (9). The NBU4+ ions are adequately stable in 9 for the MOF to impound the OH− ions. To examine the anion-exchange of OH− ions, the researchers stirred NSBU-ZIF-8 powder in an aqueous solution of NaOH and found that the included OH− ions were exchangeable. Their experiment was the first example of a MOF including free OH− ions. Other anions included in NBU4-ZIF-8-OH were likely to be HCO3− or CO32−. The existence of CO32− ions probably inhibits the complete anion-exchange with OH− ions.56
The last example involves the preparation of MOF [M(β-diketone)3Ag3]-X2·solv (10). The most promising property of this network relies on its ability to exchange the anions present in their channels. The research group selected [ZnL3Ag3]X2·solv as the host network {X = BF4−, ClO4−, CF3SO3− and PF6−} and performed ion-exchange experiments using this material (Table 1). These anions can be easily exchanged in single-crystal to single-crystal processes.
| NaBF4 | NaClO4 | NaNO3 | KPF6 | |
|---|---|---|---|---|
| [ZnIIL3Ag3](BF4)2 | Y | Y | P | |
| [ZnIIL3Ag3](ClO4)2 | N | P | P | |
| [ZnIIL3Ag3](CF3SO3)2 | Y(30 min) | Y(2 h) | Y(15 min) | Y(2 h) |
| [ZnIIL3Ag3](PF6)2 | P(24 h) | Y(3 h) | Y(2 h) |
Some preferences could also be elicited; for example (a) nitrate and perchlorate very readily displace BF4−, PF6− and triflate anions, (b) triflate is very ambulant and can be replaced by other anions and (c) triflate can be easily displaced using PF6−, while substitution of perchlorate was partial. The channel walls are covered by low-coordinated silver ions (UMCs) and these exposed centers can interact with anions. Other exchange experiments were carried out using organometallic complex anions. The anionic dinuclear organometallic complex [Re2(CO)6(μ-OH3)]− can be presented into 1D channels by exchange. In this case, the exchange was slower and after 1 day it was still only partial. This result of the group's experiments has opened the way to use their framework as a heterogeneous support for the foundation of organometallic compounds employed in homogeneous catalysis.57
2.2 Cation-exchange
Sometimes the exchange of metal ions or organic cations within anionic MOFs can modulate the chemical properties of a MOF58,59 and this can be achieved simply by merging the sample into a solution containing a certain metal salt or organic cation to generate an anionic MOF. Prevalent ion-exchange resins with cationic groups and exchangeable counter ions are still the standard ion-exchange materials.60 Porous zeolite-like metal–organic framework (ZMOF) materials with Rho and Sod topologies have also been studied. In the original ZMOFs, the negatively charged frameworks are charge-compensated by extra-framework ions inside the cavity, which can undergo ion-exchange with alkali metals.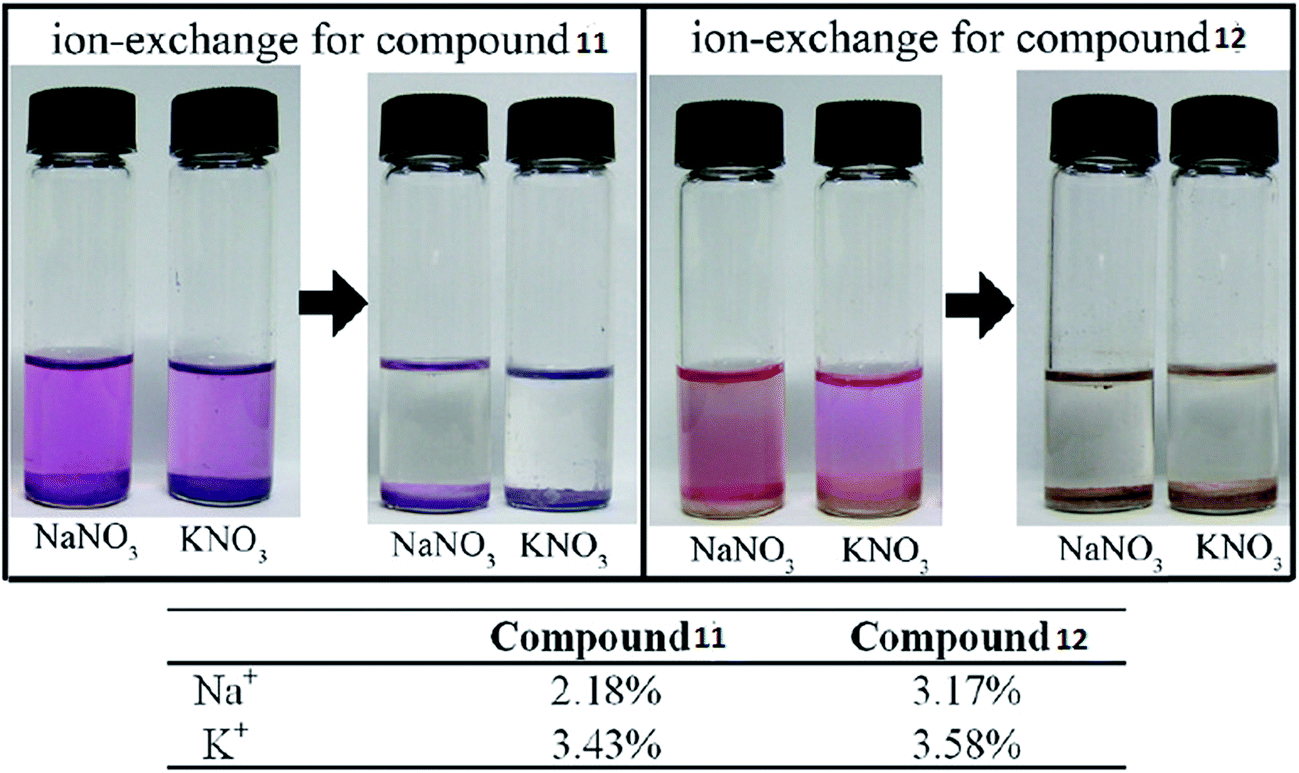 | ||
| Fig. 6 Photographs of the ion-exchange process in compounds 11 and 12. Reproduced with permission from ref. 63, copyright 2011, American Chemical Society. | ||
In another case, the first example of a microporous MOF was reported by J. Yu and co-workers. The reaction of H3BTB and InCl3 in N,N-dimethyl formamide/1,4-dioxane/H2O afforded colorless crystals of (HDMA)3[In3(BTB)4]·12DMF·22H2O (ZJU-28) (13). The ion-exchange process studied showed that the cationic Me2NH2+ molecules were readily exchanged by metal cations, such as Cu2+, Ni2+ and Eu3+. They examined the potential of 13 to encapsulate different pyridinium hemicyanine chromophores to expand non-linear optical MOF materials (Fig. 7).64
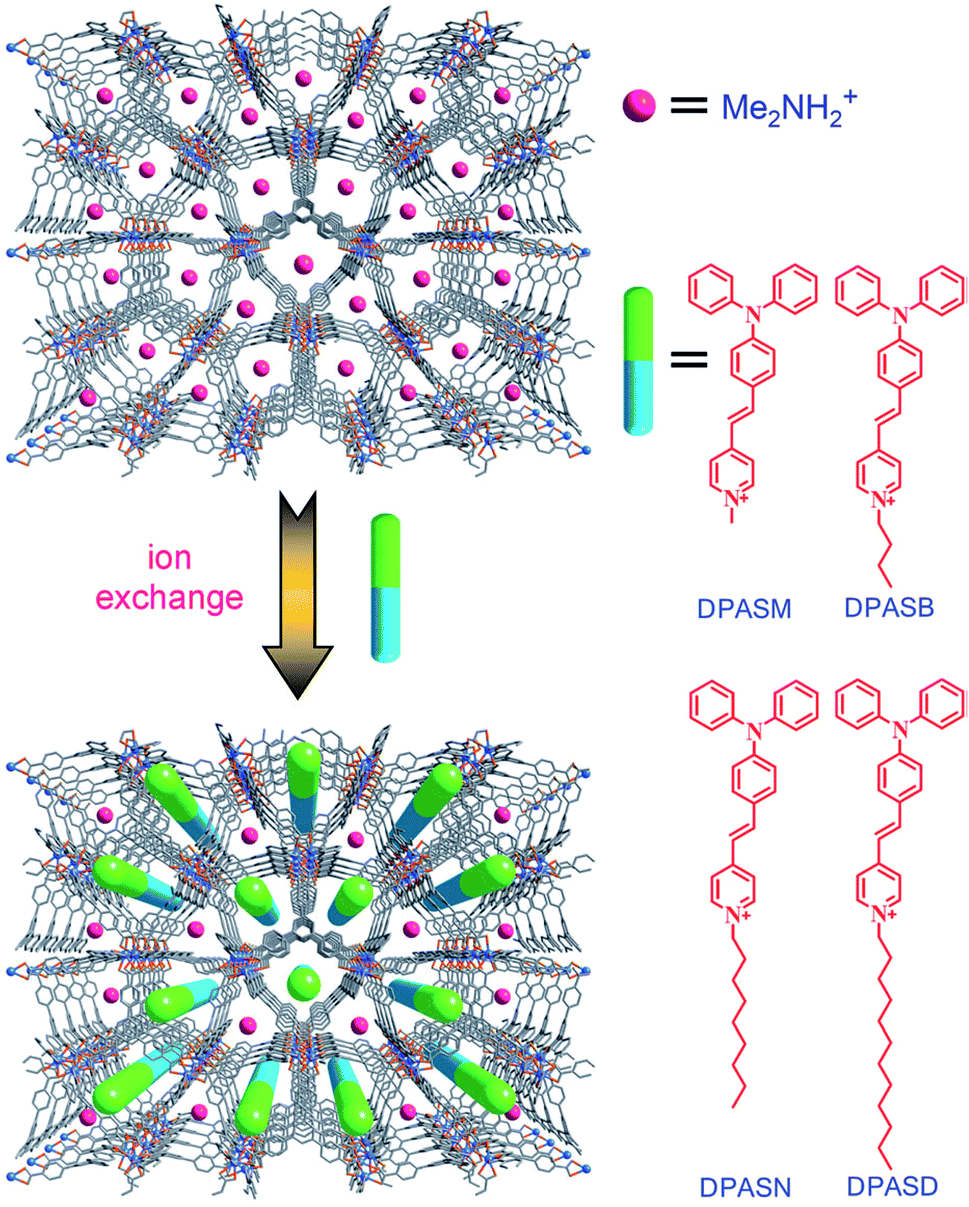 | ||
| Fig. 7 A schematic of the pyridinium hemicyanine chromophores combined into ZJU-28. Reproduced with permission from ref. 64, copyright 2012, John Wiley & Sons. | ||
Four new MOFs based on flexible V-type tetracaboxylate ligands have been synthesized by Sh. Xiong and co-workers. On the group of these MOFs, [HDMA] [In(mdip)]·2.5DMF·4H2O (14) was a 3D anionic framework that exhibits selective metal ion-exchange of HDMA+ ions with metal ions (M = Li+, Na+, K+, Mg2+, Ca2+, Sr2+, Ba2+, Cu2+ and Fe3+). Other work carried out by this group examined the selectivity of metal ion-exchange. The results indicated that Li+ ions were hardly absorbed during the ion-exchange process. Perhaps this is the main reason of the high selectivity towards Na+ and K+ observed during the metal ion-exchange process. They proposed that the metal ions interact with the carboxylic oxygen atoms in compound 14. The second group of metals used in the metal-ion exchange was Mg2+, Ca2+, Sr2+ and Ba2+. Their observations indicate that compound 14 selectively adsorbs more Mg2+ than Ca2+, Sr2+ and Ba2+. As a result, the ion-exchange selectivity of the different metal ions was ascribed to the traits of the different metal ions and their interactions with the carboxylic oxygen atoms in the framework.65
Another research study on a series of isostructural metal–carboxylate frameworks [HDMA][M2(bptc)(μ3-OH)(H2O)2] (15) and cation-exchange of one of them with M = Co has been reported. This MOF has an anionic framework and 3D channels occupied by in situ generated [HDMA]+ cations and was selected to perform ion-exchange experiments. The prepared crystals were immersed into a saturated solution of MCl (M = Li+, Na+, K+). During this process, organic cations [HDMA]+ can be selectively exchanged by alkali-metal cations. Interestingly, a comparison of the ion-exchange experiments showed that the crystal has a strange preference for Na+ rather than for Li+ and K+. After ion-exchange with alkali cations, the crystals preserve their original shape although the surface roughness seems to increase. In addition, the intensity of the N–H bands of [HDMA]+ during the exchange process was measured. After ion-exchange, the intensity of these bands became weak. All the experimental data show that alkali metal cations entered into the channels via an exchange interaction.66
Another MOF that has been studied is based on a high nuclearity cyclic-type cluster with a large internal cavity. The novel MOF is H2Na4[Cu12(OH)6(PZ)6(BTC)6]·23H2O (16) and the ion-exchange studies of this material have been reported. Because the framework in this MOF, contains large internal voids that contain in addition to solvent water molecules the Na+ alkali metal cation, the cation-exchange properties of the crystal could be investigated through facile cation-exchange experiments. Ion exchange was performed with the alkali metal cations Li+, K+ and Cs+ and transition metal cations Cu2+, Ni2+ and Mn2+.67
The synthesis of a gallium-based MOF [Ga6(1,3,5-BTC)8·6DMA·3DMF·26H2O] (Ga·MOF-1) (17) is another example that has been regarded in this context. This MOF contains an anionic porous network. Disordered positively charged ions and solvent molecules are present in the pores. A porous anionic framework capable of ion-exchange is a potential precursor for adding alkali-metal cations into the pores.68 These positively charged molecules can be exchanged with alkali-metal ions. Saturated solutions of LiNO3 and NaNO3 in DMF were separately added to the compound. Accordingly, the Li+ and Na+ ions were displaced. These results have confirmed that the presence of the cationic species within the pores make the MOF a suitable candidate for ion-exchange studies with alkali metal cations.69
Other compounds that have anionic frameworks are zeolite-like metal–organic frameworks (ZMOFs). Comparable to the prevalent inorganic zeolite, ZMOFs are anionic and show facile ionic exchange capacity. Ion-exchange studies show that the organic cations in the pores can be fully exchanged. Two important classes and topologies of ZMOF are Rho-ZMOF and Sod-ZMOF. The research group reported a novel strategy to construct ZMOFs with extra-large cavities. The extra-large cavities of {rho-ZMOF, HPP} [In48(C5N2O4H2)96(C2N3H15)24(DMF)36-(H2O)192] (18) can adapt neutral molecules in addition to the cationic guest molecules that can only be exchanged with other cations. For Na+-exchanged Rho-ZMOF formulated as [Na48+(H2O)282][In48(C5N2O4H2)96] (19), the concentration of charge carriers was higher than a typical rho zeolite. As a result, the anionic identity and the large approachable voids of Rho-ZMOF allowed the full exchange of the HPP cations with various organic and inorganic cations.70
Another research group has recently extended the design strategy that has allowed the construction of MOFs with zeolite-like topologies based on edge development. The synthesis of Rho-ZMOF, In48(C5H2N2O4)96(C7N3H15)24(C3H7NO)36(H2O)192 (20) and it's ion-exchange processes, in order to explore the potential application of this novel material, have also been investigated. They have displayed the ion-exchange of 20 in two branches: (i) metal cation-exchange: the Na+-exchange Rho-ZMOF was introduced; the ion-exchange data showed that the organic cations in the pores can be completely exchanged in an aqueous solution at room temperature. In addition, the Na+-Rho-ZMOF indicated that the large amount of water molecules residing in the cages of the exchanged structure can be completely removed at temperatures around 100 °C. (ii) Organic cation-exchange: the large size of the cavities affiliated with Rho-ZMOF, together with the negative charge of the cavity internal, affords a chance to exchange and combine cationic organic molecules. In this case, they chose to exchange acridine orange (AO); the reason of this choice was its size. It is smaller than the window dimension of Rho-ZMOF and accordingly the molecules can freely diffuse into the large cavities.71
3. Applications of the ion-exchange process in metal–organic frameworks
3.1 Applications of the anion-exchange process
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. The anion exchange experiments were performed for the ITC-3 (21) and ITC-4 (22) MOFs using OG2− as the anionic guest. The result showed that about 47.2% of the NO3− in 22 was replaced by OG2− when the ion-exchange reached equilibrium after 4 days. The charge and size of the guest are the two most important factors in the ion-exchange process. Dyes were selected as the guest to study the ion-exchange process. In this study they arranged the organic dyes in two groups; the first group contained dyes with different charges but similar size (methylene blue, MLB+; sudan I, SDI0; acid orange 7, AO7−; orange G, OG2−; and new coccine, NC3−), whereas, the dyes in the second group had different sizes, but same charges (OG2−; ponceau 6R, P6R2−; croscein scarlet 3B, CS3B2−; croscein scarlet 7B, CS7B2−; acid black 1, AB12− and methyl blue, MB2−). The separation process based on size- and charge-selective ion-exchange was mainly observed in the solutions containing two different dyes. Such selective anion-exchange, controlled by the structural characteristics of the MOFs, is advantageous and complementary to conventional ion-exchange resins because of its suitability for molecules within the ∼100 Da < Mw < ∼1000 Da range. Furthermore, the controlled release of guest anions make these P-MOFs potential candidates for use as drug carriers. The reticular synthesis and the large number of possible MOF structures make it possible to build an extensive library of hosts that can be used for guests (anionic in particular) of different sizes.72
:1 molar ratio. The anion exchange experiments were performed for the ITC-3 (21) and ITC-4 (22) MOFs using OG2− as the anionic guest. The result showed that about 47.2% of the NO3− in 22 was replaced by OG2− when the ion-exchange reached equilibrium after 4 days. The charge and size of the guest are the two most important factors in the ion-exchange process. Dyes were selected as the guest to study the ion-exchange process. In this study they arranged the organic dyes in two groups; the first group contained dyes with different charges but similar size (methylene blue, MLB+; sudan I, SDI0; acid orange 7, AO7−; orange G, OG2−; and new coccine, NC3−), whereas, the dyes in the second group had different sizes, but same charges (OG2−; ponceau 6R, P6R2−; croscein scarlet 3B, CS3B2−; croscein scarlet 7B, CS7B2−; acid black 1, AB12− and methyl blue, MB2−). The separation process based on size- and charge-selective ion-exchange was mainly observed in the solutions containing two different dyes. Such selective anion-exchange, controlled by the structural characteristics of the MOFs, is advantageous and complementary to conventional ion-exchange resins because of its suitability for molecules within the ∼100 Da < Mw < ∼1000 Da range. Furthermore, the controlled release of guest anions make these P-MOFs potential candidates for use as drug carriers. The reticular synthesis and the large number of possible MOF structures make it possible to build an extensive library of hosts that can be used for guests (anionic in particular) of different sizes.72In another study, a fluorene-based Cu(II)-MOF has been reported as a visual colorimetric anion sensor and separator based on anion-exchange [CuL2(H2O)0.5](NO3)2 (23). The anion-exchange of this compound has also been studied. During the exchange, the NO3− anions can be replaced by other anions, such as: Cl−, Br−, I−, SCN− and N3−. The compound displayed a change in color as illustrated in Fig. 8a.
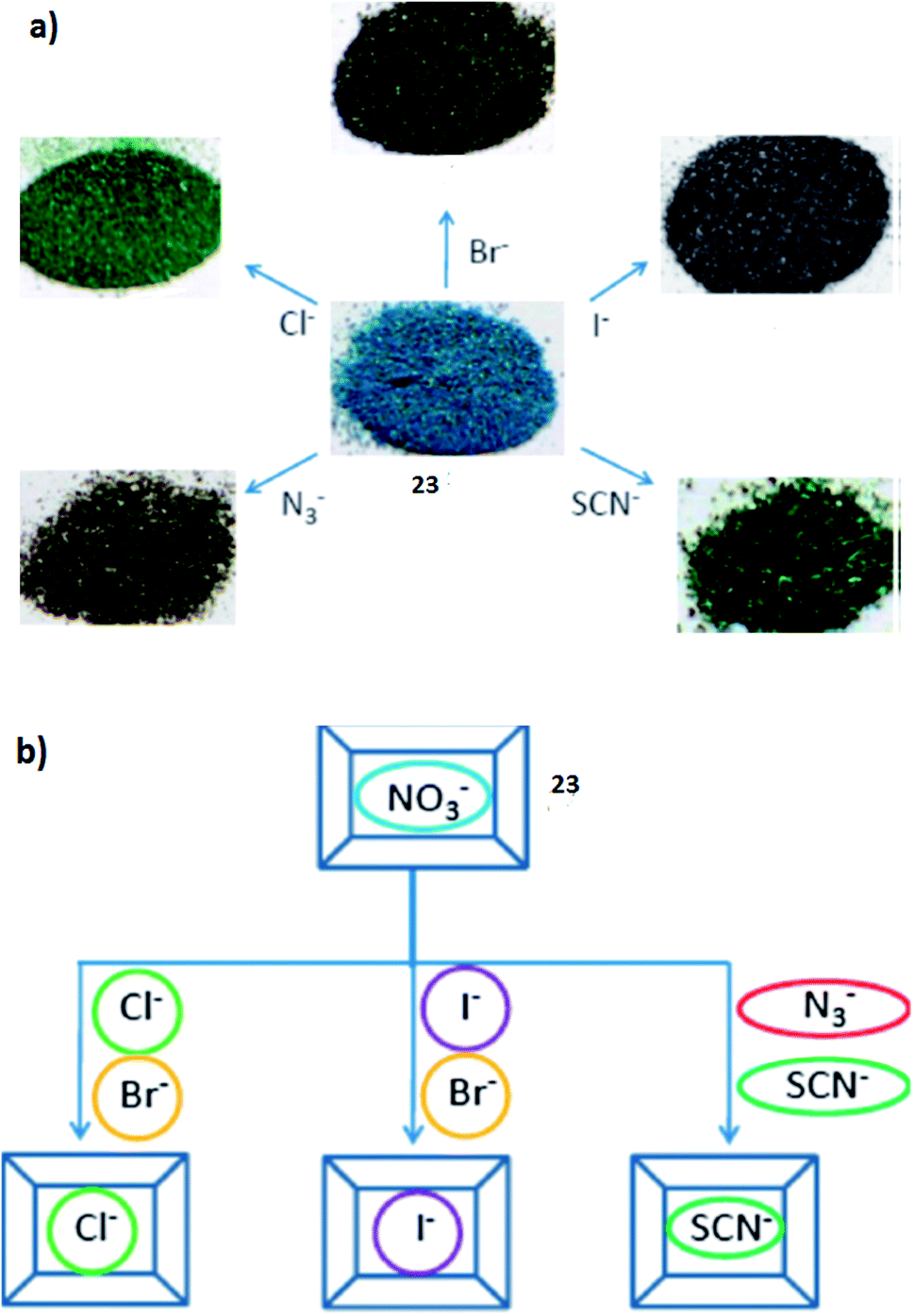 | ||
| Fig. 8 (a) The color changes of compound [CuL2(H2O)0.5](NO3)2 (23) as a result of anion-exchange. (b) The anion selectivity based on the compound. Reproduced with permission from ref. 73, copyright 2011, Royal Society of Chemistry. | ||
Compared to anion-exchange, the anion selectivity, catching a particular anion in the presence of other anionic competitors, is a more important task. As illustrated in Fig. 8b, the trigonal planar NO3− anion in compound 23 can be replaced by both spherical halide anions Cl−, Br− and I−, and linear triatomic anions SCN− and N3−, and therefore they envisioned that the compound can be used an anion separator to separate anions with similar geometry.73
The separation of anions from aqueous mixtures is another novel approach in this case. R. Custelcean, et al. presented a new approach for the separation of anions from water by their selective crystallization within MOFs functionalized with urea hydrogen-bonding groups. One of the separation approaches that has been studied is the anion-exchange of ZnCl2(BPu) (24), ZnI2(BPu) (25) and ZnBr2(BPu) (26). The most significant results of this study were that Cl−, Br− and I− were exclusively crystallized against the ClO4−, NO3− and SO42− oxo-anions, and the observed selectivity was opposite of the Hofmeister series typically governing the extraction of anions from water, whether by solvent extraction, anion-exchange with polymeric resins or coordination polymers.74
With the development of modern industry, anion pollutants have become a severe problem. Among the common anion pollutants, heavy-metal pollutants, normally in their oxo-/hydroxo anionic forms, such as Cr2O72−, MnO4−, pertechnetate (TcO4−), arsenate (AsO43−) and selenite (SeO32−, etc.) have been observed in water.75 They have been a focus of concern because they cause serious damage to human health and the environment, which is a worldwide problem that is listed as a priority by the U.S. Environmental Protection Agency.76 Recently, a few three-dimensional cationic MOFs have been used for the exchange and removal of heavy-metal pollutant anions, such as ReO4−, CrO42− and TcO4−. One of these priority pollutants is chromate (CrO42− or Cr2O72−): a known carcinogen, which originates from metal-plating, leather-tanning and other industries and is rapidly diffusing.77 However, there have been few reports of Cr2O72− anion exchange in MOFs despite the powerful carcinogenicity and extensive application of this species. Thus far, only several cationic MOFs have been reported to trap chromate or dichromate via ion-exchange. We studied all of these examples and have discussed them one by one. A 3d–4f heterometallic 3D cationic framework, {[Dy2Zn(BPDC)3(H2O)4](ClO4)2·10H2O}n (27), which exhibits high thermal stability and strong alkali resistance has been reported. It is able to capture the pollutant CrO42− unusually fast with a high capacity of 85%. According to the cationic framework and the large pores in 27, several types of anions were selected to carry out the exchange experiments. 0.05 mmol of 27 was placed into a 0.05 mmol aqueous solution of K2CrO4 (10 mL) and the entire exchange process was performed under ambient conditions with slow stirring. Simultaneously, the color of the solid in the solution changed from colorless to yellow, implying the existence of CrO42− in the solid. Similar exchange experiments were further carried out under an excess or very low concentration of K2CrO4. The results indicated that ClO4− in the channels was completely exchanged by CrO42− under an excess concentration of K2CrO4. As is known, wastewater or nuclear waste contains more than one type of pollutant anion, such as halide ions or cyanide ions; therefore, it is indispensable to investigate whether 27 selectively exchanged CrO42− from a mixture of anions. 0.05 mmol of 27 was immersed in a solution containing the mixed anions (NO3−, Cl−, Br−, I− and CrO42−) containing 0.1 mmol of each anion for 24 h. The results of the different analyses indicated that only ClO4− exchanged with CrO42−, confirming its discrimination against halide ions. Generally, the release of pollutant anions is more difficult due to the strong interaction between the metal oxo-anions and cationic frameworks.91 However, 57% and 25% CrO42− in 27 can be released upon immersion in solutions of K2CO3 and Na2SO4, respectively, and no release in other NaX solutions (X = NO3−, Cl−, Br−, I− and ClO4−) occurred.78
Another research group reported the unprecedented capacity and selectivity for trapping permanganate (MnO4−), perrhenate (ReO4−) and chromate (CrO42−) using the cationic MOF, Ag2(4,4′-bipy)2(O3SCH2CH2SO3)·4H2O (28). Unlike the strong affinity of LDHs for carbonate, 28 selectively trapped these problematic oxo-anion pollutants in record levels over all the previously reported materials. The mechanism of the high selectivity and adsorption capacity occurs via a crystal transition upon oxo-metal uptake. For a detailed investigation of the anion uptake capacity of 28, permanganate and perrhenate were chosen as models for pertechnetate since all are group 7 oxo-anions. The as-synthesized 28 was introduced into a solution containing KMnO4. In addition, the anion exchange reaction with commercially available synthetic hydrotalcite (magnesium aluminum hydroxycarbonate, Aldrich) in both the uncalcined and calcined form was carried out; after 48 h of exchange under the same conditions as in the case of 28, only 3% and 18% of the anions were adsorbed by the LDHs, respectively.
The reason for the greater uptake by their material was due to crystal transformation by the host and the stability of the oxo-anion in the resultant structure, rather than a typical equilibrium-driven anion exchange. The as-synthesized 28 may also be used for anion exchange with a much higher adsorption capacity based on a stronger interaction towards the oxo-anion pollutant displayed by the resulting crystal structure, which can be seen to take the place at each end of the EDS molecule. The incoming permanganate anion plays a crucial role in high capacity trapping by forming a thermodynamically favorable crystal structure. Indeed, the same structure is formed if the nitrate- or perchlorate-containing version of 28 is used as the starting material. In order to further demonstrate the potential application of 28 towards pertechnetate abatement, perrhenate was also investigated. The resultant exchange solution was monitored only by ICP due to the overlap between EDS and perrhenate in the UV region. ReO4− trapping by 28 was even more rapid than that of MnO4−, reaching over 90% removal from solution in only 24 h, saturating in 48 h with 95% removal. Although the crystals were not suitable for single crystal analysis, the overall adsorption capacity of 1.90 (mol mol−1) was comparable with the previous permanganate study, as expected. The adsorption capacity based on weight, however, reaches an exceptionally high value of 602 mg g−1 based on the molecular weight of ReO4− compared to that of MnO4−. Chromate exchange and adsorption capacity were also studied for 28. UV-vis showed that 25%, 33%, and 41% of the chromate was exchanged after 8, 24, and 48 h, respectively. Moreover, the selectivity of anion trapping in 28 was studied with multiple competing anions of varying excess concentration in the presence of a low concentration of the target anion pollutant. A selective reaction with 28 was introduced into an aqueous solution comprised of KMnO4 and NaNO3. The measurements of the crystals after anion exchange indicated that only MnO4− entered the cationic material to replace the EDS anions. NO3− was not present inside the solid. The selectivity towards perrhenate was also investigated with both of the competing nitrate and carbonate anions, with the same trapping behavior as permanganate. Perchlorate is another problematic anion that diffuses rapidly and widely and is emitted from rocket fuel waste along with other sources including pyrotechnics.79 A mixed anion solution indicated that perchlorate was preferred over nitrate by 28. The anion affinity for 28 thus displayed the following order, with the problematic metallate pollutants topping the list: MnO4− > ReO4− > ClO4− > CrO42− > NO3− > CO32−.80
The uptake capacities of dichromate ions in the MOFs have the potential to be improved, and the dichromate trapping processes of these MOFs are rarely known to occur in a single-crystal-to-single-crystal (SCSC) fashion. A research group reported two cationic MOFs with large nanotubular channels, [Zn2(Tipa)2(OH)]·3NO3·12H2O (29) and [Zn(Tipa)]·2NO3·DMF·4H2O (30), which show fast ion-exchange and high uptake capacity for Cr2O72− through an SCSC process. Given that 29 has a highly charged cationic layer structure, the anion exchange of oxo-anion pollutants was studied and chromate was chosen as an initial model. The anion exchange was performed under ambient conditions by simply placing crystals of 29 into the dichromate solution. The resultant exchange solution was monitored by liquid UV-vis spectroscopy and the result indicated that the Cr2O72− in solution promptly entered into the channels of 29 by exchange with NO3−. Moreover, the color of the solid in the solution changed from colorless to yellow, implying the existence of Cr2O72− in the solid. Similar ion-exchange experiments on 29 were further carried out in a solution of K2Cr2O7, where 93.3% of dichromate in the solution was taken up by 29. Subsequently, a slow decrease in the Cr2O72− concentration was observed, and no further decrease in the anion concentration was detected after 10 h and 18 h. Release tests were also carried out to evaluate the regeneration ability of the ion-exchanger. For 29, the samples were centrifuged, rinsed with water and dried in air after being submerged in a solution of dichromate. Then, the solid was placed in the same volume of solution in the presence of nitrate. Such a trapping and release process was performed for five continuous cycles and 29 still retained an approximately 87% release efficiency. Generally, it was understood that the release of the pollutant anions was more difficult due to the strong interactions between the metal oxo-anions and the cationic framework. The reversibility of 30 was also tested. Unlike 29, there was almost no dichromate released in the presence of an excess of nitrate solution. In a solution of Na2ClO4, the exchange efficiency dropped to 63% after five trapping-release cycles. Selectivity is as important for anion trapping as is capacity. Given the high-capacity and rapid ion exchange function of 29, the selective adsorption was verified. The dried sample of 29 was immersed in a solution containing mixed anions (Cl−, Br−, NO3−, and Cr2O72−). The results indicate that the adsorption of Cr2O72− was not disturbed by the other ions. Nevertheless, when an equal molar amount of CO32− was added to the abovementioned solution, the uptake efficiency of Cr2O72− was reduced sharply to approximately 17%, indicating that the CO32− ion was a strong competitor to the Cr2O72− ion. When the dried sample of 29 was immersed into a solution containing mixed anions (Cl−, Br− and NO3− at concentrations of 0.10 mmol L−1 or 0.40 mmol L−1, and Cr2O72− at a concentration of 0.01 mmol L−1), the adsorption of Cr2O72− was still at a high efficiency. Once more, the dried sample of 29 was immersed in the mixed solution (Cl−, Br−, NO3−, SO42− and ClO− at a concentration of 0.10 mmol L−1 for each anion, and Cr2O72− at a concentration of 0.01 mmol L−1) and the ion-exchange efficiency of Cr2O72− was drastically decreased. The sample of 29 (or 30) after ion-exchange was subjected to single-crystal X-ray diffraction, but only the structure of 29 containing the guest anions was obtained. It was observed that the Cr2O72− anions fill into the channels to balance the framework charge, indicating that ion-exchange occurred exactly via a single-crystal to single-crystal process. Moreover, weak C–H⋯O interactions between the framework and Cr2O72− anions were found. The phenomenon indicated that the anions, which need to go over very little resistance, are easily exchanged, and the large channels provide a fairly convenient passage for the diffusion of anions. These results can explain that 29 displays highly-efficient ion-exchange with excellent reversibility. More importantly, it demonstrates that the abovementioned adsorption capacity of Cr(VI) is correct because dichromate was identified as the principal component.81
Another example is the report that introduces an unprecedented 3D cationic MOF comprised of nanoscale cages composed of [Ag2(btr)2]2ClO4·3H2O (31), which showed fast exchange, high trapping capacity, and good selectivity for the capture and separation of Cr2O72− in water. Interestingly, the exchange of Cr2O72− in 31 was performed via a single-crystal to single-crystal (SCSC) process. Given that a great deal of ClO4− was located in the voids of the framework of 31 and that enough large channels were available for anion access, anion exchange experiments were performed using Cr2O72− as a model. When crystals of 31 were immersed in an aqueous solution of equimolar K2Cr2O7, the Cr2O72− concentration in the solution decreased by 37% and 60% after 1 h and 3 h, respectively. Subsequently, a slow decrease in the Cr2O72− concentration was observed, and there were no evident changes in anion concentration and solution color between 24 h and 48 h. This emphasized that 31 has a superior performance for Cr2O72− capture with fast exchange and high capacity. When they used double the molar amount of 31 with respect to Cr2O72− under the same conditions, 66% and 90% of Cr2O72− was exchanged after 0.5 h and 6 h, respectively. During the exchange, the solution changed from yellow to colorless. The Cr2O72− capture ability was further investigated in more dilute aqueous solutions. As shown in their article, when an excess of 31 was placed in a 14.7 ppm aqueous solution of K2Cr2O7, the crystals changed from colorless to yellow after 48 h. Their analysis indicated that the concentration of remnant Cr2O72− was 0.09 ppm, suggesting the almost complete capture of Cr2O72− (99.4%) by 31. These results implied that 31 was a promising material for the removal of low-concentrations of Cr2O72− from wastewater. In comparison with anion exchange, the anion selectivity may be more important and challenging. Selective exchange experiments were examined for mixtures of anions. When the crystals of 31 were immersed in an aqueous solution containing BF4−, CF3SO3−, NO3− and Cr2O72− for 24 h, the crystals gradually turned yellow. In the IR spectrum of the resultant crystals, only the characteristic band arising from Cr2O72− was observed, suggesting a good selectivity for Cr2O72− over the other anions. The selectivity of 31 may be attributed to the stronger interactions of Cr2O72− with the cationic framework in comparison with the other anions.82
Most MOFs are not stable in aqueous solutions and studies have shown that Zr-cluster based MOFs can be stable in aqueous solutions and are becoming promising candidates for pollutant elimination.83,84 A cationic Zr-MOF, ZJU-101 (32), (prepared from a post-synthetic process of MOF-867, Zr6O4(OH)4·(BPYDC)6 with trifluoromethanesulphonate) has been reported, which not only exhibits the highest Cr2O72− removal capacity, but can also selectively remove Cr2O72− from its aqueous solution within a very short period of time. 32 was synthesized from MOF-867 (ref. 85) through a post-synthetic modification, in which methyl groups were added to the pyridyl sites to form the cationic framework. In this study, the research group also compared the behavior of 32 with MOF-876. To evaluate and compare the adsorption activity of MOF-867 and 32, anion exchange was performed by simply placing the dried adsorbent into a solution of dichromate and the solution was stirred under ambient conditions. According to the data, which are shown in the article, after 12 h, only a very small amount of dichromate (about 22%) was removed when MOF-867 was used as adsorbent. After the crystals of 32 were immersed in the aqueous solution of dichromate, the Cr2O72− ion concentration decreased significantly by 87.8% in 5 minutes. After 10 minutes, over 96% of dichromate was removed from the aqueous solution. Apparently, 32 was superior to MOF-867 during the capture of Cr2O72− ions. Once the dichromate was removed by 32 from the aqueous solution after 10 minutes, the solution became colorless after centrifugation. It was clear from the adsorption test that, for efficient anion exchange, the balanced ions within the pores of 32 made a significant contribution to the removal of dichromate. To further confirm the adsorption capacities of the MOFs towards dichromate, adsorption isotherms were collected for MOF-867 and 32 at room temperature. As the uptake was greatly influenced by the concentration of the Cr2O72− solution, different concentrations of the Cr2O72− solution were applied to determine the uptake. The overall adsorption capacity of 32 for the dichromate reached up to 245 mg g−1, which was the highest among the porous materials used for dichromate removal reported to date. The uptake capacity of MOF-867 for dichromate was only 53.4 mg g−1, which was significantly smaller than that of 32. Given the high-capacity and fast ion-exchange function of 32, the selective adsorption tests of 32 were also carried out for a variety of anions. The dried sample of 32 was immersed in a solution of Cr2O72− together with n-fold molar excess of disturbing ions (n is equal to 0, 3, 6, 9 and 12) containing equal molar amounts of Cl−, Br−, NO3−, SO42−, I− and F−. The results indicated that the adsorption capacity of 32 for Cr2O72− was still as high as 88% when there was a 3-fold excess of disturbing ions. When there was a 12-fold excess of disturbing ions, the adsorption of Cr2O72− was still at a high efficiency of 81%. The highly selective absorption was attributed to the strong columbic attractions between the framework and Cr2O72−, and the matching of Cr2O72− with the pores.86
A three-dimensional water-stable cationic MOF pillared by a neutral ligand and with NiII metal nodes has been synthesized employing a rational design approach by A. V. Desai and co-workers. Due to the ordered arrangement of the uncoordinated tetrahedral sulfate (SO42−) ions in the channels, the compound has been employed in aqueous-phase ion-exchange applications. This system is the first example of a MOF-based system that absorbs both dichromate (Cr2O72−) and permanganate (MnO42−) ions. The water-stable, three-dimensional cationic MOF is [{Ni2(Lig)3-(SO4)(H2O)3}·(SO4)·x(G)]n (33), (G = H2O and DMF), which was built from a tripodal neutral ligand and contains free sulfate ions. The guest-free phase of 33 acts as a fast and selective adsorbent for the capture of both monovalent and divalent tetrahedral oxo-anions, namely dichromate and permanganate ions (Fig. 9). They sought to harness the ionic functionality imparted by the cationic framework to investigate the anion exchange and loading properties of 33. Due to the hydrolytic stability, robust 3D architecture, and channelized alignment of the free SO42− ions, the ability of 33 to trap dichromate ions was investigated. Crystals of 33 were dipped into an aqueous solution of K2Cr2O7 and the exchange process was monitored using different analysis methods. The partial decoloration of the solution and the change in color of the crystals showed the exchange of SO42− ions with Cr2O72−. They believed that the anion exchange process lead to the occupation of the positions of the free sulfate ions (aligned in the porous channels) by Cr2O72− ions. The group suggested that this swap was possible because one-half of the dichromate ion has a tetrahedron like geometry, even though the entire ion itself is not tetrahedral in shape. The other half of the dichromate anion could then reorganize itself in the free space as the anion position is directed towards the pore. In addition, to test the selectivity of the system for dichromate capture, 33 was dipped into an aqueous mixture of K2Cr2O7 and a salt of either ClO4−, NO3−, BF4− or CF3SO3− ions (separate experiments carried out for each ion). Careful analysis revealed that in all cases, Cr2O72− was selectively incorporated inside the porous matrix over other competing anions. These results validated the hypothesis of employing tetrahedral exchangeable anions for the targeted capture of heavy metal oxo-anions. The uptake order was estimated to be as follows: Cr2O72− > NO3− ∼ ClO4− > BF4− > CF3SO3−. To check if 33 can function as a reversible absorbent, they attempted to desorb the loaded Cr2O72− ions and replaced them with SO42− ions. Although desorption was visible to the naked eye through color changes of both the supernatant and the solid, FTIR spectra suggested the replacement of dichromate ions with SO42− ions. Furthermore, the desorbed phase of 33-Cr2O7 could be used as a Cr2O72− adsorbent, re-adsorbing the same amount of Cr2O72− desorbed in the first cycle. Similarly, 33 was probed as an adsorbent for pertechnetate ion trapping by studying its response towards its congener: the permanganate anion. Crystals of 33 were dipped in an aqueous solution of KMnO4 and the rapid decoloration of the solution with a simultaneous change in the color of the crystals from pale-green to dark-red was observed. Like in the case of Cr2O72− exchange, almost a complete decoloration was noted after 72 h, giving rise to the exchanged phase 33-MnO4. The selective entrapment of MnO4− was explored by dipping 33 into an aqueous solution of KMnO4 containing salts of either ClO4−, NO3−, BF4− or CF3SO3− ions. The FTIR spectra confirmed that competing anions were not included and suggested the selective uptake of permanganate ions. These results further confirmed the hypothesis that a tetrahedral substitutable anion acts as a facilitator for the capture of toxic-metal oxo-anions. An experiment to understand the preferential uptake between MnO4− and Cr2O72− was undertaken by dipping 33-Cr2O7 in an aqueous solution of KMnO4 and separately by dipping 33-MnO4 in a solution of K2Cr2O7. The results suggest that the framework had a higher affinity for MnO4− ions.87
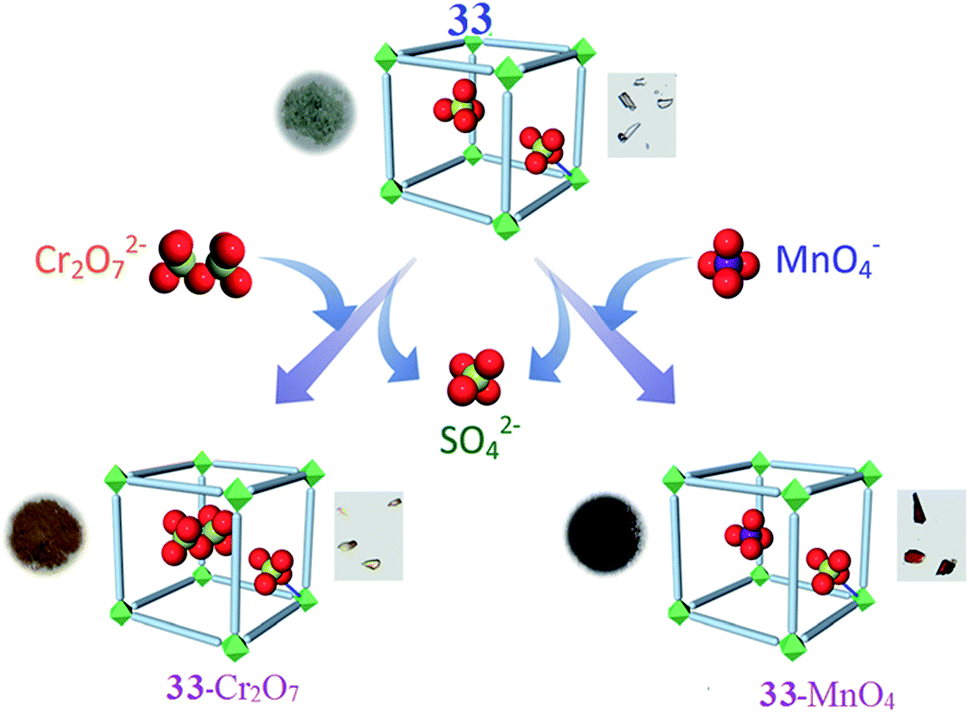 | ||
| Fig. 9 A schematic representation of the capture of heavy-metal oxo-anions by 33 with the concurrent loss of SO42− ions. Photographs of the different phases and crystals of the materials are shown with each representation. Reproduced with permission from ref. 87, copyright 2016, Wiley Online Library. | ||
 | ||
| Fig. 10 (a) The kinetics of diphenylmethane oxidation catalyzed by Cu-MOF-SiF6 (34) and Cu-MOF-NO3 (35). (b) The N2/CO2 gas adsorption–desorption isotherms and MeOH–toluene vapor adsorption–desorption isotherms of [Cu(II)(bped)2(H2O)2(SiF6)]I·4H2O. (c) MeOH and (d) EtOH vapor adsorption–desorption isotherms of [Cu(II)(bped)2(H2O)2(NO3)]I·4H2O. Reproduced with permission from ref. 89, copyright 2011, Royal Society of Chemistry. | ||
Heterogeneous catalysis has been investigated in this branch. In this prospect, reversible anion-exchange and the catalytic properties of two cationic MOFs based on Cu(I) and Ag(I) have been studied. The reported MOFs are SLUG-21 (28) and SLUG-22 (36). Both of them show reversible anion-exchange between organosulfonate and various inorganic species. As primary examples of exchange, they have focused on several inorganic anions, such as the oxo-anions of several metals, nitrate and perchlorate. As a final example of pf anion-exchange capability, they investigated 28 for permanganate and perrhenate trapping. Compound 36 was also employed in the same exchange reaction with nitrate and perchlorate to gain insight into the structure–activity properties. Cationic MOFs are potentially useful for heterogeneous catalysis due to their positive charge compared to the majority of the reported MOFs. The group found that these two materials are catalytically active in heterogeneous ketal formation. Ketalization is an important method to support carbonyl groups in organic synthesis and drug design.90 The reaction needs a Lewis acid catalyst to activate the oxygen of the carbonyl group, which permits glycol to replace the ketone group. Ketalization of 2-butanone, 2-pentanone and benzophenone by ethyl glycol was performed for both 28 and 36.91
Luminescent cationic MOFs with extra-framework anions suggest a dynamic framework and tunable luminescent behavior by exchanging these framework anions with other anions of different shape, size and coordination nature. The luminescent behavior of [{Zn(L)(MeOH)2}-(NO3)2·xG]n (39) (in which G are disordered guest molecules and the linear bichelating ligand L was synthesized by Schiff-base condensation of 4,4′-ethylenedianiline and 2-pyridine-carboxaldehyde in high yield) has been investigated. The 1D channel of the compound was filled with NO3− anions. Anion-exchange experiments using anions with a weak or non-coordinating nature, such as ClO4− and N(CN)2− (type A), and anions with a strong coordinating nature, such as N3− and SCN− (type B), were performed. The framework can easily adjust its channel dimension to encapsulate different guest anions because of its dynamic nature. The anion selectivity experiments show that the order of association of guest anions to the framework was SCN− > N3− > N(CN)2− > ClO4− (Fig. 11a).
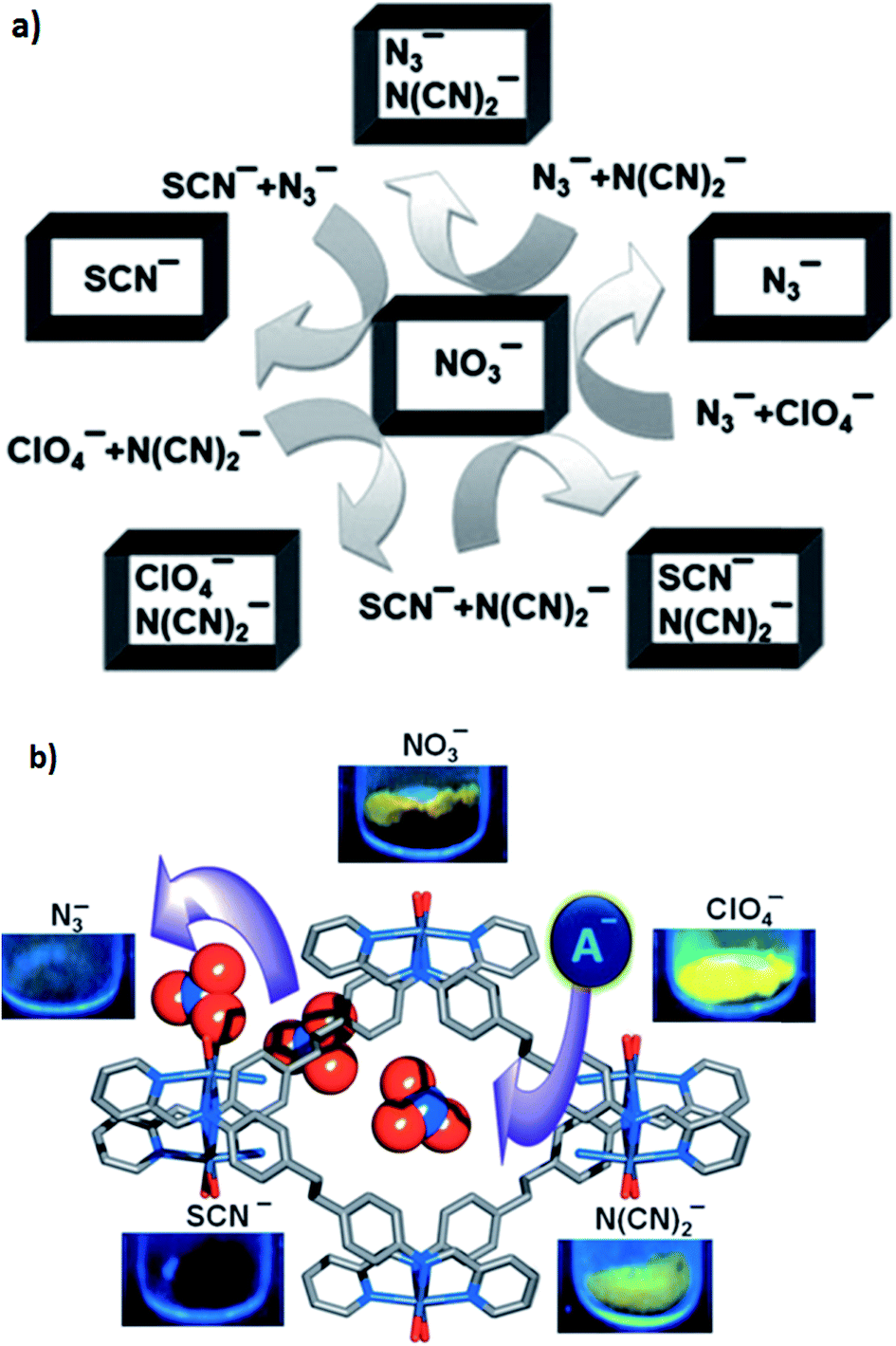 | ||
| Fig. 11 (a) A schematic representation of the anion selectivity with the combination of two anions in [{Zn(L)(MeOH)2}-(NO3)2·xG]n. (b) The effects of anion-exchange on the luminescence properties of [{Zn(L)(MeOH)2}-(NO3)2·xG]n (39). Reproduced with permission from ref. 93, copyright 2013, John Wiley & Sons. | ||
The anion-exchanged materials demonstrated amazing anion-dependent luminescent behavior (Fig. 11b). As a result for the anion-exchanged compounds of type A, a high enhancement of fluorescence was observed and on the other hand, for the anion-exchange compounds of type B, fluorescence quenching was observed.93
Compound 27 was introduced by P. F. Shi et al. previously, which acted as a luminescent probe. To further study the effect of Cr2O72− on the luminescence of 27, solid-state photoluminescent spectra were obtained. The typical luminescence peaks of Dy3+ in 27 were observed at 479 and 573 nm. With Cr2O72− continuously entering into the channels, the luminescent intensity dropped rapidly, originating from the fact that the electron transfer transitions of Cr2O72− decreased the energy transfer from BPDC2− to Dy3+. However, upon the sample containing Cr2O72− being immersed in a solution of Na2SO4 or K2CO3, the intensity may have significantly rebounded with the release of Cr2O72− upon exchange with SO42− or CO32−, which elucidated the capture and release process of Cr2O72−. Thus, 27 may be considered as a luminescent probe of Cr2O72−.78 The effect of anion-exchange on the luminescence properties was also explored using compound 31. 31 emitted a blue-violet light under UV irradiation, whereas the 31·Cr2O7 emission was almost invisible to the naked eye. The solid-state emission spectrum of 31 at room temperature displayed a broad emission band centered at 450 nm upon excitation at 345 nm, a value that was in agreement with the emission observed under UV irradiation. The emission may be attributed to ligand-to-metal charge transfer (LMCT) transition.94 However, the luminescent emission was largely suppressed after the exchange of ClO4− with Cr2O72−. The disappearance of the luminescent emission in 31 was probably because the electron-transfer transitions of Cr2O72− decreased the energy transfer from the btr ligand to AgI. Thus, 31 is considered as a luminescent probe for Cr2O72−.82
In another case, the first example of highly efficient iodine enrichment based on a Cd(II)-triazole MOF via an ion-exchange approach was reported. The report was about [Cd(4-amino)2(ClO4)2]·H2O (41) and its anion-exchange approaches. The experiments indicate that the original ClO4− anions are replaced by I3−. For detection of the enrichment of IO3− in dilute solution by anion-exchange based on the MOF, the crystals of 41 were dipped in an aqueous solution of NaIO3.96
3.2 Applications of the cation-exchange process
Notably, at low temperatures (T < 286 K), acetylene was more tightly retained by the A@42 frameworks compared with carbon dioxide. At higher temperatures (T > 286 K) the reverse situation was found, with C2H2 being eluted before CO2. They also studied the effect of ion-exchange on the separation selectivity towards benzene/cyclohexane mixtures. In the case of the original NH4@42 sample, the composition of the adsorbed benzene/cyclohexane phase reaches a 5:1 ratio, already showing a clear enrichment in the benzene component. This is further substantiated in the case of the Et3NH@42 and Li@42 materials with benzene/cyclohexane ratios of 8:1 and 12:1, respectively. The increased selectivity of the Et3NH@42 and Li@42 systems is related to the increasing bulk of the Et3NH+ and Li(H2O)4+ ions.97
As the next example, an anionic nanoporous framework material with mobile guest cations [HDMA]3[(Cu4Cl)3-(btc)8]·9DMA (43) has been reported. This MOF can perform ion-exchange with tetraalkylammonium cations, such as TMA (44), TEA (45) or TPA (46). The research group also indicated that cation-exchange within this anionic framework creates unusual pore partition effects on gas separation (CO2/N2). The results revealed that 46 with the smallest pores and volumes may be better suited for separating CO2 over N2 and showed the highest selectivity of 106.8 during CO2/N2 adsorption. Finally, they reached the conclusion that ion-exchange using organic cations to tune the pore space for gas separation is a promising course for the improvement of new functional MOFs materials.98
Another study described the heterogenization of single site transition-metal catalysts in MOFs via cation-exchange. Their efforts focused on the exchange of endogenous HDMA+ cations with cationic transition-metal complexes in anionic (HDMA)3[In3(BTB)4]·12DMF·22H2O (ZJU_28) (Fig. 12) (50). Furthermore, the Rh-containing MOF 50 was shown to be a recyclable catalyst for alkene hydrogenation and demonstrated developed catalytic performance related to its homogeneous counterpart under certain conditions.100
 | ||
| Fig. 12 The proposed heterogenization of single-site transition-metal catalysts in ZJU-28 (50) via cation-exchange. Reproduced with permission from ref. 100, copyright 2013, American Chemical Society. | ||
An anionic MOF, [HDMA]2[Zn2(BDC)3(DMA)2]·6DMF (51), has been investigated in several studies to date because of its cation-exchange ability and subsequent applications. We will discuss this MOF in the following sections, but here we discuss the results of its catalytic properties. The prepared MOF has an anionic framework with [HDMA+] cations in the channels. These cations are exchanged with Ni2+ (52), Na+ (53), Li+ (54), (TEA+) (55) and (TPA)+ (56) during the experiments. Using this strategy, they were able to prepare cation-exchanged anionic MOFs as a novel heterogeneous catalytic system for the solvent-free Knoevenagel condensation reaction between benzaldehyde and malononitrile. The catalytic activity of the compound 51 apohost framework was studied. They thought that the catalytic activity of 51 was as a result of its anionic framework. Thus, in order to examine this idea, the activated samples of 52, 53 and 54 after the post-synthetic exchange were studied too. The obtained results showed that the catalytic activity decreased in the order of 51 > 54 > 53 > 52. As it was clear, the ionization potential decreased in the following order: Ni2+ > Li+ > Na+ > HDMA+. Therefore, by increasing the ionization potential of the cations in the channels of 51, the acidic properties of the metal ions increased and the basic properties of the anionic framework decreased. In order to confirm this supposition, they performed cation-exchange processes of HDMA+ with the larger organic cations of 55 and 56. The experiment proved this hypothesis too and indicated that the catalytic activities decreased in the order of 56 > 55 > 51. Consequently, they showed that with a simple post-synthetic cation-exchange in anionic MOFs, some new solids with different catalytic activities were produced.101
In this section, another novel application of these anionic MOFs as heterogenous catalysts is introduced. S. Beheshti et al. were able to prepare a cation-exchanged anionic MOF as a new heterogeneous catalytic system for the solvent-free Micheal addition of pyrrole to electron-deficient β-nitro styrenes. In this case, using cation-exchange as post-modification in the anionic MOF, [HDMA]2[Zn2(BDC)3(DMA)2]·6DMF (51), the catalytic activity was improved. The [HDMA+] cations in MOF were replaced with (TEA)+ (55) or (TPA)+ (56) via cation-exchange. The catalytic activity of the compound 51 apohost framework was studied in the Micheal addition reaction. They attributed the activity of 51 to its anionic framework. Thus, in order to approve this hypothesis, activated samples of 55 and 56 were used after post-synthetic cation-exchange. The results of applying the apohost frameworks of 55 and 56 in the Micheal reaction confirmed this idea and indicated that the catalytic activity decreased in order of 56 > 55 > 51.102
Hydrogen adsorption. As we have insinuated, ZMOFs are anionic and have readily exchangeable extra framework cations. F. Nouar and co-workers showed that ZMOFs can be regarded as a platform for systematic studies on the effect of different structural factors on H2 interactions with porous metal organic materials. In their study, |(HPP2+)24|[In48(HImDC)96] (HPP-rho-ZMOF) (57) was reported as the parent framework, where the Na+ ions replaced the extra-framework cations. The group reported the effect of several extra-framework cations (HDMA+, Li+, Mg2+) on the H2 sorption energetics and uptake. Li+ cations have a small ionic radius compared to most inorganic cations; this results in a reduced framework density and large enough voids for H2 storage. The other cation, Mg2+, is also ionized because of its high dependency for H2. The Li+-exchanged material, Li-rho-MOF (58), can store slightly more H2 molecules per In. It is important to note that the slightly higher value for 58 is in contrast to results obtained in the previous Li+-exchanged MOFs, which show no proof for strong H2–Li+ interactions.103
A more detail study has been carried out in this case. Herein, they illustrated a systematic study of the solvothermal conditions used for optimizing the synthesis of the two mentioned ZMOF structures (Rho (59) and Sod (60)). Both the Rho and Sod ZMOF materials were prepared basically following the procedure published elsewhere, in which (H3ImDC), In(NO3)3·XH2O, (DMF), CH3CN, (HPP), and HNO3 were consecutively added to obtain a mixture. These materials were exchanged with Li+, Na+, K+ and Cs+ cations. The H2 sorption of the as-prepared and exchanged materials was discussed too. The ion-exchange degree of these three cations (Na+, K+, Li+) in 60 were measured. The results showed that the ion-exchange degree increased with the cation size. In regard to the H2 adsorption, their experiments show that Na+Me-sod ZMOF adsorbs less H2 than the non-exchanged sod-ZMOF, although the exchanged material has a higher surface area. This behavior suggests that the weaker adsorption of H2 by Na+ can be compensated by its smaller cation size, which results in a higher surface area and pore volume of the exchanged material. In comparison with the sample exchanged only once with Na+, the sample exchanged four continuous times with Li+ showed very similar results with H2 uptake. The K+-exchanged sample was not crystalline enough to extract any conclusion about its association for H2.104
Enhancement of the isosteric heat of adsorption for H2 in the Li-exchanged rho-ZMOF is an interesting instance that has been investigated by S. Yang and co-workers. Synthesis of three anionic frameworks (NOTT-206-solv) (61), (NOTT-200-solv) (62) and (NOTT-208-solv) (63), and their organic cation replacement with Li+ (NOTT-209-solv) (64) have been reported. Both 63 and 64 showed reversible H2 sorption isotherms, which are consistent with their larger pores. Furthermore, they reported that 63 and 64 have identical framework structures, the enhancement in the H2 storage capacities can be linked to the cations, H2PPZ2+ or Li+, animating in the channels. Their findings indicate that the gas storage properties of the charged MOFs are controllable by the choice of the counter-ions; different cations within the pores may act as a code for the enhancement of H2 storage properties.105
The other description was about an anionic MOF material built from In(III) centers and H4L ligands. The framework presented hysteretic hydrogen adsorption with piperazinium (H2PPZ)2+ dications in its pores. The paper demonstrated the synthesis of 1-PPZ-solv MOF (65) and its exchange reaction product, Li-PPZ-solv (66). During the exchange, the H2PPZ2+ dications were replaced by Li+ (Fig. 13). The MOF 65 showed a considerable kinetic trap (hysteresis) for H2 and N2 adsorption, whereas 66 showed an increase in both pore volume and a notably higher isosteric heat of adsorption for H2 compared with 65. Their results exhibited a cation-dependent hysteretic H2 sorption with the hysteresis tunable by post-synthetic modification via cation-exchange.106
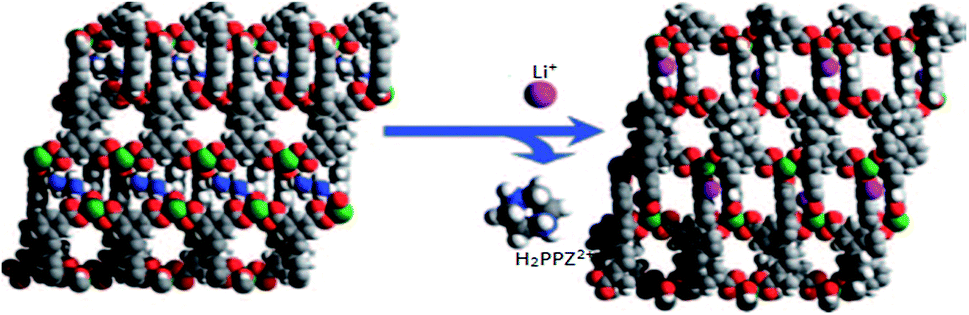 | ||
| Fig. 13 Views of framework structures of 1-PPZ-solv (65) and Li-PPZ-solv (66). The H2ppz2+ dications in channel B of 65 can be completely exchanged with Li+ ions in channel C of 66. Reproduced with permission from ref. 106, copyright 2009, Nature publishing group. | ||
Do-MOF (67) was introduced by K. Mulfort et al. The combination of an octa-oxygen ligand with a Zn(II) source and the diol-containing strut, after two days of heating, results into colorless block crystals of 67. Do-MOF is a non-catanated hydroxyl functionalized MOF and exchange of the hydroxyl protons for Li+ and Mg2+ cations was investigated. The replacement of Li+ by a more highly charged cation may increase the heat of adsorption, and therefore they also examined an Mg2+ containing version. In this study, the H2 uptake was also measured and its diminution after the exchange was reported. In their experiments, the low-pressure adsorption of H2 by 67 was reversible at 77 K and reached 1.23 wt% at 1 atm. 67-Li exhibited only a slightly greater uptake (1.32 wt% at 1 atm). Nevertheless, the increase corresponds to two additional H2 per Li+. Their finding was broadly consistent with computational predictions that an exposed lithium cation on carbon or MOF materials can (depending on pressure) directly bind up to six H2 molecules.107
In the bunch of ZMOFs that have adsorption properties, another ZMOF has been reported. ([In80(Himdc)160]80−)n (Usf-ZMOF) (68) was introduced by Y. Liu, et al. The anionic character, extra-large cavities, large openings and chemical stability in an aqueous solution of Usf-ZMOF allowed the full exchange of the 1,2-H2DACH cations with different organic and inorganic cations, including Li+, Na+ and Mg2+. In order to detect the gas sorption properties of 68, the extra framework cations present in the as-synthesized compound were fully exchanged with DMA cations. Gas sorption experiments were performed on DMA-exchanged 68 and the fully evacuated framework exhibited permanent microporosity. H2 sorption studies were also performed and the results showed a relatively enhanced isosteric heat of sorption at lower loadings that can be related to the charge nature of the cavities.108
We have already discussed the gas separation of [HDMA+]3[(Cu4Cl)3-(btc)8]·9DMA (43). Another application of this MOF that has been reported after ion exchange is gas adsorption. Cation-exchange within the anionic framework created unusual pore partition effects on the gas adsorption (N2, CO2 or H2). The H2 adsorption observed for the MOF and exchanged-MOF was measured. The authors noted that the H2 adsorptive dependence on the framework was not just concerned with the surface area, but was also concerned with the suitable pore volume.98
The last example, examined by us, was the MOF materials {(A)[In(Ln)]·solv}∞, (A = 1/2H2PPZ2+ or HDMA+, solv = DMF, CH3CN and H2O) (69) that have been prepared from In(III) with tetracarboxylate isophtalate-based ligands. The MOFs were comprised of organic cations, H2PPZ2+ or Me2NH2+. The organic cations within the as-synthesized materials could be exchanged with Li+ ions (Fig. 14). The gas adsorption of H2 and N2 has been studied in detail. Thus, an increase in the H2 capacity and overall porosity were observed on going from the desolvated MOF incorporating HDMA+ cations to desolvated MOF incorporating Li+. They also demonstrated that by varying the pore gate and the isophthalate-based bridging ligand, the desolvated framework materials display either non-porous, or hysteretic, or reversible N2 sorption properties, respectively. The group thus reported that the desolvated Li+-exchanged framework materials did not show adsorption/desorption hysteresis with H2, but enhanced H2 adsorption properties in terms of both the valencies and adsorption enthalpies.109
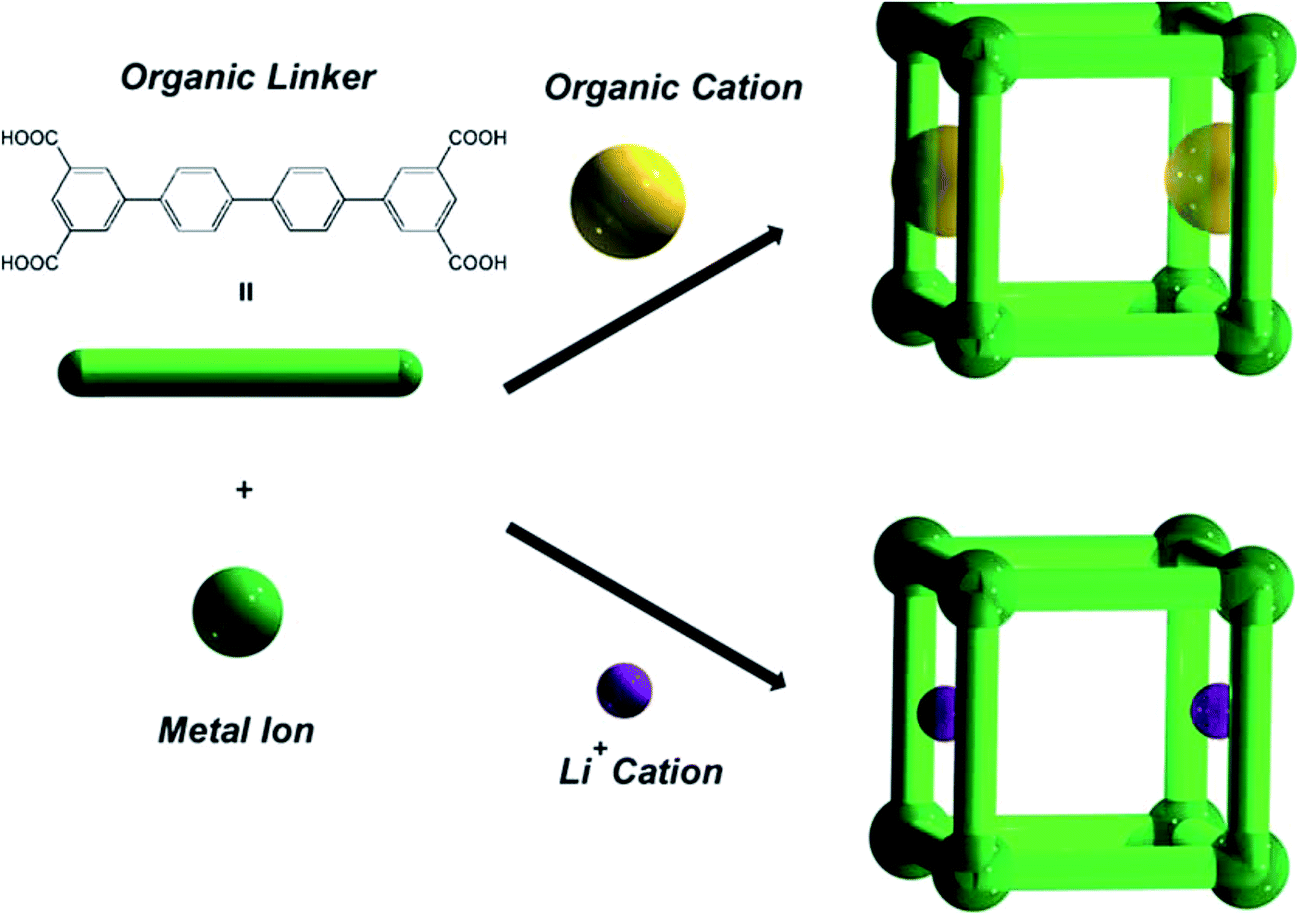 | ||
| Fig. 14 A schematic representation of the construction of modulated MOFs in {(A)[In(Ln)]·solv}∞ (69) with different pore gates. Reproduced with permission from ref. 109, copyright 2011, Royal Society of Chemistry. | ||
Carbon dioxide adsorption. The CO2 adsorption properties are another application considered via cation-exchange. Our studied paper is about Bio-MOF, Zn8(ad)4(BPDC)60.2HDMA (70) and the replacement of its HDMA+ cations with other organic cations such as TMA, TEA and TBA. The researchers also demonstrated that the post-synthetic exchange of the extra framework cations within the anionic Bio-MOF-1 can be used as a means to systematically modify its pore dimension and metrics. They efficiently used this strategy to optimize its CO2 adsorption properties. To determine the effects of the pore modifications on the CO2 capacity of this MOF, they studied the CO2 adsorption of 70 and the exchanged MOFs. They interestingly animadverted on the contemplation that the CO2 capacity of the MOFs did not depend on the pore volume and BET surface area. At all the temperatures studied, compound 70 showed the lowest CO2 capacity compared to the exchanged-MOFs. They found that TEA@70 had the largest CO2 capacity at each temperature and TBA@70 was nearly as effective as TMA@70. It was notable that TBA@70 was a more effective CO2 sorbent than 70, even though its pore volume and surface area were almost half as large.110
In the group of ZMOFs, CO2 adsorption has been obtained for the Sod topologies only. A research group that published their results in this case, reported that ion-exchanged sod-ZMOFs (71) by alkali-metals resulted in improved CO2 adsorption performance compared with the as-prepared ZMOF. In this study, ion-exchange of 71 by different alkali metals such as Li+, Na+, K+ and Cs+ cations was carried out to study the effects of ion-exchange alkali metal on CO2 adsorption. As a result of their experiments, they reported that all the ion-exchanged 71 samples displayed improved CO2 adsorption capacity over the as-prepared sample. The highest CO2 capacity was achieved by K+-sod-ZMOF (72). They concluded that the CO2 adsorption capacity in ion-exchanged sod-ZMOF appeared to depend not only on the size and charge of the balancing cations, but also on the dispensation of the cations located in the framework structures.111
Nitrogen adsorption. NOTT MOFs including NOTT-206-solv (61), NOTT-200-solv (62) and NOTT-208-solv (63) and their Li+-containing framework (64) were discussed in the previous section on H2 adsorption. N2 adsorption has been also considered using these materials. By replacing the HDMA+ cations with Li+ ions, both of the channels released and accordingly, the adsorption of N2 in NOTT-207 showed a typical type-I isotherm with little hysteresis. Therefore, using a combination of Li+ with NOTT-207, the internal surface area became fully available for N2 uptake. Modulation of the gas adsorption properties with different pore gates in NOTT-MOF materials has been finally demonstrated (Fig. 15).105
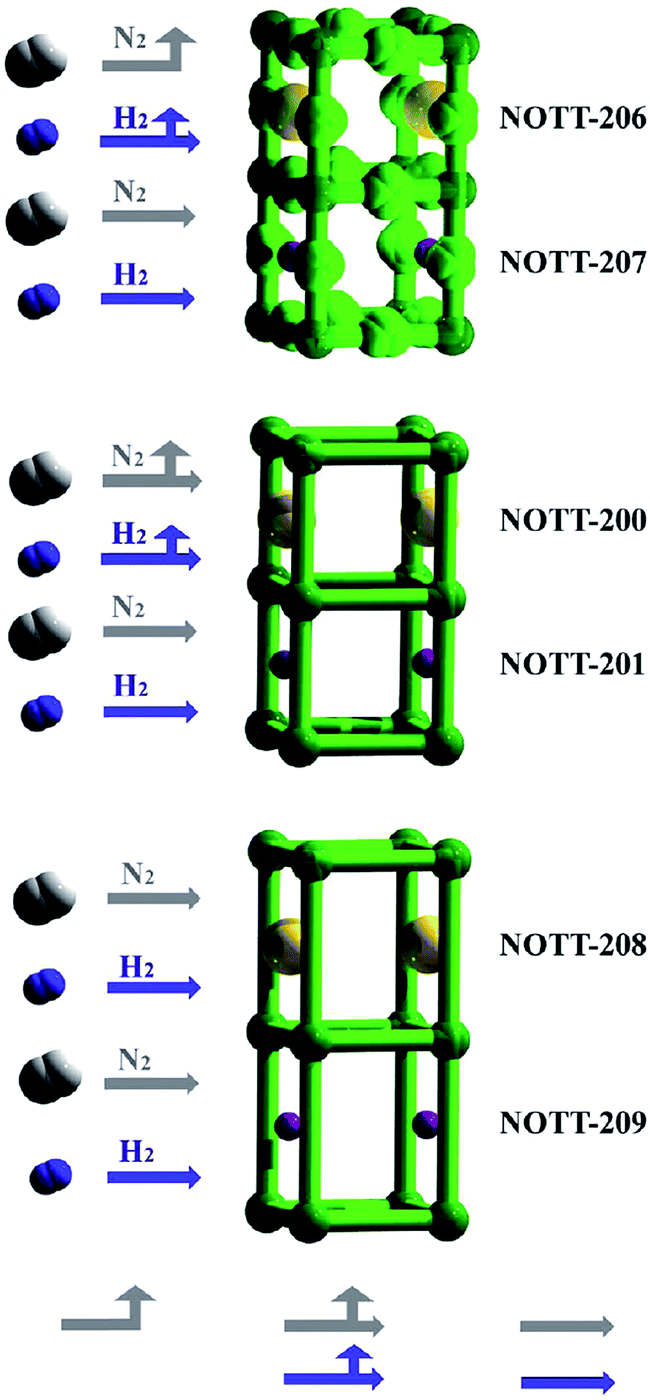 | ||
| Fig. 15 Modulation of the adsorption properties with different pore gates in NOTT-MOFs. Reproduced with permission from ref. 105, copyright 2011, American Chemical Society. | ||
Another MOF that has been introduced and explicated N2 adsorption is compound 34. We have previously discussed its gas separation properties and the gas adsorption application of this MOF is now considered. In the mentioned study, N2 adsorption measurements were performed on the exchanged systems and the results showed changes in the adsorption isotherms due to the ion-exchange processes. Indeed on going from NH4+ to Et3NH+, a lowering of the adsorption capacity and pore surface was observed.97
Some novel synthesized ZMOFs, such as sod-ZMOF derived from 4,6-PmDc [In-(C6N2O4H2)2Na0.36K1.28](NO3)0.64(H2O)2.1 (73), also possess this property. The reaction between 4,6-PmDC and In(NO3)3·5H2O in a solution of DMF and water afforded pale yellow homogeneous crystals with polyhedral morphology, referred to as sod-ZMOF. Complete exchange of the K+ cations with smaller Li+ cations in this ZMOF was demonstrated. In addition, gas adsorption experiments were performed on the Li+-exchanged sample. The result of the experiments demonstrated that the Li+-exchanged and fully depleted sod-ZMOF, displaying permanent microporosity, as evidenced by the reversible type I N2 and Ar adsorption isotherms. The dual character of ZMOFs, anionic frameworks and/or containing large accessible cavities, forms a suitable platform to evaluate the effect of pore size and/or intra-/extra framework charge density on the hydrogen uptake and its sorption energetics. Accordingly, hydrogen sorption studies were conducted on both compounds at 78 and 87 K at atmospheric pressure using this compound 73.112
A zinc-based MOF {(HDMA)2[Zn3(BDC)4]·DMF·H2O} (74) was synthesized by a research group and its cation-exchange processes and N2 adsorption isotherms have been investigated. In order to study the cation-exchange process in a crystal-to-crystal transformation, initially they examined the stability of the compound's crystallinity in various solvents; however, the MOF only retained its crystallinity in DMF. A post-synthetic cation-exchange process of the MOF was carried out using Cu2+ (75), Li+ (76) and Na+ (77). During the exchange, HDMA+ cations were replaced by Cu2+, Li+ and Na+. The sorption properties of 74 were investigated using N2 adsorption isotherms. They reported that compound 74 showed a typical type V adsorption curve and also mentioned that this strange behavior can be attributed to the blocked channels of 74 by the large, non-removable HDMA+ cations.113 A comparison of the N2 isotherms obtained for compound 74 and 76 show a change in the adsorption isotherm from type V in compound 74 to type I in compound 76. This behavior was because of the existence of small Li+ cations in the channels of 76, which were not an impediment for entrance of N2 molecules compared with the blocked channels in 74 by the large, non-removable HDMA+ cations. Thus compound 76 showed type I adsorption, indeed the channels of 74 only opened at high pressure of N2 gas.114
A solid-type MOF constructed from heterometallic alkali metal is the last example that will be described by us in the N2 adsorption area. The obtained MOFs were [(CH3)3NH][NaVO(BTC)4/3]·0.5DMF (78) and {[(CH3)4N]3LiVO(BTC)2} (79), which represent the first MOF constructed from V/alkali metal mixed metal-oxo clusters. The effective free volumes of these compounds and their high architectural stability actuated the research group to follow cation-exchange experiments. The organic amine cations within the channels can be replaced by transition metal ions through a cation-exchange process. The Cu(II) ion was used as a suitable probe for the exchange experiments considering its size, stability and instinct. An N2 adsorption measurement was subsequently carried out for the fully activated sample of sod-ZMOF, which was achieved by dipping the crystals in Cu2+ ion-containing acetone for 12 h, followed by heating at 80 °C for 12 h in a vacuum. The N2 sorption isotherm reveals characteristic type II behavior. The analysis of the isotherm revealed a specific surface area SBET = 122.5 m2 g−1 (Langmuir, 196.4 m2 g−1). A maximum N2 uptake of 101 m2 g−1 (at standard temperature and pressure, STP) was reached at 1 atm. They also measured the gas adsorption isotherms of fresh samples of 78 using the same method. However, the maximum N2 uptake was very small at STP as the guest amine cations reside in the frameworks and the amine cations could not be removed with the neutral solvent. An analogous N2 adsorption measurement was carried out for compound 79. The analysis of the isotherm revealed a specific surface area SBET = 316 m2 g−1 (Langmuir, 368 m2 g−1). A maximum N2 uptake of 129 m2 g−1 (at STP) was reached at 1 atm.115
Methane adsorption. Although, the physical adsorption of methane on MOFs has amused much interest, limited achievement has been obtained in storing methane under ambient conditions. The only in depth study on methane storage in anionic MOFs via cation-exchange was carried out by K. Akhbari et al. Their study started from post-synthesis cation-exchange of [HDMA]2-[Zn2(BDC)3(DMA)2]·6DMF (51). We discussed this MOF in the Catalytic properties section. Herein, the CH4 adsorption properties were considered. The existence of HDMA+ organic cations in the channels of the MOF with an anionic framework established the possibility of their post-synthetic cation-exchange with metal ions such as Ni2+ (52), Na+ (53), Li+ (54), Cu2+ (80) and K+ (81). The methane adsorption capacities of all of these materials were also measured. The results showed an increase in the CH4 adsorption capacity going from 51 to the last MOF (81), and Li+-exchanged MOF (54) with the highest surface area had the highest methane adsorption capacity. Nevertheless, it was noted, by post-synthetic cation-exchange of 51 with some metal ions and particularly with Li+, that we can successfully modulate the surface area and methane adsorption capacity. Another interesting result was reported that K+-exchanged MOF (81) did not have any available pores for the adsorption of methane. On the other hand, the lowest N2 gas sorption in 51 and the same surface area of 51 and 81 indicated that the MOF material of 51 did not allow N2 gas molecules to enter its channels, but the channels in 51 showed different behavior upon facing methane gas molecules and showed a low CH4 adsorption capacity. Therefore, the HDMA+ cations in 51 act as a gate in an opposite way to N2 gas molecules, permitting the CH4 gas molecules to enter their channels.116
Other materials adsorption. The adsorption of volatile organic compounds in porous MOFs is one of the studied cases that was not comprised in the different gases group. Accordingly, F. J. Ma et al. achieved the functionalization of MOF (NENU-3) (82) with (Cu3(BTC)2) frameworks and POMs. They further modified the inclusion state of these materials via ion-exchange. A complete exchange of (CH3)4N+ by K+ was observed without losing the MOF structure (NENU-28) (83). For the VOCs adsorption studies, they selected methanol, ethanol, 1-propanol, 2-propanol, cyclohexane, benzene and toluene. The adsorption isotherms of methanol showed a corresponding development in the storage capacity on 83. By continuing their experiments, they found that the exchange of alkali metal cations may also bring on an increase in the adsorption capacity when comparing the data of 82. The results of the Li+-exchanged MOF (84) also confirmed their deduction. Alkali metal exchanged 82 and 84 bring an extra interaction with the adsorbed molecules, which leads to a significant enhancement in adsorption. At the end of the study, it was reported that 83 displays the best adsorption for VOCs. In addition, the data obtained from N2 adsorption studies were also reported in their work, and an increasing adsorption capacity of 82 upon K+-exchange was suggested.117
The selective adsorption of dyes via ion-exchange is also notable. In this case, IFMC-2 [HDMA]4[(Zn4dttz6)Zn3]·15DMF·4.5H2O (85) has been used. The Ln(III)-loaded MOF material Ln3+@85 and the adsorption of cationic dyes through ion-exchange were also investigated. IFMC-2 (85) has an anionic framework, which is filled with [HDMA]+ ions and guest molecules. The [HDMA]+ ions can be exchanged with Ln3+ cations. The organic cations in 85 can be exchanged with cationic dyes due to the anionic framework. The results of the paper suggest that the dye molecules in dye@85 can be gradually released in the presence of NaCl. Therefore, releasing the dyes were an ion-exchange process in this study.118
The 3D lanthanide anionic MOF {K5[Ln5(IDC)4(ox)4]}n·(20H2O)n, (Ln = Gd (86), Tb (87), and Dy (88)) show luminescent properties that are induced by cation-exchange. The luminescent studies revealed that the luminescent properties of 86–88 can be changed through the exchange of the guest K+ ions with various cations. To examine the possibility of modifying the luminescent properties through cation-exchange, a solid sample of 87 was soaked in DMF containing various metal cations. The luminescent intensity in the presence of 3 equiv. of Ca2+ ions was about twice as strong as that found without Ca2+ ions. However, the luminescent intensities did not change or increase in the presence of 3 equiv. of Na+, NH4+, Mg2+, Sr2+, Ba2+, Zn2+, Cd2+, Hg2+ and Pb2+ cations. When 1–3 equiv. of transition-metal ions, such as Mn2+, Fe2+, Co2+, Ni2+ and Cu2+, were added to the emulsion of 87 in DMF, the luminescent intensity was not enhanced but rather weakened dramatically or even quenched. As a result, they noted that compound 87 was the first lanthanide-based MOF to be used as a promising Ca2+ ion-selective luminescent probe and it can also show a remarkable increase in the luminescent emission of Tb(III) upon the addition of Ca2+ ions, while the exchange of K+ ions with other cations did not modulate the luminescence emission of this compound.119
Another polymer that shows a cation-exchange role in the luminescent sensing of aqueous metal ions is [NH4]2[ZnL′]·6H2O (89), which has been studied by Sh. Liu and co-workers. Polymer 89 was soaked in aqueous solutions of MClx {M = Na+, K+, Mg2+, Ca2+, Mn2+, Ni2+, Co2+ and Cu2+} to form a metal-ion infused phase (M@89). The research group indicated that the luminescence intensity of M@89 was highly dependent on the nature of the doped metal ion. For polymer 89, there are two types of suitable counter ions for cation-exchange: (i) Zn(II) ions and (ii) [NH4]+ ions. Their suggestion shows that in the Cu@89 sample, cation-exchange occurs between (NH4+) and Cu(II), rather than between Zn(II) and Cu(II). In this study, the cation-exchange materials also performed as potential luminescent sensors of aqueous metal ions.120
The last example in this section is MOF_COOH, [Zn3(Httca)2(4,4′-bpy)(H2O)2]n (90), which was prepared by J. Cao et al. The uncoordinated carbonyl groups in the channels of 90 can act as post-synthetic modification sites for cation-exchange. During the exchange, Na+ cations were loaded into the pores of 90. As a matter of fact, they reported that the MOFs have several advantages that make them useful for sensitizing lanthanide cations. Thus, they also loaded lanthanide cations into the pores via a post-synthetic cation-exchange (Ln = Sm3+, Eu3+, Dy3+ or Tb3+). In addition, photoluminescence studies were performed on each sample of 81, Na+@81 and Ln3+@90. It is significant in their study that the Ln3+-loaded 90 sample exhibited very interesting photoluminescence properties. These materials showed a very similar emission to Na+@MOF-COO−, but with different intensity. The results of their experiments indicated that the microporous metal–organic open framework, reported in the paper, was not suitable for the sensitization of Sm3+, Dy3+ and Eu3+ emitters.121
One more MOF has been reported with this ability. Zh. Chen et al. reported [HDMA][Eu(H2O)2(BTMIPA)]·2H2O (97) and studied the cation-exchange between [HDMA]+ and metal ions. In this study, Fe3+ and Al3+ were initially exchanged with the [HDMA]+ cations in the channels and the framework of 97 may have been faced with the exchange between Fe3+/Al3+ and Eu3+. A decrease in the photoluminescence intensity of the Al3+-loaded sample was observed. Therefore, the cation-exchange resulted in a complex that can selectively sense Fe3+ and Al3+ ions through fluorescence quenching and enhancement. To further prove that the fluorescence quenching and enhancement were caused by a cation-exchange process, detailed studies on the luminescence properties of 97 in the presence of Fe3+ and Al3+ were carried out. With increasing time, the photoluminescence intensity decreased for the Fe3+-loaded sample, but increased for the Al3+-loaded sample. This result indicates that, according to the replacement of cations in channels with Fe3+ and Al3+, they had a notable effect on the fluorescence emissions.123
 | ||
| Fig. 16 A comparison of the SHG-intensities of different materials. Reproduced with permission from ref. 124, copyright 2007, John Wiley & Sons. | ||
The last application of ion-exchange processes that has been noticed in our study is in two-photon-pumped dye lasers, which are very important because of their various applications. J. Yu and co-workers connected them to cation encapsulation in an anionic MOF. Generally, they demonstrated a new two-photon-pumped micro-laser by encapsulating a cationic pyridinium hemicyanine dye into an anionic MOF. Thus, Bio-MOF-1 (Zn8(Ad)4(BPDC)6O·2HDMA) (99) was synthesized, in which the HDMA+ cations were located in the channels that allowed the introduction of the cationic dye DMASM via an ion-exchange process. On the other hand, most of the HDMA+ cations in the as-synthesized 99 were replaced by the DMASM cations in 99 × DMASM (Fig. 17). The enhanced fluorescence of 99 × DMASM was also observed. Such a notable emission enhancement showed the pore confinement of DMASM molecules within 99.125 A summary of all the points discussed about MOF ion-exchange are shown in Table 2.
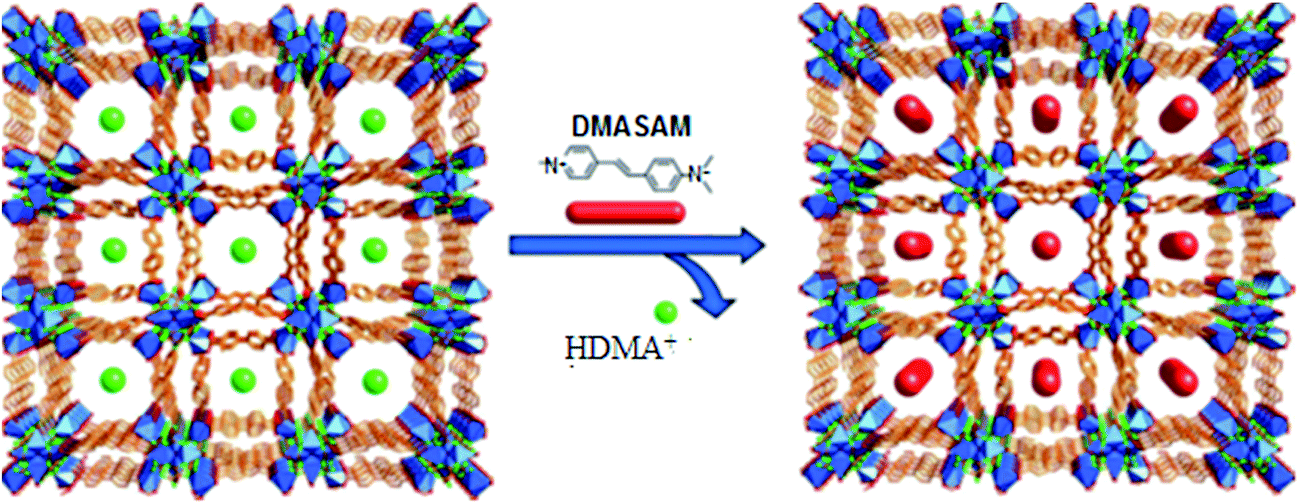 | ||
| Fig. 17 Encapsulation of DMASM dye into (Zn8(Ad)4(BPDC)6O·2HDMA) (99). Reproduced with permission from ref. 125, copyright 2013, Nature publish group. | ||
| MOFs formula | Type of exchange | Type of exchanged species | The resulting properties or applications | Ref. |
|---|---|---|---|---|
| [Ni2(C28H52N10)]3[BTC]·6C5H5N·36H2O (1) | Anionic | I3− anions in the channels | — | 49 |
| [{Cd(H2O)3}34(N4C6H12)17]Cl68·46H2O·68DMF (2) | Anionic | SCN− by Cl− | — | 50 |
| [Co(4-pyrdpm)3AgBF4] (3) | Anionic | — | — | 52 |
| [Co(4-pyrdmp)3AgOTf]; (4-pyrdpm) (4) | Anionic | PF6− by triflate | — | 52 |
| [Ni(timpt)2](ClO4)2 (5) | Anionic | ClO4− by NO3−/ClO4− by NaBF4− | — | 53 |
| {[Ag3(1,3,5-Tris)2X2]X}n (X = ClO4− (6)) | Anionic | ClO4− by NO3− | — | 54 |
| {[Ag3(1,3,5-Tris)2X2]X}n (X = NO3− (7)) | Anionic | NO3− by ClO4− | — | 54 |
| {[Cu(2-(2-Pyridyl))2](ClO4)(H2O)1/2}n (8) | Anionic | ClO4− by C6H5COO− | — | 55 |
| (NBU4)m(A)n{Zn(mim)2}6 (9) | Anionic | OH− by HCO3− or CO32− | — | 56 |
| [M(β-Diketone)3Ag3]-X2·solv (10) [ZnL3Ag3]X2·solv as host network {X = BF4−, ClO4−, CF3SO3−, PF6−} | Anionic | BF4−, ClO4−, CF3SO3−, PF6− by each other | — | 57 |
| [M3X2]·[HDMA]2·8DMA (M = Co (11)) | Cationic | HDMA+ by Na+ | — | 63 |
| [M3X2]·[HDMA]2·8DMA (M = Mn (12)) | Cationic | HDMA+ by Na+ | — | 63 |
| (HDMA)3[In3(BTB)4]·12DMF·22H2O (ZJU-28) (13) | Cationic | Me2NH2+ by Cu2+, Ni2+ and Eu3+ | — | 64 |
| [HDMA][In(mdip)]·2.5DMF·4H2O (14) | Cationic | HDMA+ by (M = Li+, Na+, K+, Mg2+, Ca2+, Sr2+, Ba2+, Cu2+, Fe3+) | — | 65 |
| [HDMA][M2(bptc)(μ3-OH)(H2O)2] (15) | Cationic | [HDMA]+ by Li+, Na+ and K+ | — | 66 |
| H2Na4[Cu12(OH)6(PZ)6(BTC)6]·23H2O (16) | Cationic | Na+ by Li+, K+ and Cs+/Cu2+, Ni2+ and Mn2+ | — | 67 |
| [Ga6(1,3,5-BTC)8·6DMA·3DMF·26H2O] (Ga·MOF-1) (17) | Cationic | Ga+ by Li+ and Na+ | — | 69 |
| [In48(C5N2O4H2)96(C2N3H15)24(DMF)36-(H2O)192] (18) | Cationic | The cationic guest molecules by other cations | — | 70 |
| [Na48+(H2O)282][In48(C5N2O4H2)96] (19) | Cationic | — | — | 70 |
| In48(C5H2N2O4)96(C7N3H15)24(C3H7NO)36(H2O)192 (20) | Cationic | Organic cations by Na+/acridine orange (AO) | — | 71 |
| [In3O(pba)3(ndc)1.5](NO3) (21) | Anionic | — | Separation process {for use as drug carriers} | 72 |
| [In3O(pba)3(bpdc)1.5](NO3) (22) | Anionic | NO3− by OG2− | Separation process {for use as drug carriers} | 72 |
| [CuL2(H2O)0.5](NO3)2 (23) | Anionic | NO3− by Cl−, Br−, I−, SCN− and N3− | Could be an anion separator to separate these anions | 73 |
| ZnCl2(BPu) (24) | Anionic | Cl− by ClO4−, NO3− and SO42− | Separation of anions from water | 74 |
| ZnI2(BPu) (25) | Anionic | I− by ClO4−, NO3− and SO42− | Separation of anions from water | 74 |
| ZnBr2(BPu) (26) | Anionic | Br− by ClO4−, NO3− and SO42− | Separation of anions from water | 74 |
| {[Dy2Zn(BPDC)3(H2O)4](ClO4)2·10H2O}n (27) | Anionic | ClO4− by CrO42− | Separation of pollutant anions from water; considered as a luminescent probe | 78 |
| Ag2(4,4′-bipy)2(O3SCH2CH2SO3)·4H2O (28) | Anionic | EDS− by MnO4−, ReO4−, and CrO42− | Separation of pollutant anions from water | 80 |
| [Zn2(Tipa)2(OH)]·3NO3·12H2O (29) | Anionic | NO3− by Cr2O72− | Separation of pollutant anions from water | 81 |
| [Zn(Tipa)]·2NO3·DMF·4H2O (30) | Anionic | NO3− by Cr2O72− | Separation of pollutant anions from water | 81 |
| [Ag2(btr)2]2ClO4·3H2O (31) | Anionic | ClO4− by Cr2O72− | Separation of pollutant anions from water – considered as a luminescent probe | 82 |
| ZJU-101 (32) | Anionic | NO3− by Cr2O72− | Separation of pollutant anions from water | 86 |
| [{Ni2(Lig)3-(SO4)(H2O)3}·(SO4)·x(G)]n (33) | Anionic | SO42− by Cr2O72− and MnO4− | Separation of pollutant anions from water | 87 |
| [Cu(II)(bped)2(H2O)2(SiF6)]I·4H2O (Cu-MOF-SiF6) (34) | Anionic | SiF62− by NO3− | Catalytic activity in the selective oxidation of benzylic compounds/MeOH adsorption, desorption/EtOH adsorption, desorption | 89 |
| [Cu(II)(bped)2(H2O)2(NO3)]I·4H2O (Cu-MOF-NO3)h (35) | Anionic | — | Catalytic activity in the selective oxidation of benzylic compounds/MeOH adsorption, desorption/EtOH adsorption, desorption | 89 |
| Ag2(4,4′-bipy)2(O3SCH2CH2SO3)·4H2O (28) | Anionic | Organosulfonate by various inorganic species | Catalytically active in heterogeneous ketal formation | 91 |
| Cu2(4,4′-bipy)2(O3SCH2CH2SO3)·3H2O (36) | Anionic | Organosulfonate by various inorganic species | Catalytically active in heterogeneous ketal formation | 91 |
| [Ln(TTP)2]·(CF3SO3)3·C3H6O·5H2O (Ln = Eu (37)) | Anionic | CF3− by ClO4− and SCN− | Luminescent properties | 92 |
| [Ln(TTP)2]·(CF3SO3)3·C3H6O·5H2O (Ln = Gd (38) | Anionic | CF3− by ClO4− and SCN− | — | 92 |
| [{Zn(L)(MeOH)2}-(NO3)2·xG]n (39) | Anionic | NO3− by ClO4− and N(CN)2− (type A) and N3− and SCN− (type B) | Having tunable luminescent behavior | 93 |
| [Ln(bipyNO4)](Tfo)3·x·solvent (Ln = Tb, Dy, Ho, Er) (40) | Anionic | [Tfo]− by POM− | Preserving slow magnetic relaxation | 95 |
| [Cd(4-Amino)2(ClO4)2]·H2O (41) | Anionic | ClO4− by the I3− | Used for the detection of the enrichment of IO3− in dilute solutions | 96 |
| NH4[Cu3-(μ3-OH)(μ3-4-cpy)3] (NH4@1) (42) | Cationic | NH4+ by Li+, Na+, K+, Ca2+/2, La3+/3, Et3NH+ and Me3NH+ | N2 adsorption/separation selectivity by the adsorption of gases such as N2, CH4, CO2, C2H2 and harmful vapors (benzene, cyclohexane) | 97 |
| [HDMA]3[(Cu4Cl)3-(btc)8]·9DMA (43) | Cationic | HDMA+ by organic cations | Gas separation (CO2/N2)/N2, CO2 or H2 adsorption | 98 |
| [HDMA]3[(Cu4Cl)3-(btc)8]·9TMA (44) | Cationic | HDMA+ by organic cations | Gas separation (CO2/N2))/N2, CO2 or H2 adsorption | 98 |
| [HDMA]3[(Cu4Cl)3-(btc)8]·9TEA (45) | Cationic | HDMA+ by organic cations | Gas separation (CO2/N2))/N2, CO2 or H2 adsorption | 98 |
| [HDMA]3[(Cu4Cl)3-(btc)8]·9TPA (46) | Cationic | HDMA+ by organic cations | Gas separation (CO2/N2))/N2, CO2 or H2 adsorption | 98 |
| [Zn17thb14(μ4-O)4(H2O)(HDMA)]·Me2NH2·xguest (47) | Cationic | Zn2+ by Pd2+ and Ag+ | — | 99 |
| [Pd17thb14(μ4-O)4(H2O)(HDMA)]·Me2NH2·xguest (48) | Cationic | Me2NH2+ by Pd+ or Ag+ | Catalytic properties | 99 |
| [Ag17thb14(μ4-O)4(H2O)(HDMA)]·Me2NH2·xguest (49) | Cationic | Me2NH2+ by Pd+ or Ag+ | Catalytic properties | 99 |
| (HDMA)3[In3(BTB)4]·12DMF·22H2O (ZJU_28) (50) | Cationic | HDMA+ by cationic transition metal complexes | Used as a recyclable catalyst for alkene hydrogenation | 100 |
| [HDMA]2[Zn2(BDC)3(DMA)2]·6DMF (51) | Cationic | [HDMA+] by Ni2+ (53), Na+ (54), Li+ (55), (TEA+) (56) and (TPA)+ (57) | Catalytic activity | 101 and 102 |
| |(HPP2+)24|[In48(HImDC)96] (HPP-rho-ZMOF) (57) | Cationic | (HPP2+) by HDMA+, Li+ (59), Mg2+ | H2 adsorption | 103 |
| Rho_ZMOF (59) | Cationic | By Li+, Na+, K+ and Cs+ | H2 adsorption | 104 |
| Sod_ZMOF (60) | Cationic | By Li+, Na+, K+ and Cs+ | H2 adsorption | 104 |
| {[Hdma)(H3O)][In2(L)2]·4DMF·5H2O}∞ (61) | Cationic | Organic cation by Li+ | N2 adsorption | 105 |
| {[(H2ppz][In2(L)2]·3.5DMF·5H2O}∞ (62) | Cationic | Organic cation by Li+ | N2 adsorption | 105 |
| {[(H2ppz][In2(L)2]·4DMF·5.5H2O}∞ (63) | Cationic | Organic cation by Li+ | H2 adsorption/H2 storage/N2 adsorption | 105 |
| {Li1.4(H3O)0.6][In2(L)2]·4-acetone·11H2O}∞ (64) | Cationic | — | H2 adsorption/H2 storage/N2 adsorption | 105 |
| {[(H2ppz][In2(L)2]·3.5DMF·5H2O}∞ (65) | Cationic | H2PPZ2+ by Li+ | H2 and N2 adsorption | 106 |
| {[Li1.5(H3O)0.5][In2(L)2]·4-acetone·11H2O}∞ (66) | Cationic | — | H2 adsorption | 106 |
| Do-MOF (67) | Cationic | Hydroxyl protons by Li+ and Mg2+ | H2 adsorption | 107 |
| ([In80(Himdc)160]80−)n (Usf-ZMOF) (68) | Cationic | [1,2-H2dach]+ by Li+, Na+ and Mg2+ | H2 adsorption | 108 |
| {(A)[In(Ln)]·solv}∞, (A = 1/2H2PPZ2+ or HDMA+, solv = DMF, CH3CN and H2O) (69) | Cationic | H2PPZ2+ or Me2NH2+ by Li+ | H2 and N2 adsorption | 109 |
| Zn8(ad)4(BPDC)60.2HDMA (70) | Cationic | HDMA+ by TMA+, TEA+ and TBA+ | CO2 adsorption | 110 |
| Sod-ZMOFs (71) | Cationic | By alkali-metals such as: Li+, Na+, K+ (73) and Cs+ | CO2 adsorption | 111 |
| 4,6_PmDc [In-(C6N2O4H2)2Na0.36K1.28](NO3)0.64(H2O)2.1 (73) | Cationic | K+ by Li+ | N2 and Ar adsorption | 112 |
| {(HDMA)2[Zn3(BDC)4]·DMF·H2O} (74) | Cationic | HDMA+ by Cu2+ (76), Li+ (77) and Na+ (78) | N2 adsorption | 114 |
| [(CH3)3NH][NaVO(BTC)4/3]·0.5DMF (78) | Cationic | The organic amine cations by transition metal ions | N2 adsorption | 115 |
| {[(CH3)4N]3LiVO(BTC)2} (79) | Cationic | The organic amine cations by transition metal ions | N2 adsorption | 115 |
| [Zn2(BDC)3(DMA)2]·6DMF (51) | Cationic | HDMA+ by Ni2+ (53), Na+ (54), Li+ (55), Cu2+ (81) and K+ (82) | CH4 adsorption | 116 |
| (C4H12N)2[Cu12(BTC)8·12H2O][HPW12O40]·25H2O (82) | Cationic | (CH3)4N+ by K+ and Li+ | VOCs adsorption/N2 adsorption | 117 |
| K2[Cu12(BTC)8·12H2O][HPW12O40]·28H2O (83) | Cationic | — | VOCs adsorption/N2 adsorption | 117 |
| Li2[Cu12(BTC)8·12H2O][HPW12O40]·28H2O (84) | Cationic | — | VOCs adsorption/N2 adsorption | 117 |
| [HDMA]4[(Zn4dttz6)Zn3]·15DMF·4.5H2O (85) | Cationic | [HDMA]+ by Ln3+ | The adsorption of cationic dyes/luminescent properties: suitable for the sensitization of Tb3+ and Dy3+ ions rather than Eu3+ and Sm3+ emitters | 118 |
| {K5[Ln5(IDC)4(ox)4]}n·(20H2O)n, (Ln = Gd (86)) | Cationic | K+ ions with various cations | Luminescent properties | 119 |
| {K5[Ln5(IDC)4(ox)4]}n·(20H2O)n, (Tb (87)) | Cationic | K+ ions with various cations | Luminescent properties | 119 |
| {K5[Ln5(IDC)4(ox)4]}n·(20H2O)n, (Dy (88)) | Cationic | K+ ions with various cations | Luminescent properties | 119 |
| [NH4]2[ZnL′]·6H2O (89) | Cationic | [NH4]+ by Na+, K+, Mg2+, Ca2+, Mn2+, Ni2+, Co2+ and Cu2+ | The luminescent sensing of aqueous metal ions | 120 |
| [Zn3(Httca)2(4,4′-bpy)(H2O)2]n (90) | Cationic | The uncoordinated carbonyl groups located in the channels by Na+ and (Ln = Sm3+, Eu3+, Dy3+ or Tb3+) | Photoluminescence properties | 121 |
| LnL′′ (91) (Ln = La (92), Y (93), Eu (94), Tb (95) and Gd (96)) {L′′ = tetrakis[4-(carboxyphenyl)oxamethyl]methane} | Cationic | [HDMA]+ by a number of metal ions | The detection of ions | 122 |
| [HDMA][Eu(H2O)2(BTMIPA)]·2H2O (97) | Cationic | Firstly, [HDMA]+ by Fe3+ and Al3+, secondly Fe3+/Al3+ by Eu3+ | The detection of ions | 123 |
| [(HDMA)2Cd3(C2O4)4]·MeOH·2H2O (98) | Cationic | [HDMA]+ by NH4+, Na+ and K+ | Effect SHG intensities | 124 |
| (Zn8(Ad)4(BPDC)6O·2HDMA) (99) | Cationic | HDMA+ by DMASM cations | Enhanced fluorescence | 125 |
4. Conclusion
Metal organic frameworks (MOFs) constitute an important field of study due to their ease of synthesis and high surface areas in certain cases. Moreover, a wide range of MOFs can be employed to exhibit various abilities, which are because of their construction. MOFs include a framework with different materials in their channels. These frameworks can be anionic, cationic or neutral. In some cases, various ions fill the channels, which can be exchanged with other organic or inorganic ions. Therefore, the synthesis, characterization and ion-exchange properties of MOFs have become important fields of study. Thus, for improvement in this field of study, it is necessary to reach a better understanding of these materials and their ion-exchange mechanisms. In this study, we have summarized most of the reports on ion-exchange experiments that occur in the pores of MOFs reported to date. There are two types of ion-exchanges (cation-exchange and anion-exchange) that we have discussed separately. Active cations have been successfully introduced into a series of anionic MOFs via cation-exchange reactions. Zeolite-like MOFs (ZMOFs) also have an anionic framework and remain in this group. ZMOFs include two topologies that have been named Rho and Sod. Most of the cation-exchange processes are observed in the Rho-topology. We also conclude that metal ions with higher charge and coordination numbers are more susceptible to construct cationic metal–ligands framework. The selectivity of this exchange depends mainly on the size of the pore openings, the electrostatic interactions between the anionic framework and the cationic guest and finally the concentration of the ions in solution. In addition, cationic frameworks exhibit anion-exchange in their pores too. One important observation is that several experiments described represent a certain single-crystal-to-single-crystal transformation. Another important observation is that most of the experiments indicate anion-exchange against the generally reported trends for anion coordinative ability. Discussing the important applications that appear after the exchange process is another important part of this study. For both, cation- and anion-exchange, we classified these applications to five groups: separation processes, catalytic properties, adsorption, luminescence properties and others. In regard to anion-exchange, we noticed that MOFs have also been studied for the removal of hazardous organic materials from water. These compounds have attracted a considerable amount of attention due to their high surface area, uniform pore size, controllable structures, and tailorable functions. These cationic coordination compounds can serve as promising anion exchange materials through the selective exchange of charge balancing anions in the framework with other anions, leading to tunable changes in their physical properties. The charge-balancing anions usually occupy the framework voids and are sometimes just weakly coordinated or even uncoordinated to the metal centers, potentially allowing the capture and separation of anions through anion exchange. Because of these reasons, there are some examples of separating different anions from water that we collected in our study. Moreover, comparing cation-exchange with anion-exchange, we have shown that the adsorption of different gases with only the anionic framework has been observed and this was due to their cation-exchange experiments. In this context, H2 and N2 were used for further experiments. In the subject of H2 adsorption, the efficient frameworks that have been utilized several times were Rho-ZMOFs, whereas, the Sod-topology of ZMOFs were only used for CO2 adsorption. A wide range of gas adsorption was observed in the case of the exchanged materials bearing Li+ cations. The subject of CH4 adsorption has a few reports published to date and it has ambient reportage. Working on more applications, particularly the gas adsorption of the exchanged-materials of MOFs, can be worth aspiring to. It seems that post-synthetic ion-exchange and the cation-exchange process are suitable methods for modulating the properties of metal organic frameworks.Abbreviations
| CPs | Coordination polymers |
| (BOF-1,1) | [Ni2(C28H52N10)]3[BTC]·6C5H5N·36H2O |
| Bismacrocyclic nickel(II) | [Ni2(C26H52N10)-(Cl)4]·H2O |
| BTC | 1,3,5-Benzenetricarboxylate |
| DMF | N,N-Dimethyl formamide |
| (MOF-Co/AgBF4-1) | [Co(4-pyrdpm)3AgBF4] |
| (MOF-Co/AgOTf-1) | [Co(4-pyrdmp)3AgOTf] |
| (4-pyrdpm) | 5-(4-Pyridyl)-4,6-dipyrrinato |
| timpt | 2,4,6-Tris[4-(imidazole-1-ylmethyl)phenyl]-1,3,5-triazine |
| 1,3,5-tris | 1,3,5-Tris(pyrazol-1-yl)benzene |
| 2-(2-pyridyl) | 2-(2-Pyridyl)-5-(4-pyridyl)-1,3,4-oxadiazole |
| ZIF | Zeolitic imidazolate framework |
| β-diketone | 1,3-Bis(4′-cyanophenyl)-1,3-propanedionato |
| H4X | Tetrapodal ligand tetrakis[4-(carboxy phenyl)oxamethyl]methan acid |
| H3BTB | 4,4′,4′′-Benzene-1,3,5-triyl-tribenzoic acid |
| H4mdip | 5,5′-Methylenediisophtalic acid |
| H4bptc | 3,3′,4,4′-Biphenyltetracarboxylic acid |
| PZ | Pyrazolate |
| HPP | 1,3,4,6,7,8-Hexahydro-2H-pyrimido[1,2-a]pyrimidine |
| ina | Isonicotinate |
| pba | 4-(Pyridine-4-yl)benzoate |
| bdc | Terephthalate |
| NH2-bdc | 2-Aminoterephthalate |
| ndc | 2,6-Naphthalenedicarboxylate |
| bpdc | 4,4′-Biphenyldicarboxylate |
| ITC-3 | [In3O(pba)3(ndc)1.5](NO3) |
| ITC-4 | [In3O(pba)3(bpdc)1.5](NO3) |
| BPu | N,N′-Bis(m-pyridyl)urea |
| DMA | Dimethylamine |
| BPDC | 4,4′-Dicarboxylate-2,2′-dipyridine anion |
| LDH | Layered double hydroxide |
| EDS | 1,2-Ethanedisulfonate |
| Tipa | Tris(4-(1H-imidazol-1-yl)phenyl)amine |
| btr | 4,4′-Bis(1,2,4-triazole) |
| H2bpydc | 2,2′-Bipyridine-5,5′-dicarboxylate |
| Lig | Tris(4-(1H-imidazol-1-yl)phenyl)amine |
| bped | meso-1,2-Bis(4-pyridyl)-1,2-ethandiol |
| SLUG-22 | Cu2(4,4′-bipy)2(O3SCH2CH2SO3)·3H2O |
| TfO | Triflate |
| bipyNO | 4,4′-Bypyridyl-N,N′-dioxide |
| POM | Polyoxometalates |
| 4-amino | 4-Amino-3,5-bis(4-pyridyl-3-phenyl)-1,2,4-triazole |
| 4-cpy | Carboxypyrazolato |
| DMA | N,N-Dimethylamide |
| TMA | Tetramethylammonium |
| TEA | Tetraethylammonium |
| TPA | Tetrapropylammonium |
| HImDC | Imidazoledicarboxylate |
| H3ImDC | 4,5-Imidazoledicarboxylic acid |
| H2thb | 2,5-Thiophenedicarboxylate |
| NOTT-206-solv | {[(Hdma)(H3O)][In2(L)2]·4DMF·5H2O}∞ |
| NOTT-200-SOLV | {[(H2ppz)][In2(L)2]·3.5DMF·5H2O}∞ |
| NOT-208-solv | {[(H2ppz)][In2(L)2]·4DMF·5.5H2O}∞ |
| ppz | Piperazine |
| NOT-209-solv | {Li1.4(H3O)0.6][In2(L)2]·4-acetone·11H2O}∞ |
| H4L | Tetracarboxylate isophthalic acid |
| 1-ppz-solv | {[(H2ppz)][In2(L)2]·3.5DMF·5H2O}∞ |
| 1-Li-solv | {[Li1.5(H3O)0.5][In2(L)2]·4-acetone·11H2O}∞ |
| Himdc | 4,5-Imidazoledicarboxylate |
| 1,2-dach | 1,2-Diaminocyclohexane |
| TBA | Tetrabutylammonium |
| ad | Adeninate |
| BPDC | Biphenyl dicarboxylate |
| 4,6-PmDc | 4,6-Pyrimidicarboxylate |
| HMDA+ | Dimethylamonnium |
| NENU-3 | (C4H12N)2[Cu12(BTC)8·12H2O][HPW12O40]·25H2O |
| NENU-28 | K2[Cu12(BTC)8·12H2O][HPW12O40]·28H2O |
| BDC2− | 1,4-Benzenedicarboxylate |
| H3dttz | 4,5-Di(1H-tetrazol-5-yl)-2H-1,2,3-triazole |
| IDC3− | Imidazole-4,5-dicarboxylate |
| L′ | 1,2,4,5-Benzenetetracarboxylate |
| H4ttca | 1,1′:2′,1′′-Terphenyl-4,4′,4′′,5′-tetracarboxylic acid |
| L′′ | Tetrakis[4-(carboxyphenyl)oxamethyl]methane |
| SHG | Second harmonic generation |
| BPDC | Biphenyldicarboxylate |
| DMASM | 4-[p-(Dimethylamino)styryl]-1-methylpyridinium |
| H4BTMIPA | 5,5′-Methylenebis(2,4,6-trimethyl sophtalic acid) |
| LDH | Layered double hydroxide |
Acknowledgements
The authors would like to acknowledge the financial support of University of Tehran for this research under grant number 01/1/389845.References
- K. Akhbari and A. Morsali, Coord. Chem. Rev., 2010, 257, 1972–1981 Search PubMed.
- S. Hojaghani, K. Akhbari, M. H. Sadr and A. Morsali, Inorg. Chem. Commun., 2014, 44, 1–5 CrossRef CAS.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC.
- K. Akhbari, S. Beheshti, A. Morsali, V. Yilmaz and O. Büyükgüngör, J. Mol. Struct., 2014, 1074, 279–283 CrossRef CAS.
- T. Uemura and S. Kitagawa, Chem. Lett., 2005, 34, 132–137 CrossRef CAS.
- K. Akhbari, A. Morsali and P. Retailleau, Ultrason. Sonochem., 2013, 20, 1428–1435 CrossRef CAS PubMed.
- S. Wu, X. Shen, B. Cao, L. Lin, K. Shen and W. Liu, J. Mater. Sci., 2009, 44, 6447–6450 CrossRef CAS.
- K. Akhbari, M. Hemmati and A. Morsali, J. Inorg. Organomet. Polym., 2011, 21, 352–359 CrossRef CAS.
- X. P. Sun, S. J. Dong and E. K. Wang, J. Am. Chem. Soc., 2005, 127, 13102–13103 CrossRef CAS PubMed.
- M. Du, C.-P. Li, C.-S. Liu and S.-M. Fang, Coord. Chem. Rev., 2013, 257, 1282–1305 CrossRef CAS.
- M. Lin Hu, A. Morsali and L. Aboutorabi, Coord. Chem. Rev., 2011, 225, 2821–2859 Search PubMed.
- K. Akhbari and A. Morsali, Inorg. Chem. Acta, 2009, 363, 1692–1700 CrossRef.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed.
- M. Y. Masoomi and A. Morsali, Coord. Chem. Rev., 2012, 256, 2921–2943 CrossRef CAS.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, CrystEngComm, 2012, 14, 3001–3004 RSC.
- C. De Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef PubMed.
- S. Noro, R. Kitaura, M. Kondo, S. Kitagawa, T. Ishii, H. Matsuzaka and M. Yamashita, J. Am. Chem. Soc., 2002, 124, 2568–2583 CrossRef CAS PubMed.
- A. M. Shultz, A. A. Sarjeant, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2011, 133, 13252–13255 CrossRef CAS PubMed.
- L. J. Murray, M. Dinco and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- J. L. Rowsell and O. M. Yaghi, Angew. Chem., 2005, 44, 4670–4679 CrossRef CAS PubMed.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2005, 127, 6506–6507 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- J. Lee, O. K. Farha, J. Roberts, k. a. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- R. Zou, H. Sakurai and Q. Xu, Angew. Chem., 2006, 45, 2542–2546 CrossRef CAS PubMed.
- S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitgawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS PubMed.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2011, 112, 869–932 CrossRef PubMed.
- R. C. Huxford, J. D. Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS PubMed.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS PubMed.
- F. Shahangi and K. Akhbari, Inorg. Chem. Acta, 2015, 436, 1–6 CrossRef.
- F. Shahangi and K. Akhbari, RSC Adv., 2015, 5, 50778–50782 RSC.
- M. Moeinian and K. Akhbari, J. Solid State Chem., 2015, 225, 459–463 CrossRef CAS.
- K. S. Min and M. P. Suh, J. Am. Chem. Soc., 2000, 122, 6834–6840 CrossRef CAS.
- K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 9604–9605 CrossRef CAS PubMed.
- T. K. Maji, R. Matsuda and S. Kitagawa, Nat. Mater., 2007, 6, 142–148 CrossRef CAS PubMed.
- Z. Z. Lin, F. L. Jiang, D. Q. Yuan, L. Chen, Y. F. Zhou and M. C. Hong, Eur. J. Inorg. Chem., 2005, 1927–1931 CrossRef CAS.
- M. Lalonde, W. Bury, O. Karagiaridi, Z. Brown, j. t. Hupp and O. K. Farha, J. Mater. Chem. A, 2013, 1, 5453–5454 CAS.
- C. K. Brozek and M. Dinca, Chem. Soc. Rev., 2014, 43, 5457–5467 RSC.
- H. Fei, J. F. Cahill, K. A. Prather and S. M. Cohen, Inorg. Chem., 2013, 52, 4011–4016 CrossRef CAS PubMed.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- S. Das, H. Kim and K. Kim, J. Am. Chem. Soc., 2009, 131, 3814–3815 CrossRef CAS PubMed.
- A. Nalaparaju and J. Jiang, J. Phys. Chem., 2012, 116, 6925–6931 CAS.
- J. Fan, L. Gan, H. Kawaguchi, W. Y. Sun and W. X. Tang, Chem.–Eur. J., 2003, 9, 3965–3973 CrossRef CAS PubMed.
- N. Gimenon and R. Vilar, Coord. Chem. Rev., 2006, 250, 3161–3189 CrossRef.
- D. X. Wang, H. Y. He, X. H. Chen, S. Y. Feng, Y. Z. Niu and D. F. Sun, CrystEngComm, 2010, 12, 1041–1043 RSC.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1996, 118, 295–296 CrossRef CAS.
- H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 2004, 126, 15845–15851 Search PubMed.
- Q. Fang, G. Zhu, M. Xue, J. Sun, Y. Wei, S. Qiu and R. Xu, Angew. Chem., Int. Ed., 2005, 44, 3845–3848 CrossRef CAS PubMed.
- X. Z. Kiang, Y. H. Lee, S. Lobkovsky and E. B. Emmott, J. Am. Chem. Soc., 2000, 122, 8376–8391 CrossRef.
- S. R. Halper, L. Do, J. R. Stork and S. M. Cohen, J. Am. Chem. Soc., 2006, 128, 15255–15267 CrossRef CAS PubMed.
- S. Y. Wan, Y. T. Huang, Y. Z. Li and W. Y. Sun, Microporous Mesoporous Mater., 2004, 73, 101–108 CrossRef CAS.
- M. Shu, C. Tu, W. Xu, H. Jin and J. Sun, Cryst. Growth Des., 2006, 6, 1890–1896 CAS.
- J. Chen, C. P. Li, J. Shang and M. Du, Inorg. Chem. Commun., 2012, 15, 172–175 CrossRef CAS.
- M. Sadakiyo, H. Kasai, K. Kato, M. Takata and M. Yamaudi, J. Am. Chem. Soc., 2014, 136, 1702–1705 CrossRef CAS PubMed.
- L. Carlucci, G. Ciani, S. Maggini, D. H. Proserpio and M. Visconti, Chem.–Eur. J., 2010, 16, 12328–12341 CrossRef CAS PubMed.
- M. L. Feng, D. N. Kong, Z. L. Xie and X. Y. Huang, Angew. Chem., 2008, 74, 8623–8626 CrossRef PubMed.
- P. Wang, J. P. Ma, Y. B. Dong and R. Q. Huang, J. Am. Chem. Soc., 2007, 129, 10620–10621 CrossRef CAS PubMed.
- R. Mark and W. N. Findley, Polym. Eng. Sci., 1978, 18, 6–15 CAS.
- J. An and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 5578–5579 CrossRef CAS PubMed.
- L. Carlucci, G. Ciani, S. Maggini, D. M. Proserpio and M. Visconti, Chem.–Eur. J., 2010, 16, 12328–12341 CrossRef CAS PubMed.
- T. F. Liu, J. Lu, C. Tian, M. Cao, Z. Lin and R. Cao, Inorg. Chem., 2011, 50, 2264–2271 CrossRef CAS PubMed.
- J. Yu, Y. Cui, C. Wu, Y. Yang, Z. Wang, M. O'Keeffe, B. Chen and G. Qian, Angew. Chem., 2012, 51, 1–5 CrossRef.
- S. Xiong, S. Li, S. Wang and Z. Wang, CrystEngComm, 2011, 13, 7236–7245 RSC.
- R. X. Yao, X. Xu and X. M. Zhang, Chem. Mater., 2012, 24, 303–310 CrossRef CAS.
- H. Zhang, Y. Lu, Z. Zhang, H. Fu, Y. Li, D. Volkmer, D. Denysenko and E. Wang, Chem. Commun., 2012, 48, 7295–7297 RSC.
- D. Banerjee, S. J. Kim, H. Wu, W. Xu, L. A. Borkowski, J. Li and J. B. Parise, J. Am. Chem. Soc., 2011, 50, 208–212 CAS.
- D. W. Breck and R. E. Krieger, Struct. Chem., 1984, ix, 771 Search PubMed.
- Y. Liu, V. C. Kravtsov, R. Larsen and M. Eddaoudi, Chem. Commun., 2006, 1488–1590 RSC.
- M. Eddaoudi, J. F. Eubank, Y. Liu, V. C. Kravtsov, R. W. Larsen and J. A. Brant, Stud. Surf. Sci. Catal., 2007, 170B, 2021–2029 CrossRef CAS.
- X. Zhao, X. Bu, T. Wu, S. T. Zheng, L. Wang and P. Feng, Nat. Commun., 2013, 3344, 1–9 Search PubMed.
- J. P. Ma, Y. Yu and Y. B. Dong, Chem. Commun., 2012, 48, 2946–2948 RSC.
- R. Custelcean, T. J. Havelock and B. A. Moyer, Inorg. Chem., 2006, 45, 6446–6452 CrossRef CAS PubMed.
- S. R. Oliver, Chem. Soc. Rev., 2009, 38, 1868–1881 RSC.
- US Department of Health and Human Services, Toxicological Profile for Chromium, Public Health Service Agency for Toxic Substances and Disease Registry, Washington, DC, 1991 Search PubMed.
- E. A. Katayev, Y. A. Ustynyuk and J. L. Sessler, Coord. Chem. Rev., 2006, 250, 3004–3037 CrossRef CAS.
- P. F. Shi, B. Zhao, G. Xiong, Y. L. Hou and P. Cheng, Chem. Commun., 2012, 48, 8231–8233 RSC.
- E. T. Urbansky, Environ. Sci. Pollut. Res. Int., 2002, 9, 187–192 CrossRef CAS PubMed.
- H. Fei, M. R. Bresler and S. R. Oliver, J. Am. Chem. Soc., 2011, 133, 11110–11113 CrossRef CAS PubMed.
- H. R. Fu, Z. X. Xu and J. Zhang, Chem. Mater., 2015, 27, 205–210 CrossRef CAS.
- X. Li, H. Xu, F. Kong and R. Wang, Angew. Chem., Int. Ed., 2013, 52, 13769–13773 CrossRef CAS PubMed.
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Férey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9267–9271 CrossRef CAS PubMed.
- H. Furukawa, F. Gandara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- K. M. Choi, H. M. Jeong, J. H. Park, Y.-B. Zhang, J. K. Kang and O. M. Yaghi, ACS Nano, 2014, 8, 7451–7457 CrossRef CAS PubMed.
- Q. Zhang, J. Yu, J. Cai, L. Zhang, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2015, 51, 14732–14734 RSC.
- A. V. Desai, B. Manna, A. Karmakar, A. Sahu and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 7811–7815 CrossRef CAS PubMed.
- F. X. L. Xamena, O. Casanova, R. G. Tailleur, H. Garcia and A. Corma, J. Catal., 2008, 225, 220–227 Search PubMed.
- S. Wang, L. Li, J. Zhang, X. Yuan and C. Y. Su, Mater. Chem., 2011, 21, 7098–7104 RSC.
- B. K. Banik, M. Chapa, J. Marquez and M. Cardona, Tetrahedron Lett., 2005, 46, 2341–2343 CrossRef CAS.
- H. Fei, D. L. Rogow and S. R. J. Oliver, J. Am. Chem. Soc., 2010, 132, 7202–7209 CrossRef CAS PubMed.
- Q. Y. Yang, K. Li, J. Luo, M. Pan and C. Y. Su, Chem. Commun., 2011, 47, 4234–4236 RSC.
- B. Manna, A. K. Chaudhari, B. Joorder, A. Karmaker and S. K. Ghosh, Angew. Chem., 2013, 52, 998–1002 CrossRef CAS PubMed.
- X. P. Li, J. Y. Zhang, M. Pan, S. R. Zheng, Y. Liu and C. Y. Su, Inorg. Chem., 2007, 46, 4617–4625 CrossRef CAS PubMed.
- J. J. Baldovi, E. Coronado, A. G. Arino, C. Gamer, M. G. Marques and G. M. Espallargas, Chem.–Eur. J., 2014, 20, 10695–10702 CrossRef CAS PubMed.
- Q. K. Liu, J. P. Ma and Y. B. Dong, Chem. Commun., 2011, 47, 7185–7187 RSC.
- E. Q. Procopio, F. Linares, C. Montoro, V. Colombo, A. Maspero, E. Barea and J. A. R. Navarro, Angew. Chem., 2010, 122, 7456–7469 Search PubMed.
- Y. X. Tan, Y. P. He and J. Zhang, Chem. Commun., 2011, 47, 10647–10649 RSC.
- Y. X. Tan, Y. P. He and J. Zhang, Chem. Commun., 2014, 50, 6153–6156 RSC.
- D. T. Genna, A. G. Wong-Fey, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed.
- S. Beheshti and A. Morsali, RSC Adv., 2014, 4, 41825–41830 RSC.
- S. Beheshti and A. Morsali, RSC Adv., 2014, 4, 37036–37040 RSC.
- F. Nouar, J. Eckert, J. F. Eubank, P. Forster and M. Eddaoudi, J. Am. Chem. Soc., 2009, 131, 2864–2870 CrossRef CAS PubMed.
- G. Calleja, J. A. Botas, M. S. Sanchez and M. G. Orcajo, Int. J. Hydrogen Energy, 2010, 35, 9916–9923 CrossRef CAS.
- S. Yang, G. S. B. Martin, J. J. Titman, A. J. Blake, D. R. Allan, N. R. Champness and M. Schroder, Inorg. Chem., 2011, 50, 9374–9384 CrossRef CAS PubMed.
- S. Yang, X. Lin, A. J. Blake, G. S. Walker, P. Hubberstay, N. R. Champness and M. Schroder, Nat. Chem., 2009, 1, 487–493 CrossRef CAS PubMed.
- K. L. Mulfort, O. K. Farha, C. L. Stern, A. A. Sarjeant and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 3866–3868 CrossRef CAS PubMed.
- Y. Liu, V. C. Kravtsov and M. Eddaoudi, Angew. Chem., 2008, 47, 8446–8449 CrossRef CAS PubMed.
- S. Yang, S. K. Callear, A. J. Ramirez-Cuesta, W. I. F. David, J. Sun, A. J. Blake, N. R. Champness and M. Schroder, Faraday Discuss., 2011, 151, 19–36 RSC.
- J. An and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 5578–5579 CrossRef CAS PubMed.
- C. Chen, J. Kim, D. A. Yang and W. S. Ahn, Chem. Eng. J., 2011, 168, 1134–1139 CrossRef CAS.
- D. F. Sava, V. C. Kravtsov, F. Nouar, L. Wojtas, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 3768–3770 CrossRef CAS PubMed.
- X. R. Hao, X. L. Wang, Z. M. Su, K. Z. Shao, Y. H. Zhao, Y. Q. Lan and Y. M. Fu, Dalton Trans., 2009, 40, 8562–8566 RSC.
- Z. J. Li, S. K. Khani, K. Akhbari, A. Morsali and P. Retailleau, Microporous Mesoporous Mater., 2014, 199, 93–98 CrossRef CAS.
- T. Z. Zhang, Z. M. Zhang, Y. Lu, H. Fu and E. B. Wang, CrystEngComm, 2013, 15, 459–462 RSC.
- K. Akhbari and A. Morsali, Dalton Trans., 2013, 42, 4786–4789 RSC.
- F. J. Ma, S. X. Liu, D. D. Liang, G. J. Ren, F. Wei, Y. G. Chen and Z. M. Su, Solid State Chem., 2011, 184, 3034–3039 CrossRef CAS.
- J. S. Qin, S. R. Zhang, D. Y. Du, P. Shen, S. J. Bao, Y. Q. Lan and Z. M. Su, Chem.–Eur. J., 2014, 20, 5625–5630 CrossRef CAS PubMed.
- W. G. Lu, L. Jiang, X. L. Feng and T. B. Lu, Inorg. Chem., 2009, 48, 6997–6999 CrossRef CAS PubMed.
- S. Liu, J. Li and F. Luo, Inorg. Chem. Commun., 2010, 13, 870–872 CrossRef CAS.
- J. Cao, Y. Gao, Y. Wang, C. Du and Z. Liu, Chem. Commun., 2013, 49, 6897–6899 RSC.
- S. Dang, E. Ma, Z. M. Sun and H. Zhang, Mater. Chem., 2011, 22, 16920–16926 RSC.
- Z. Chen, Y. Sun, L. Zhang, D. Sun, F. Liu, Q. Meng, R. Wang and D. Sun, Chem. Commun., 2013, 49, 11557–11559 RSC.
- Y. Liu, G. Li, X. Li and Y. Cui, Angew. Chem., 2007, 119, 6417–6420 CrossRef.
- J. Yu, Y. Cui, H. Xu, Y. Yang, Z. Wang, B. Chen and G. Qian, Nat. Commun., 2013, 3719, 1–7 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |