
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient construction of biologically important functionalized polycyclic spiro-fused carbocyclicoxindoles via an asymmetric organocatalytic quadruple-cascade reaction†
Wen Ren‡
a,
Xiao-Yan Wang‡b,
Jing-Jing Lia,
Mao Tiana,
Jie Liu*a,
Liang Ouyanga and
Jin-Hui Wang*c
aState Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Collaborative Innovation Center for Biotherapy, Chengdu 610041, China. E-mail: liujie2011@scu.edu.cn; Fax: +86-028-8550-3817; Tel: +86-028-8550-3817
bAnalytical & Testing Center, Sichuan University, Chengdu 610064, China
cCollege of Pharmacy, Xinjiang Medical University, Urumqi 830011, China. E-mail: wangjinhui2015@sina.com
First published on 12th January 2017
Abstract
An efficient construction of highly functionalized polycyclic spiro-fused carbocyclicoxindoles has been developed via an asymmetric organocatalytic quadruple domino reaction of (E)-3-(2-hydroxybenzylidene)oxindole derivatives and two molecules of α,β-unsaturated aldehyde under quadruple iminium–enamine–iminium–enamine catalysis. The complex cascade products bearing a spiro quaternary center and five contiguous stereocenters were obtained in moderate to high yields (up to 90%) with good diastereoselectivities (up to 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and excellent ee values (up to 99% ee). The structure and absolute configuration of the products were confirmed by NMR spectroscopy and single crystal X-ray analysis. In addition, the biological study showed that these compounds had moderate antitumor activities in the micromolar range.
:1) and excellent ee values (up to 99% ee). The structure and absolute configuration of the products were confirmed by NMR spectroscopy and single crystal X-ray analysis. In addition, the biological study showed that these compounds had moderate antitumor activities in the micromolar range.
Introduction
Spirooxindoles have become a privileged skeleton and exist in a large number of natural products and pharmaceutically important molecules such as spirotryprostatin A, pteropodine, elacomine, horsfiline, marcfortine A, cirinalins A, NF54 and MptpB.1 They have various types of bioactivity such as progesterone receptor modulators, anti-HIV, anticancer, antitubercular, and MDM2-p53 interaction inhibitors (Fig. 1). Due to these facts, considerable effort has been devoted to developing efficient protocols to access these interesting motifs over the past years.2 Among those successful examples, isatins or 3-substituted 2-oxindoles have been employed and represent a broadly successful strategy in the design and synthesis of spiro-cyclic frameworks.3 This strategy can allow efficient double cascade reactions. Since the pioneering work of Enders involving triple cascade reactions,4 more and more researchers gradually realized that the organocatalytic cascade reactions were a useful synthetic tool for the construction of complex chiral polycyclic derivatives. As a consequence, some multicomponent quadruple cascade reactions involving the formation of four C–C bonds and generating multiple stereocenters also have been reported.5 However, among them, fewer examples have been reported that construct spiro-fused polycyclic frameworks. Therefore, the development of exquisite higher-order cascade reactions in the construction of more complex spiro-fused polycyclic frameworks with stereocontrolled spiro quaternary centers and multiple chiral centers is highly attractive and in great demand.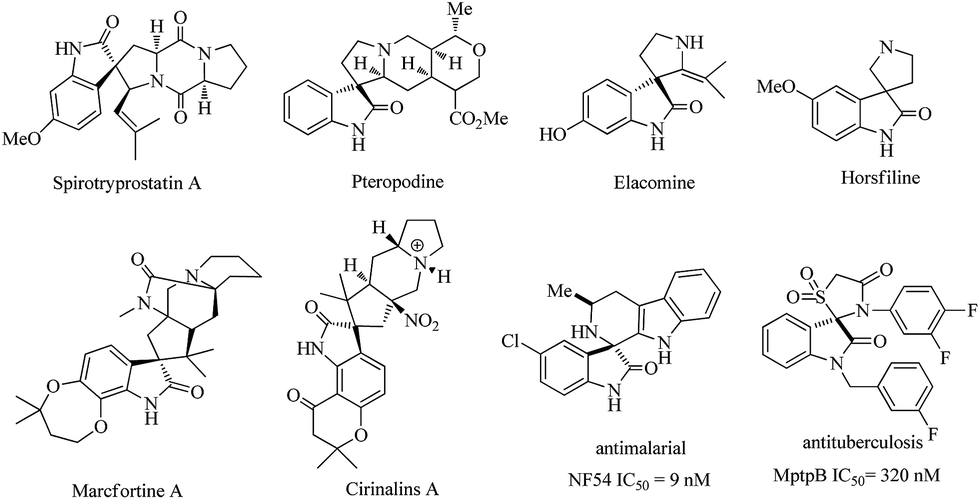 | ||
| Fig. 1 Examples of natural products, pharmaceutical molecules containing spiro-fused cyclic frameworks. | ||
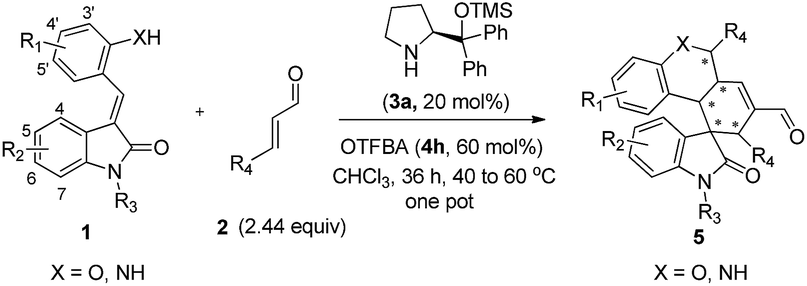
Alkylideneoxindoles (R groups are usually phenyl, alkyl, ester) as prochiral electrophiles have been used in catalytic asymmetric synthesis of spirooxindoles via different cyclizations such as 1,3-dipolar cycloadditions, Diels–Alder reactions, and cyclopropanation.3d–f,k,p–s,6 In contrast, fewer examples that construct spirooxindoles from alkylideneoxindoles involving quadruple cascade reactions have been reported. This most likely owed to the insufficient reactive sites on the alkylideneoxindoles and the fact organocatalytic domino reactions are unable to proceed. The careful design of substrate by introducing multifunctional group on the substrates could serve as a good starting point to form novel transformations in both reaction sequences and complete the domino reactions. Recently, many double or triple cascade reactions have been initiated by oxa-Michael.7 Secondary amines, such as proline and derivatives, were employed in these reactions by their iminium-ion and enamine activation modes for an efficient activation of both donor- and acceptor-type molecules. Inspired by these successes, we reasoned that the introduction of a hydroxy group on the alkylideneoxindoles, as a donor in Michael reaction, might successively perform as nucleophiles and electrophiles, and facilitate the quadruple cascade reactions (Scheme 1). In addition, the quadruple cascade reactions are atom-, time-, step-economic, and can often imitate biosynthetic pathways, all which make them increasingly attractive for the organic chemistry community.8 Herein, we envisioned a simple and efficient protocol for quadruple cascade reaction through iminium–enamine–iminium–enamine sequential activation initiated by oxa-Michael addition of phenolic hydroxyl to α,β-unsaturated aldehyde, as outlined in Scheme 1, to provide a straightforward protocol for the synthesis of polycyclic spiro-fused carbocyclicoxindoles with a spiro quaternary center and five contiguous stereocenters.
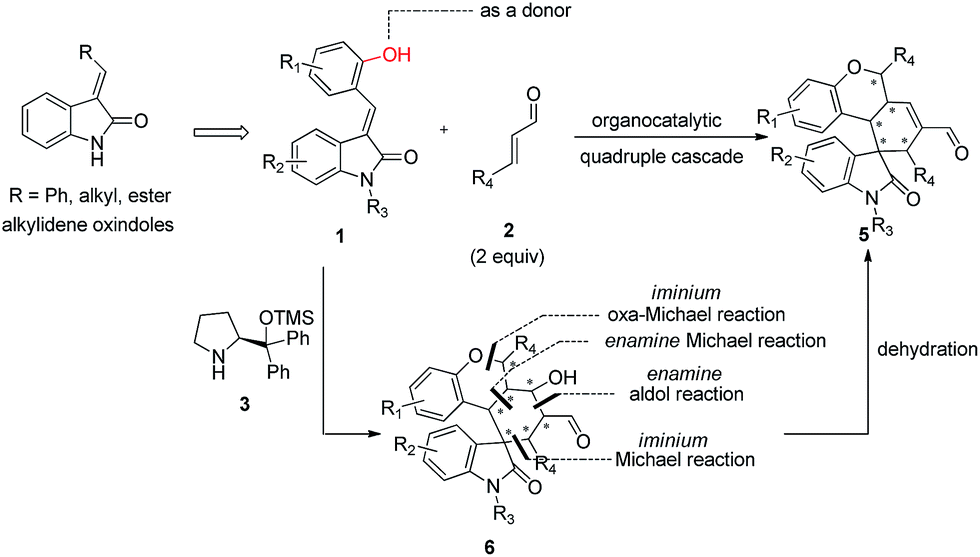 | ||
| Scheme 1 Organocatalytic quadruple cascade reaction through iminium–enamine–iminium–enamine sequential activation initiated by oxa-Michael addition to construct polycyclic spiro-fused carbocyclicoxindoles. | ||
Results and discussion
At the outset of the study, the multifunctional substrate 1a was easily prepared via a Knoevenagel reaction of oxindole and salicylaldehyde.9 Then the quadruple domino reaction of (E)-3-(2-hydroxybenzylidene)oxindole 1a (1.0 equiv.) and excess crotonaldehyde 2a (2.44 equiv.), as a simple model substrate was investigated to establish the feasibility of the strategy and optimize the reaction conditions. By using chloroform as solvent at 60 °C for 12 h, the catalysis of (S)-diphenylprolinol TMS ether 3a (20 mol%) and acetic acid (20 mol%) gave satisfactory results, and the desired polycyclic spiro-fused carbocyclicoxindoles 5aa, bearing five contiguous stereocenters, was obtained in 60% yield (Table 1, entry 1). Although the diastereoselectivity was only 1:1, the isolation of the quadruple domino product 5aa by flash chromatography revealed that the two diastereomer with 98% ee and 90% ee could be separated from each other. The reaction with 3a without the acid additive gave no reaction for two days. Other different secondary amine catalysts such (S)-diphenylprolinol TES ether 3b and (S)-diphenylprolinol 3c were also estimated, no superior result was obtained (Table 1, entries 2–3). To obtain higher yields and diastereoselectivities, some other acids, which probably promoted the formation of iminium ion, were screened (Table 1, entries 4–11). The 2-(trifluoromethyl)benzoic acid 4h was shown to be suitable for this reaction, affording the product with 2:1 diastereoselectivity (Table 1, entry 10). While stronger acids, such as TFA 4c, made reaction sluggish with only trace product (Table 1, entry 5). Subsequently, we performed a short solvent screening (Table 1, entries 12–21). Whereas the reaction in anisole gave a product of reversed configuration (Table 1, entry 16), no conversion could be detected when the reaction was performed in protic and polar solvents (Table 1, entries 12–15, 19–21). Toluene showed a comparable yield and enantiomeric excess of product of 5aa to chloroform but the diastereoselectivity was decreased (Table 1, entry 18). To our delight, we found that the diastereoselectivity could be improved by increasing the ratio of additive 4h and catalyst 3a to 3:1 (Table 1, entry 22 the detail see the ESI†). Lower catalyst loading led to a significant decrease in the yields while higher amounts did not give any remarkable improvement (data not shown). Best results were afforded by adjusting the temperature and prolonging reaction time (Table 1, entry 25). Based on the above results, the optimal condition was found: the reaction was conducted at 40 °C for 24 h, then heated up to 60 °C for 12 h, and the quadruple domino product 5aa was obtained in 80% yield with 8:1 diastereoselectivity and >99% ee (major).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Additive | Yieldb [%] | drc | eed [%] |
| a Unless otherwise noted, reactions were performed with 0.1 mmol 1a and 0.244 mmol (20 μL) 2a in the presence of catalyst 3 (20 mol%) and the additive acid or phenol 4 (20 mol%) in 1.0 mL of solvent at 60 °C for 12 h.b The values are yields of isolated product after silica gel column chromatography.c Determined by 1H NMR on the crude reaction mixture.d Determined by chiral HPLC.e The ratio of catalyst 3a (20 mol%) and additive 4h (60 mol%) was 1:3.f The reaction was conducted at 40 °C for 60 h.g The reaction was conducted at 25 °C for 168 h.h The reaction was conducted at 40 °C for 24 h, then heated up to 60 °C for 12 h. MTBE = methyl tert-butyl ether; EA = ethyl acetate; n.r. = no reaction. n.d. = not detected. |
||||||
| 1 | 3a | CHCl3 | 4a | 60 | 1:1 |
98; 90 |
| 2 | 3b | CHCl3 | 4a | 55 | 1:1 |
92; 90 |
| 3 | 3c | CHCl3 | 4a | Traces | n.d. | n.d. |
| 4 | 3a | CHCl3 | 4b | Traces | n.d. | n.d. |
| 5 | 3a | CHCl3 | 4c | Traces | n.d. | n.d. |
| 6 | 3a | CHCl3 | 4d | Traces | n.d. | n.d. |
| 7 | 3a | CHCl3 | 4e | 63 | 1:1 |
98; 93 |
| 8 | 3a | CHCl3 | 4f | 65 | 1:1 |
98; 94 |
| 9 | 3a | CHCl3 | 4g | 61 | 1:1 |
98; 91 |
| 10 | 3a | CHCl3 | 4h | 68 | 2:1 |
>99; 94 |
| 11 | 3a | CHCl3 | 4i | 58 | 1:1 |
97; 93 |
| 12 | 3a | MeOH | 4h | n.r. | n.d. | n.d. |
| 13 | 3a | EtOH | 4h | n.r. | n.d. | n.d. |
| 14 | 3a | THF | 4h | n.r. | n.d. | n.d. |
| 15 | 3a | Dioxane | 4h | n.r. | n.d. | n.d. |
| 16 | 3a | Anisole | 4h | 44 | 1:2 |
>99; 92 |
| 17 | 3a | MTBE | 4h | n.r. | n.d. | n.d. |
| 18 | 3a | Toluene | 4h | 55 | 1:1 |
>99; 92 |
| 19 | 3a | MeCN | 4h | n.r. | n.d. | n.d. |
| 20 | 3a | EA | 4h | n.r. | n.d. | n.d. |
| 21 | 3a | DMF | 4h | n.r. | n.d. | n.d. |
| 22e | 3a | CHCl3 | 4h | 83 | 3:1 |
99; 85 |
| 23f | 3a | CHCl3 | 4h | 72 | 6:1 |
97; n.d. |
| 24g | 3a | CHCl3 | 4h | 67 | 5:1 |
>99; n.d. |
| 25h | 3a | CHCl3 | 4h | 80 | 8:1 |
>99; n.d. |
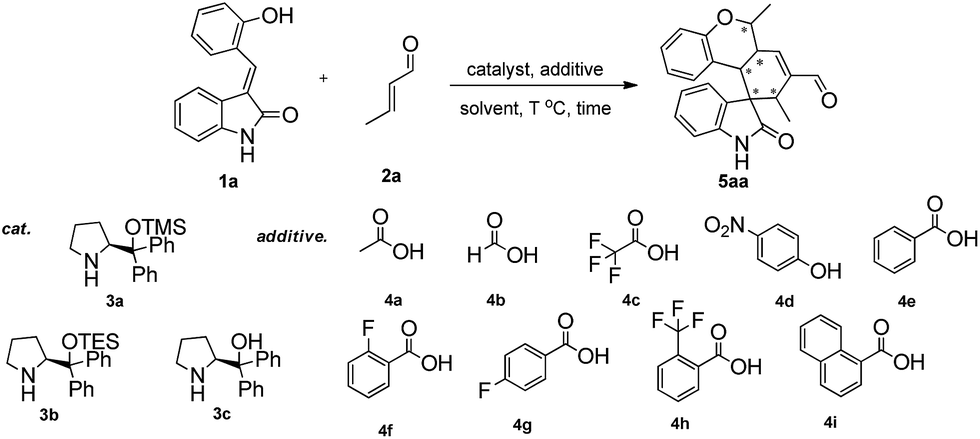
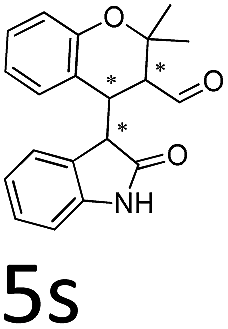
Having established the optimal reaction conditions, we evaluated the scope of the quadruple cascade reaction (Table 2). Different alkylideneoxindoles derived from various substituted oxindole and substituted salicylaldehyde proceeded smoothly, and the corresponding products could be isolated in moderate to high yields (up to 90%) with good diastereoselectivities (up to 8:1) and excellent ee values (up to 99% ee) (Table 2, entries 1–15). The electronic properties of the substituents at the aromatic ring had no obvious effect on enantioselectivity, but affected the yields and diastereoselectivity strongly. Generally, alkylideneoxindoles with halogen-substituted groups gave higher yields and diastereoselectivities than other substituted groups (Table 2, entries 2, 4–6, 11, 12 vs. 3, 7, 8). The steric hindrance of the substituents also influenced the diastereoselectivity. The introduction of more sterically naphthalene ring to alkylideneoxindoles proceeded smoothly but provided the domino product of other configuration (Table 2, entry 9). Moreover, incorporating methyl or phenyl groups on the N1 of alkylidene oxindole led to an improvement in diastereoselectivities to 10:1 and 12:1, respectively, but the yields of the reaction dropped dramatically (Table 2, entries 13, 14). Interestingly, the Michael addition could be realized when an amine group as a donor was introduced at 2′ position of alkylideneoxindole. The tetrahydroquinoline framework 5oa incorporating a spirooxindole motif was obtained in 83% yield and 99% ee (Table 2, entry 15). The reaction of other aliphatic α,β-unsaturated aldehydes with representative alkylideneoxindole 1a was also examined (Table 2, entries 16–19). These reactions were performed under the same conditions, and gave the corresponding products in moderate to high yields with moderate diastereoselectivities and excellent ee values (Table 2, entries 16–18). Notably, for 3-methyl-2-butenal was employed, only intermediate 5s was obtained (Table 2, entry 19). This may be caused by steric hindrance of 2e. On the other hand, we hope to introduce more substitution diversity into spirocyclic products. After the Michael–Michael reaction of 1a and 1 equiv. of 2e conducted at 40 °C for 24 h, from which intermediate was generated and isolated. Then 1 equiv. of 2a was added to continue the cascade Michael-aldol condensation sequences. The polycyclic spiro-fused carbocyclicoxindoles 5sa was directly obtained with excellent enantioselectivity while the yields were fair due to more possible reaction pathways (Table 2, entry 20).
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 | 2 | 5 | Yieldb (%) | drc | eed (%) |
| a Unless otherwise noted, reactions were performed with 0.1 mmol 1 and 0.244 mmol 2 in the presence of the catalyst 3a (20 mol%) and the additive 4h (60 mol%) in CHCl3 1 mL at 40 °C for 24 h and then heated up to 60 °C for 12 h, X = O.b The values are yields of isolated product after silica gel column chromatography.c Determined by 1H NMR on the crude reaction mixture.d Determined by chiral HPLC.e X = NH.f 2 equiv. of 2e was involved in the reaction at 40 °C for 24 h to generate 5s.g 1 equiv. of 2e was employed in the reaction at 40 °C for 24 h to generate 5s, and then 1 equiv. of 2a was added in the reaction at 60 °C for 12 h to generate 5sa. | ||||||
| 1 | R1 = R2 = R3 = H, 1a | R4 = CH3, 2a | 5aa | 80 | 8:1 |
>99 |
| 2 | R1 = 3′-F, R2 = R3 = H, 1b | R4 = CH3, 2a | 5ba | 75 | 6:1 |
>99 |
| 3 | R1 = 4′-OCH3, R2 = R3 = H, 1c | R4 = CH3, 2a | 5ca | 65 | 5:1 |
>99 |
| 4 | R1 = 5′-F, R2 = R3 = H, 1d | R4 = CH3, 2a | 5da | 72 | 7:1 |
98 |
| 5 | R1 = 5′-Cl, R2 = R3 = H, 1e | R4 = CH3, 2a | 5ea | 65 | 6:1 |
98 |
| 6 | R1 = 5′-Br, R2 = R3 = H, 1f | R4 = CH3, 2a | 5fa | 70 | 7:1 |
98 |
| 7 | R1 = 5′-NO2, R2 = R3 = H, 1g | R4 = CH3, 2a | 5ga | 35 | 4:1 |
98 |
| 8 | R1 = 5′-CH3, R2 = R3 = H, 1h | R4 = CH3, 2a | 5ha | 75 | 5:1 |
>99 |
| 9 | 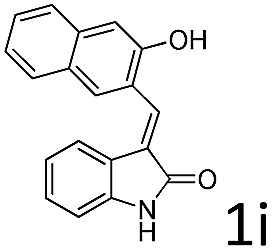 |
R4 = CH3, 2a | 5ia | 65 | 1:4 |
>99 |
| 10 | R2 = 5-CH3, R1 = R3 = H, 1j | R4 = CH3, 2a | 5ja | 67 | 6:1 |
98 |
| 11 | R2 = 5-F, R1 = R3 = H, 1k | R4 = CH3, 2a | 5ka | 80 | 6:1 |
96 |
| 12 | R2 = 6-Cl, R1 = R3 = H, 1l | R4 = CH3, 2a | 5la | 90 | 7:1 |
>99 |
| 13 | R1 = R2 = H, R3 = CH3, 1m | R4 = CH3, 2a | 5ma | 31 | 10:1 |
94 |
| 14 | R1 = R2 = H, R3 = Ph, 1n | R4 = CH3, 2a | 5na | 45 | 12:1 |
92 |
| 15e | R1 = R2 = R3 = H, 1o | R4 = CH3, 2a | 5oa | 83 | 2.5:1 |
>99 |
| 16 | R1 = R2 = R3 = H, 1a | R4 = H, 2b | 5pa | 55 | 1:2.5 |
64 |
| 17 | R1 = R2 = R3 = H, 1a | R4 = Et, 2c | 5qa | 60 | 3:1 |
>99 |
| 18 | R1 = R2 = R3 = H, 1a | R4 = n-Pr, 2d | 5ra | 78 | 4:1 |
>99 |
| 19f | R1 = R2 = R3 = H, 1a | 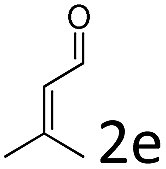 |
 |
55 | >20:1 |
87 |
| 20g | R1 = R2 = R3 = H, 1a | 2e/2a | 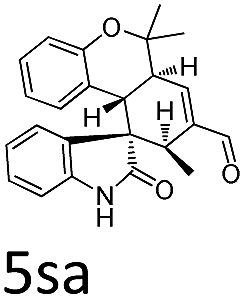 |
50 | 5:1 |
97 |
Further examination of the substrate focused on the alkylideneindanedione 1t. As illustrated in Scheme 2, the one pot quadruple cascade reaction of substrate 1t and 2a proceed smoothly and gave the polycyclic spiro-fused carbocyclicoxindoles 5ta with a spiro quaternary center and four chiral centers.
 | ||
| Scheme 2 Further expanding the scope to alkylideneindanedione. | ||
The absolute configuration of 5ka was assigned by CuKa X-ray single crystal diffraction analysis (Fig. 2),10 and the stereochemistry of other products 5aa–5ha, 5ja–5oa, 5qa–5sa was assigned by analogy and specific rotation values. To account for the stereoselectivity of this transformation, we propose a plausible mechanism, as depicted in Scheme 3. As starting material 1k, there is a facial selectivity in the first oxo-Michael addition step of the cascade. The iminium-activated crotonaldehyde 2a is attacked on its Re face by the hydroxyl group (TS-A) generating intermediate enamine B, then reacts in a Michael addition to give cyclic intermediate C, which can again act as nucleophiles to another equivalent of the activated α,β-unsaturated aldehyde from the Re face under the control of catalyst (TS-D). The third Michael addition step leads to intermediate E, which is unstable and easily reacts through an intramolecular aldol condensation under the reaction conditions, providing product 5ka and regenerating the catalyst.
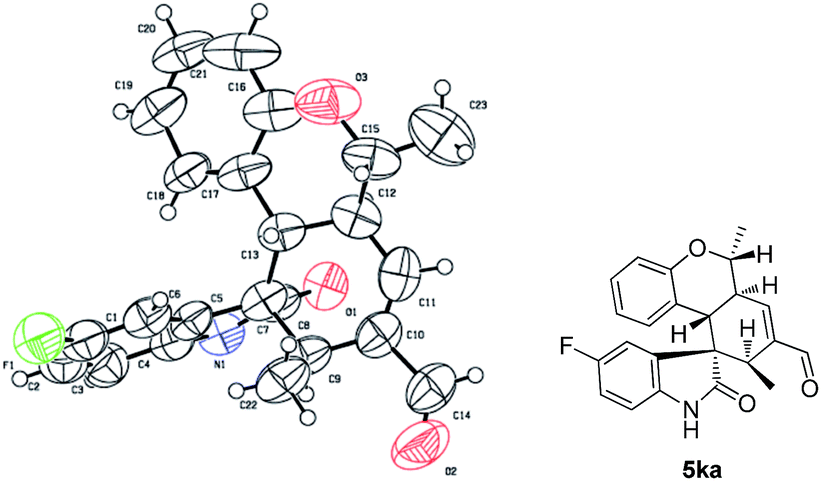 | ||
| Fig. 2 X-ray structure of enantio-pure 5ka. | ||
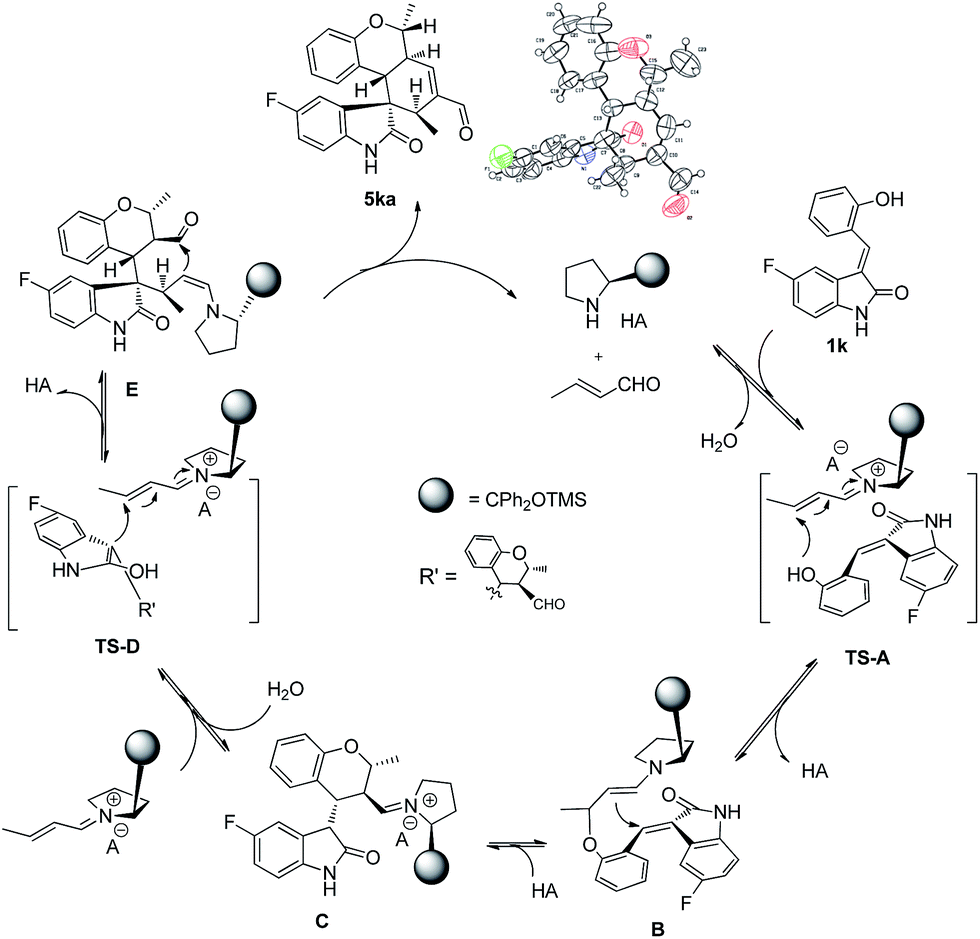 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Finally, in order to identify potential bioactivity of these novel compounds, some selected polycyclic spiro-fused carbocyclicoxindoles 5 were investigated for inhibition of cell proliferation against human cancer cell line. The preliminary results reveal that compound 5ia performed broad-spectrum antitumor activities against MCF-7, MDA-MB-231, H1975 and FaDu cancer cell lines with IC50 values in the low micromolar range (MCF-7 IC50 = 9.2 μM, MDA-MB-231 IC50 = 11.5 μM, H1975 IC50 = 14.1 μM and FaDu IC50 = 13.7 μM). These results indicates this type of polycyclic spiro-fused carbocyclicoxindoles derivatives holds great potential in synthetic and medicinal chemistry.
Conclusions
In conclusion, we have designed a novel type of multifunctional synthons (E)-3-(2-hydroxybenzylidene) oxindole derivatives. They have been successfully employed in an organocatalytic quadruple cascade reaction with two molecules of α,β-unsaturated aldehyde through iminium–enamine–iminium–enamine sequential activation. A range of polycyclic spiro-fused carbocyclicoxindoles bearing one spiro quaternary center and five contiguous stereocenters were delivered in moderate to high yields (up to 90%) with good diastereoselectivities (up to 8:1) and excellent ee values (up to 99% ee). Further expanding the scope of substrate demonstrated the spiro-fused indanedione framework also could be assembled in an effective manner. The absolute configurations of the products were unambiguously confirmed by single crystal X-ray crystallographic analyses and the possible mechanism was proposed for understanding the stereochemistry experimentally observed. Moreover, the results of anti-tumor activity screening indicated that this type of polycyclic spiro-fused carbocyclicoxindole derivatives may find medicinal applications after further structural modulation and biological studies.
Experimental section
General information
NMR spectra were recorded on the commercial 400 MHz spectrometer. The chemical shifts were recorded in ppm relative to tetramethylsilane (TMS) and with the solvent resonance as the internal standard. 1H NMR data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet, dt = doublet of triplet, dq = doublet of quartet), coupling constants (Hz), integration. 13C NMR data were collected at 100 MHz with complete proton decoupling. Enantiomeric excesses (ee) were determined by chiral HPLC analysis on DAICEL CORPORATION AS-H and AD-H columns in comparison with the authentic racemates. Optical rotations were reported as follows: [α]TD (c: g/100 mL, in solvent). ESI-HRMS spectra were recorded on a commercial apparatus and methanol was used to dissolve the sample. Solvents were commercially available and used directly without further purification. Several substituted oxindoles were prepared according to the literatures and references therein. The MTT method was used to evaluate the antiproliferative activities of the synthesized polycyclic spiro-fused carbocyclicoxindole derivatives. These cell lines (MCF-7, MDA-MB-231, H1975, FaDu) were obtained from ATCC.General procedure for asymmetric organocatalytic quadruple-cascade reaction of alkylideneoxindoles with enal (5aa–5sa)
α,β-Unsaturated aldehydes 2 (0.244 mmol) was added to a dry tube containing a suspension of substituted (E)-3-(2-hydroxybenzylidene)oxindole 1 (0.1 mmol), α,α-L-diphenylprolinol trimethylsilyl ether 3a (20 mol%), 2-(trifluoromethyl)benzoic acid 4h (60 mol%) and CHCl3 1 mL. The mixture was stirred at 40 °C for 24 h and then heated up to 60 °C for 12 h. The reaction mixture was subjected to silica gel column chromatography to yield the corresponding product.Acknowledgements
We appreciate the National Natural Science Foundation of China (No. 81573290, U1603123 and 81202403). We also thank Sichuan University Analytical & Testing Center for NMR analysis.Notes and references
- (a) A. P. Antonchick, C. Gerding-Reimers, M. Catarinella, M. Schürmann, H. Preut, S. Ziegler, D. Rauh and H. Waldmann, Nat. Chem., 2010, 2, 735 CrossRef CAS PubMed; (b) S. M. Rajesh, S. Perumal, J. C. Menendez, P. Yogeeswari and D. Sriram, Med. Chem. Commun., 2011, 2, 626 RSC; (c) V. V. Vintonyak, K. Warburg, H. Kruse, S. Grimme, K. Hübel, D. Rauh and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 5902 CrossRef CAS PubMed; (d) J. J. Badillo, N. V. Hanhan and A. K. Franz, Curr. Opin. Drug Discovery Dev., 2010, 13, 758 CAS; (e) G. Kumari, Nutan, M. Modi, S. K. Gupta and R. K. Singh, Eur. J. Med. Chem., 2011, 46, 1181 CrossRef CAS PubMed; (f) K. Ding, Y.-P. Lu, Z. Nikolovska-Coleska, S. Qiu, Y.-S. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. M. Wang, J. Am. Chem. Soc., 2005, 127, 10130 CrossRef CAS PubMed; (g) K. Ding, Y. Lu, Z. Nikolovska-Coleska, G. Wang, S. Qiu, S. Shangary, W. Gao, D. Qin, J. Stukey, K. Krajewski, P. P. Rollera and S. Wang, J. Med. Chem., 2006, 49, 3432 CrossRef CAS PubMed; (h) M. Tsuda, Y. Kasai, K. Komatsu, T. Sone, M. Y. Tanaka and J. Kobayashi, Org. Lett., 2004, 6, 3087 CrossRef CAS PubMed; (i) T. J. Greshock, A. W. Grubbs, P. Jiao, D. T. Wicklow, J. B. Gloer and R. M. Williams, Angew. Chem., Int. Ed., 2008, 47, 3573 CrossRef CAS PubMed.
- For recent reviews: (a) N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165 RSC; (b) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef CAS PubMed; (c) D.-J. Cheng, Y. Ishihara, B. Tan and C. F. Barbas III, ACS Catal., 2014, 4, 743 CrossRef CAS; (d) R. Rios, Chem. Soc. Rev., 2012, 41, 1060 RSC; (e) A. Moyano and R. Rios, Chem. Rev., 2012, 112, 6104 CrossRef PubMed; (f) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed.
- For selected examples of double and triple cascade reactions to spiro-fused cyclic frameworks containing 2-oxindoles: (a) L.-L. Wang, L. Peng, J.-F. Bai, Q.-C. Huang, X.-Y. Xu and L.-X. Wang, Chem. Commun., 2010, 46, 8064 RSC; (b) B. Tan, N. R. Candeias and C. F. Barbas III, Nat. Chem., 2011, 3, 473 CAS; (c) K. Albertshofer, K. E. Anderson and C. F. Barbas III, Org. Lett., 2012, 14, 5968 CrossRef CAS PubMed; (d) G. Bencivenni, L.-Y. Wu, A. Mazzanti, B. Giannichi, F. Pesciaioli, M.-P. Song, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7200 CrossRef CAS PubMed; (e) C. Cassani, X. Tian, E. C. Escudero-Adán and P. Melchiorre, Chem. Commun., 2011, 47, 233 RSC; (f) Q. Wei and L.-Z. Gong, Org. Lett., 2010, 12, 1008 CrossRef CAS PubMed; (g) Y.-B. Lan, H. Zhao, Z.-M. Liu, G.-G. Liu, J.-C. Tao and X.-W. Wang, Org. Lett., 2011, 13, 4866 CrossRef CAS PubMed; (h) Y.-M. Li, X. Li, F.-Z. Peng, Z.-Q. Li, S.-T. Wu, Z.-W. Sun, H.-B. Zhang and Z.-H. Shao, Org. Lett., 2011, 13, 6200 CrossRef CAS PubMed; (i) B. Wu, G.-G. Liu, M.-Q. Li, Y. Zhang, S.-Y. Zhang, J.-R. Qiu, X.-P. Xu, S.-J. Ji and X.-W. Wang, Chem. Commun., 2011, 47, 3992 RSC; (j) X.-W. Dou and Y.-X. Lu, Chem.–Eur. J., 2012, 18, 8315 CrossRef CAS PubMed; (k) Y.-M. Cao, X.-X. Jiang, L.-P. Liu, F.-F. Shen, F.-T. Zhang and R. Wang, Angew. Chem., Int. Ed., 2011, 50, 9124 CrossRef CAS PubMed; (l) X. Companyó, A. Zea, A.-N. R. Alba, A. Mazzanti, A. Moyano and R. Rios, Chem. Commun., 2010, 46, 6953 RSC; (m) A.-N. R. Alba, A. Zea, G. Valero, T. Calbet, M. Font-Bardia, A. Mazzanti, A. Moyano and R. Rios, Eur. J. Org. Chem., 2011, 1318 CrossRef CAS; (n) W.-B. Chen, Z.-J. Wu, Q.-L. Pei, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2010, 12, 3132 CrossRef CAS PubMed; (o) X.-H. Chen, Q. Wei, S.-W. Luo, H. Xiao and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 13819 CrossRef CAS PubMed; (p) F. Pesciaioli, P. Righi, A. Mazzanti, G. Bartoli and G. Bencivenni, Chem.–Eur. J., 2011, 17, 2842 CrossRef CAS PubMed; (q) A. Noole, N. S. Scucman, M. A. Kabeshov, T. Kanger, F. Z. Macaev and A. V. Malkov, Chem.–Eur. J., 2012, 18, 14929 CrossRef CAS PubMed; (r) W.-S. Sun, G.-M. Zhu, C.-Y. Wu, L. Hong and R. Wang, Chem.–Eur. J., 2012, 18, 6737 CrossRef CAS PubMed; (s) K. Jiang, Z.-J. Jia, S. Chen, L. Wu and Y.-C. Chen, Chem.–Eur. J., 2010, 16, 2852 CrossRef CAS PubMed.
- D. Enders, M. R. M. Hüttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861 CrossRef CAS PubMed.
- (a) M. Rueping and C. M. R. Volla, RSC Adv., 2011, 1, 79 RSC; (b) A. Raja, B.-C. Hong and G.-H. Lee, Org. Lett., 2014, 16, 5756 CrossRef CAS PubMed; (c) B.-C. Hong, P. Kotame, C.-W. Tsai and J.-H. Liao, Org. Lett., 2010, 12, 776 CrossRef CAS PubMed; (d) C. Joie, K. Deckers, G. Raabe and D. Enders, Synthesis, 2014, 46, 1539 CrossRef PubMed; (e) T. Chanda, S. Chowdhury, B. J. Ramulu, S. Koley, R. C. F. Jones and M. S. Singh, Tetrahedron, 2014, 70, 2190 CrossRef CAS; (f) P. Kotame, B.-C. Hong and J.-H. Liao, Tetrahedron Lett., 2009, 50, 704 CrossRef CAS; (g) N. Erdmann, A. R. Philipps, I. Atodiresei and D. Enders, Adv. Synth. Catal., 2013, 355, 847 CrossRef CAS; (h) L.-Y. Liu, Y.-N. Zhu, K.-M. Huang, B. Wang, W.-X. Chang and J. Li, Eur. J. Org. Chem., 2014, 4342 CrossRef CAS; (i) F.-L. Zhang, A.-W. Xu, Y.-F. Gong, M.-H. Wei and X.-L. Yang, Chem.–Eur. J., 2009, 15, 6815 CrossRef CAS PubMed; (j) D. Enders, A. Greb, K. Deckers, P. Selig and C. Merkens, Chem.–Eur. J., 2012, 18, 10226 CrossRef CAS PubMed; (k) M. Rueping, K. Haack, W. Ieawsuwan, H. Sundén, M. Blanco and F. R. Schoepke, Chem. Commun., 2011, 47, 3828 RSC; (l) D. Enders, C. Wang, M. Mukanova and A. Greb, Chem. Commun., 2010, 46, 2447 RSC; (m) K. Jiang, Z.-J. Jia, X. Yin, L. Wu and Y.-C. Chen, Org. Lett., 2010, 12, 2766 CrossRef CAS PubMed.
- N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165 CAS.
- A.-B. Xia, C. Wu, T. Wang, Y.-P. Zhang, X.-H. Du, A.-G. Zhong, D.-Q. Xu and Z.-Y. Xu, Adv. Synth. Catal., 2014, 356, 1753 CrossRef CAS.
- (a) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854 CrossRef CAS PubMed; (b) B. M. Trost, Science, 1991, 254, 1471 CAS; (c) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40 CrossRef CAS PubMed; (d) P. G. Bulger, S. K. Bagal and R. Marquez, Nat. Prod. Rep., 2008, 25, 254 RSC; (e) J. Kim and M. Movassaghi, Chem. Soc. Rev., 2009, 38, 3035 RSC.
- For the synthesis of multifunctional substrates 1, see the ESI.†.
- Crystallographic data of 5ka reported in this manuscript have been deposited with Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-1040723.†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1040723. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra24910h |
| ‡ These authors made equal contributions to this work. |
| This journal is © The Royal Society of Chemistry 2017 |