
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Redox-responsive release of active payloads from depolymerized nanoparticles†
Li-Ping Lv *ab,
Shuai Jianga,
Alper Inan‡
a,
Katharina Landfestera and
Daniel Crespy*ac
*ab,
Shuai Jianga,
Alper Inan‡
a,
Katharina Landfestera and
Daniel Crespy*ac
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128, Mainz, Germany. E-mail: crespy@mpip-mainz.mpg.de
bDepartment of Chemical Engineering, School of Environmental and Chemical Engineering, Shanghai University, Shangda Road 99, 200444, Shanghai, P. R. China. E-mail: liping_lv@shu.edu.cn
cDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong 21210, Thailand
First published on 25th January 2017
Abstract
The difference in the reactivity of two monomers, aniline (ANI) and 2,5-dimercapto-1,3,4-thiadiazole (DMcT), was employed to design nanoparticles with completely different nanostructures. The monomers were simultaneously polymerized by tandem oxidative polymerization occurring in the miniemulsion droplets. DMcT is also a corrosion inhibitor and its polymer can be depolymerized by reduction, which avoids the unwanted release of the payload DMcT when the capsules are not activated. The redox-responsive release profile of DMcT from the composite particles is controlled by the morphology of the particles and it was investigated for monolithic, multi-hollow, and yolk–shell structures. These PANI/PDMcT composite particles may find potential application in Li–S batteries or in the self-healing systems for corrosion protection.
Introduction
Stimuli-responsive release of active agents is of great importance for an extrinsic self-healing system because the system can intelligently release their payloads upon an environmental change and adapt itself to the defect. The organosulfur 2,5-dimercapto-1,3,4-thiadiazole (DMcT) is a suitable active payload because of its important applications such as in anti-corrosion and in Li–S batteries. Moreover, DMcT has been reported to be an efficient corrosion inhibitor for copper and steel due to the presence of N, S, and thiadiazole ring in the compound,1–3 and it is also attractive for Li–S batteries due to its high theoretical capacity (362 mA h g−1).4–6 Another interesting characteristic of DMcT is that the thiol groups at both ends of DMcT molecules can be bonded upon oxidation to form a polymer poly(2,5-dimercapto-1,3,4-thiadiazole) (PDMcT), and then can be reversibly depolymerized to DMcT monomers upon reduction. Note that PDMcT can be regarded as a self-immolative polymer because its complete degradation results in only functional agents, such as DMcT, and no other by-products are generated.7,8However, due to its low conductivity and sluggish redox kinetics, the use of DMcT usually requires an additional support in practical applications. In Li–S batteries, low conductivity blocks electron transport, whereas the slow redox kinetic hampers the performance of the electrodes and finally the cyclability and capability of the battery. Therefore, when used as an electrode for Li–S battery, DMcT is often mixed with conducting polymers such as polyaniline (PANI).5,6 PANI, on one hand, provides the electronic connection for the electrode, whereas on the other hand, it accelerates the redox kinetics of DMcT during charging and discharging processes.5,6,9
For anti-corrosion applications, conducting materials are also required due to the insulating characteristics of DMcT. Conducting polymers, such as PANI, exhibit interesting anti-corrosion properties.10–12 PANI capsules loaded with a corrosion inhibitor 3-nitrosalicylic acid were found to yield a redox responsive self-healing behavior for corrosion protection.13 The decrease in the electrode potential of the corroded metal-reduced PANI capsules and the consequent permeability change in the capsule shell resulted in the release of corrosion inhibitors, which then terminated the corrosion process.13 In another report, PDMcT immobilized on a conducting matrix was electrochemically reduced to DMcT, which exhibited inhibiting effects on the oxygen reduction reaction at the Cu cathode. The release of DMcT was realized by the depolymerization of PDMcT through galvanic coupling to a reducing metal.14 Therefore, combining PANI with PDMcT could provide an anti-corrosive system with higher efficiency. PANI provides conducting property to the system and also accelerates the redox reactions of DMcT during its application.5,15,16 Furthermore, PANI has an anti-corrosive property in addition to the corrosion inhibitor DMcT. Moreover, the PANI/PDMcT should also hinder the unwanted release of DMcT because its release is achieved only by the reductive depolymerization of PDMcT.14,17
Although many studies on PANI/PDMcT composites have been reported, to the best of our knowledge, there has been no investigation on the redox-responsive release of DMcT from PANI capsules or particles. The capsule or particle structure of PANI/PDMcT is supposed to be a more sufficient architecture that may facilitate their functions for anticorrosion or Li–S batteries application. It is known that the release of payloads from redox responsive nanocontainers can be tuned by the chemistry of the matrix and thickness of the shell.18–22 In both the cases, the release is achieved by increasing the permeability of the nanocontainers upon chemical or electrochemical triggering. Another manner to control the release of payloads is to control the nanoarchitectures of the carrier matrix.23 For this, block and random copolymers with the corrosion inhibitor 2-mercaptobenzothiazole (MBT) bonded to the side chains of the polymer via disulfide bonds were synthesized by RAFT and free-radical polymerization, respectively. Using the miniemulsion-solvent evaporation technique,24,25 redox-responsive capsules with different morphologies were obtained. The release of MBT was triggered by adding a reducing agent to the capsule dispersions. The block copolymer nanocapsules exhibited a faster release of MBT compared to the random copolymer nanocapsules. The difference was attributed to the microphase separation between the two blocks in block copolymers nanocapsules, which induced a lamellar structure on the surface of the capsules. Therefore, the MBT moiety was aggregated on the capsule surface and the accessibility of the reducing agent to the disulfide bonds was then improved. Release of MBT led to a faster cleavage of other disulfide bonds in the capsules shell, and therefore, a faster release of payloads encapsulated in the capsule core was observed.
Therefore, the release behaviors of payloads from these copolymer capsules were influenced by the different morphologies of the capsules. However, the intrinsic materials that form the capsules in the aforementioned case were also different, i.e. the capsules were made from random or block copolymers. In the present study, by changing the type of initiator for the polymerization of ANI and DMcT, PANI/PDMcT particles with different morphologies were synthesized via miniemulsion polymerization. The stimuli-responsive release of DMcT from the PANI particles was investigated by tuning only the nanoarchitectures, i.e. the morphology, with the particle matrix formed from the same materials. Based on the promising property of DMcT as an electrode for Li–S batteries and anti-corrosive agent, the study of its redox responsive release may provide a fundamental insight on the potential application of these PANI/PDMcT composite particles.
Experimental
Materials
Aniline (ANI) was purchased from Sigma Aldrich and distilled under vacuum prior to use. Ammonium persulfate (APS, 98%, ACROS Organics), 2,5-dimercapto-1,3,4-thiadiazole (DMcT, 98%, Aldrich), dithiothreitol (DTT, 99%, Alfa Aesar), ethyl acetate (EA, 99.97%, Fisher Chemical), ethylbenzene (EB, 99.8%, ACROS Organics), hydrogen peroxide solution (H2O2, 34.5–36.5%, Sigma Aldrich), sodium dodecyl sulfate (SDS, 99%, Sigma Aldrich), and poly(vinyl alcohol) (PVA, 98–99% hydrolyzed, Alfa Aesar) with a molecular weight Mw of 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 450 g mol−1 (measured by gel permeation chromatography, calibrated with poly(ethylene oxide) standards, 80% 0.1 M NaNO3 in water/20% MeOH as eluent) were used as received. Distilled water was used throughout the experiments if not specifically mentioned.
450 g mol−1 (measured by gel permeation chromatography, calibrated with poly(ethylene oxide) standards, 80% 0.1 M NaNO3 in water/20% MeOH as eluent) were used as received. Distilled water was used throughout the experiments if not specifically mentioned.
Analytical methods
UV-vis absorption spectra were obtained using a Perkin Elmer Lambda 25 UV/vis spectrometer. The morphology of all the PANI/PDMcT particles was examined using a Gemini 1530 (Carl Zeiss AG, Oberkochem, Germany) scanning electron microscope (SEM) operating at 0.45 kV and a transmission electron microscope (TEM, Jeol 1400) operating at an accelerating voltage of 120 kV. To prepare SEM samples, a purified particle dispersion was dropped onto a silicon wafer and dried at room temperature. All the TEM samples were prepared by dropping the particle dispersions onto a copper grid (300 square mesh) coated with a carbon layer. The FTIR spectra of all the samples were obtained by a Perkin-Elmer spectrum BX FT-IR spectrometer using KBr pellets. All the spectra were acquired in the range of 400–4000 cm−1. Thermogravimetric analysis (TGA) of the PANI/PDMcT particles was carried out using a TGA 851 thermogravimetric analyzer at a heating rate of 5 °C min−1 in a N2 atmosphere. The monomer conversion of aniline during oxidation polymerization was detected by 1H NMR spectroscopy at room temperature using a console Avance 300 with d-DMSO as the solvent.Synthesis of PANI/PDMcT nanoparticles with different morphologies
The PANI/PDMcT particles were prepared according to a previously reported procedure.26 In brief, 5.38 mmol of aniline, 0.33 mmol of DMcT, and 9.79 mmol of ethylbenzene were first mixed, and then added to 6 mL of an aqueous solution of SDS (62.5 mg). After stirring for 1 h under 1000 rpm at room temperature, the mixture was ultrasonicated for 240 s at a 90% amplitude (Branson sonifier W450) under ice cooling. After emulsification, 16.5 g of an aqueous solution of PVA (10 wt%) was added to the miniemulsion followed by the dropwise addition of the oxidant solution of APS, APS/H2O2, or H2O2 solution with known amount, as shown in Table 1. The dispersion was then subjected to stirring at room temperature for 20 h, then PANI/PDMcT-1, 2, and 3 particles were purified by washing with water, and centrifuged several times until the supernatant was clear. All particles were then collected by centrifuging at 8000 rpm for 5 min. In the case of PANI/PDMcT-3, particles were floating on the surface of the dispersion after centrifugation. Therefore, the bottom part of the dispersion was removed. All the purified PANI/PDMcT dispersions were then diluted with water to achieve a solid content of 0.49 wt%. The particle size of all the samples was determined by SEM and TEM by counting at least 100 particles.| Entry | Dispersed phase | Continuous phase | Initiator | Morphology | |||||
|---|---|---|---|---|---|---|---|---|---|
| ANI [mL] | DMcT [mg] | EB [mL] | SDS [mg] | H2O [mL] | APS [g] | H2O [mL] | H2O2 (34 wt%) [mL] | ||
| a Aniline:initiator = 1:1 (mol:mol). |
|||||||||
| PANI/PDMcT-1 | 0.5 | 50 | 1.2 | 62.5 | 6 | 1.23 | 2 | 0 | Yolk–shell capsule |
| PANI/PDMcT-2 | 0.62 | 1 | 0.27 | Multi-hollow nanoparticle | |||||
| PANI/PDMcT-3 | 0 | 0 | 0.54 | Solid nanoparticle | |||||
To synthesize PDMcT, 0.9 g of DMcT monomer was dissolved in a 90 mL mixture of EtOH/H2O (1:1, v/v). After this, 4.1 g of APS dissolved in 30 mL of EtOH/H2O mixture (1:1, v/v) was added dropwise to the monomer solution under stirring. The reaction was carried out overnight at room temperature. The obtained light yellow precipitate was washed with a hot EtOH/H2O (1:1, v/v) solution to remove the unreacted DMcT, and then dried at room temperature.4
Redox responsive release of DMcT from PANI/PDMcT nanoparticles
To study the redox responsive release of DMcT, 0.45 mL of each purified PANI/PDMcT particle dispersion (0.49 wt%) was first mixed with 4 mL of EA in a glass vial. Then, the mixture was stirred for several hours and was kept still for several minutes until phase separation between water and EA was observed. After this, the EA phase was removed followed by the addition of 4 mL of fresh EA. The process was repeated several times until no obvious signal of DMcT (∼345 nm) was detected in the EA phase by UV-vis spectroscopy. Finally, EA was removed, the PANI/PDMcT samples were first dried in air, and then degassed with argon for 5 min.To study the release of DMcT from the PANI/PDMcT particles, a DTT/EA solution with different concentrations of DTT was prepared. EA used for the release experiments was degassed with argon. The redox responsive release of DMcT was then conducted by adding 4 mL of the DTT/EA solution to the as-prepared PANI/PDMcT dry samples. It was found that after drying, the PANI/PDMcT samples stuck to the inner surface of the glass vials. Therefore, the EA solution dissolved with the released DMcT was directly taken for the UV-vis measurements during the release procedure.
The theoretical molar ratio of DTT to DMcT was set to 0:1, 1:1, 10:1, and 100:1, respectively. After different time intervals, 0.3 mL of the upper EA was retrieved for the UV-vis measurements, and then filled back into the vial. The amount of released DMcT was determined by monitoring the peak λmax ∼ 345 nm by UV-vis spectroscopy. A calibration curve was obtained by measuring a series of DMcT/EA solutions with known concentrations (Fig. S1†). No change in the absorption peak was observed for 6 h after the addition of DTT (DTT:DMcT = 100:1). This value for the maximum absorption was considered as the value for the 100% release of DMcT. According to the maximum amount of DMcT released, as determined by UV-vis spectroscopy and monomer conversion of aniline after polymerization measured by 1H NMR spectroscopy, the weight ratios of PDMcT compared to those of PANI were estimated to be 3.7% and 7.0% in the sample PANI/PDMcT-1 and 2, respectively.
To exclude the influence of DTT absorption on the released DMcT in UV-vis spectroscopy, the UV-vis spectra of DTT and oxidized DTT were obtained, as shown in Fig. S4a.† DTT did not exhibit obvious UV-vis absorption. However, a sharp peak in the range of 250–345 nm was observed for the oxidized DTT, which was assigned to the formation of cyclo-DTT.27,28 Using the multipeak function of the software Origin 8.5, the absorption from 250–390 nm was fitted to two peaks, i.e. the peak of the oxidized DTT and that of the released DMcT. As shown in Fig. S5 (ESI†), the fitting curve coincided with the measured curve of the released sample quite well. The absorption intensity of DMcT at around 345 nm was almost not influenced by the signal of the oxidized DTT. Therefore, the absorption intensity around 345 nm was used as the absorption of DMcT for the calculation of the release profile without further normalization of the spectra for all the released samples.
Results and discussion
Synthesis of PANI/PDMcT nanoparticles with different morphologies
The preparation of PANI/PDMcT nanoparticles was conducted by the oxidative polymerization of ANI and DMcT in the miniemulsion droplets. ANI and DMcT monomers were first mixed with the solvent EB and emulsified in an aqueous solution of a surfactant to create dispersed droplets composed of ANI, DMcT, and EB. The polymerization of ANI and DMcT was then started upon the addition of the oxidant such as ammonium persulfate (APS), hydrogen peroxide (H2O2), or a mixture of both. The molar ratio of ANI to the initiator was kept at 1:1, as shown in Table 1. The oxidative polymerization is displayed in Fig. 1a. The color of the dispersions changed with time after the addition of the oxidant, indicating the progress of the polymerization process (Fig. S2a, ESI†). After the reaction, the dispersions of PANI/PDMcT-1–3 particles exhibited a dark green, brown, and yellow color, respectively (Fig. S2b, ESI†).
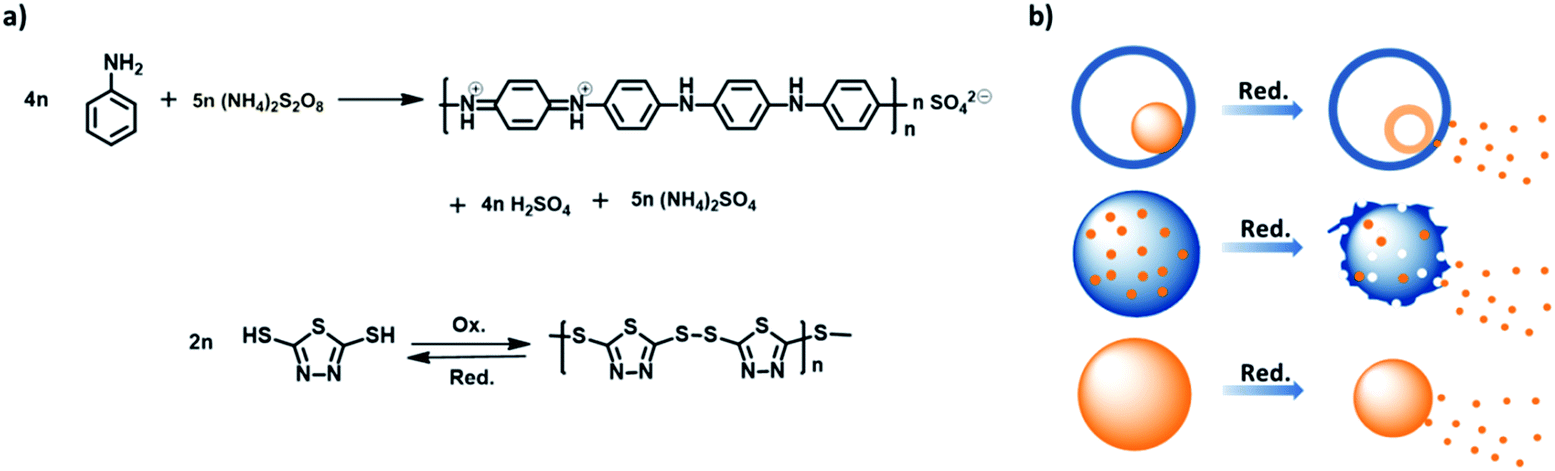 | ||
| Fig. 1 (a) Reaction equation for the oxidative polymerization of aniline and polymerization/depolymerization of DMcT; (b) schematic of the release of DMcT from PANI/PDMcT particles with different morphologies. PANI and PDMcT are indicated in blue and orange, respectively. | ||
The morphology of the obtained PANI/PDMcT nanoparticles was investigated by electron microscopy. As shown in Fig. 2, three different nanoparticles displayed a spherical morphology with a diameter of 730 ± 200, 290 ± 100, and 240 ± 50 nm, respectively for the samples PANI/PDMcT-1, 2, and 3 (see Fig. 2a–c). Interestingly, different internal morphologies were observed for PANI/PDMcT-1, 2, and 3 by TEM. PANI/PDMcT-1 particles exhibited a yolk–shell structure, i.e. a nanoparticle encapsulated by but not merged to a surrounding shell. The yolk particle inside the PANI shell was estimated to be 340 ± 80 nm in size. In contrast, the PANI/PDMcT-2 and PANI/PDMcT-3 particles exhibited a multi-hollow and monolithic structure, respectively. The difference in the morphology and size can be attributed to the difference in the polymerization rates for the monomers ANI and DMcT in the presence of different oxidants as well as the phase separations occurring during the formation of the polymers.
 | ||
| Fig. 2 SEM (a–c) and TEM (d–f) images of PANI/PDMcT-1 (a and d), PANI/PDMcT-2 (b and e), and PANI/PDMcT-3 (c and f). The insets are the images with high magnification. | ||
According to previous reports, the polymerization of ANI was much faster with the initiator APS than that with H2O2.29 The conversion of ANI was quite low when H2O2 was used alone for the polymerization.30 Indeed, an iron(II) catalyst is usually required to initiate the polymerization of ANI when H2O2 was used as an oxidant.31 Moreover, it was reported that the redox kinetics of DMcT is quite sluggish at room temperature. The assistance of conducting polymers, such as polyaniline, polypyrrole, or polythiophene, is known to accelerate the polymerization kinetics of DMcT.6,15,16 Therefore, in the case of PANI/PDMcT-1, where only APS was used as the initiator, ANI monomer was quickly polymerized in a few minutes to form PANI chains that precipitated at the interface of the miniemulsion droplets.26 The polymerization of DMcT was then accelerated by the formed PANI and yielded the particles inside the formed PANI shell. Finally, yolk–shell structure of PANI/PDMcT-1 was obtained.
In the case of PANI/PDMcT-2, due to the combined use of APS and H2O2, the polymerization of ANI and DMcT was supposed to be slower due to the lower oxidation potential of H2O2 (1.8 V) as compared to that of APS (2.1 V). Moreover, compared to the ionic character of APS, the less polar and small molecule of H2O2 may diffuse faster in the ANI/DMcT phase, and then oxidize DMcT once ANI was polymerized. Therefore, PANI and PDMcT were formed roughly at the same time in each of the dispersed droplets and precipitated before the phase separation of PANI and PDMcT was completed. As a result, particles composed of mixed PANI and PDMcT were generated. After the reaction, the evaporation of EB from the particles left holes inside the particle and multi-hollow structures were formed. For the PANI/PDMcT-3 sample, where only H2O2 was used as the oxidant, both the polymerization of ANI and DMcT were slow. As a result, the polymerization rate of DMcT was also slow due to the lack of PANI, which can accelerate the redox reaction of DMcT.
To detect the monomer conversion of ANI in the PANI/PDMcT-1–3 samples, 1H NMR spectroscopy was used to track the amount of ANI as the polymerization proceeded (Fig. S3†). EB present in the dispersed phase was used as the internal standard for the integration of the NMR signals. A higher and faster monomer conversion was observed for PANI/PDMcT-1 compared to those for PANI/PDMcT-2 and 3. After 20 h of reaction, around 50% of ANI was found to be polymerized, whereas only 6% of ANI was converted into PANI/PDMcT-3, indicating the slow reaction kinetics of ANI with H2O2 as the sole initiator. In the PANI/PDMcT-2 sample, where both APS and H2O2 were used, the ANI conversion was slower and lower (28% after 20 h) than that of PANI/PDMcT-1 (50% after 20 h), whereas much faster and higher than that of PANI/PDMcT-3 (6% after 20 h). The polymerization of ANI/DMcT was also confirmed by FT-IR spectroscopy, as shown in Fig. S6b (ESI†). The characteristic peaks around 1496 cm−1 and 1561 cm−1 were assigned to the benzenoid and quinonoid rings of the PANI chain, respectively. The characteristic absorption peaks around 1047 cm−1 and 1381 cm−1 assigned to C–S and S–S of PDMcT (Fig. S6a, ESI†), respectively, were not obviously observed due to the small amount of PDMcT compared to that of PANI in the sample. TGA also indicated the formation of PANI in the PANI/PDMcT samples because the complete degradation of DMcT and PDMcT was already found after the temperature was increased to 700 °C (Fig. S7, ESI†).
Note that although nanoparticles with yolk/shell structure were already reported,32 the yolk/shell structures composed solely of polymers are rare due to difficulty in their synthesis. Among the reported polymer/polymer yolk/shell particles, the utilization of inorganic templates, such as silica, was often carried out.33–35 Another method is related to the use of organic latex particle as a support36 or the polymerization-induced self-assembly and reorganization process.37 However, to develop a feasible method to obtain polymeric yolk/shell particles is still challenging in polymer chemistry. In the present study, the yolk–shell particles were obtained using a template-free and one pot miniemulsion polymerization procedure. Moreover, the morphology could be changed by simply tuning the oxidant used for the polymerization of monomers.
Fig. 3 shows the electron microscopy images of PANI/PDMcT-1 and 2 samples after extraction with EA, which is a good solvent for DMcT. A similar morphology was observed for PANI/PDMcT-1 before (Fig. 2d) and after (Fig. 3a) washing with EA. In contrast, PANI/PDMcT-2 exhibited more pores, as indicated from both the particle surface (Fig. 3c) and inner part (Fig. 3b) of the particles compared to the particles before washing (Fig. 2b and e), indicating the removal of unreacted DMcT monomer and oligomer of PANI and PDMcT. However, PANI/PDMcT-3 sample was found to be mostly dissolved in EA. According to a previous report, the polymerization of aniline was not completed even after 40 h using H2O2 as an oxidant in the presence of catalyst Fe2+.27 As abovementioned, the monomer conversion of ANI in the PANI/PDMcT-3 sample was only 6%, as determined by 1H NMR spectroscopy, after the reaction for 20 h. Therefore, the polymerization of DMcT was even slower without the assistance of PANI6,15,16 and low-molecular weight polymer was assumed to be formed in PANI/PDMcT-3.17,38 Therefore, the release study in EA is difficult for PANI/PDMcT-3 and only PANI/PDMcT-1 and 2 were used.
 | ||
| Fig. 3 TEM (a and b) and SEM (c) images of PANI/PDMcT-1 (a) and PANI/PDMcT-2 (b and c) after extraction by EA. The insets are the images with higher magnifications. | ||
Redox responsive release of DMcT from PANI/PDMcT nanoparticles
Stimuli, such as pH change, temperature, and redox signal, are often utilized for the controlled release of payloads.39 Among these, the redox stimuli are a very important stimulus because they can be used to trigger an action, such as the release of payloads, either in cells or during the corrosion process.40–42 The materials, synthesized to yield activation via oxidation and reduction, usually contain disulfide linkage43–46 and are based on organometallic19,47,48 or semiconducting polymers.13,26 Unlike most of the redox responsive systems based on disulfide linkages, in which the release of payloads is achieved by the decomposition of a system due to the cleavage of disulfide bonds, the systems based on polyferrocenylsilanes19,49,50 and semiconducting polymers13,26 usually display their release through reversible morphology transformation caused by the transition between the ferrocenium/ferrocene pair or different oxidation states of semiconducting polymers upon a redox trigger. In this case, a combination of the two semiconducting polymers was used. Because of the distinct morphologies displayed by the PANI/PDMcT particles, their release behaviors were interesting to investigate. The release of DMcT from the PANI/PDMcT particles was supposed to be achieved upon the reduction of the particles. The PDMcT exhibits reversible polymerization and depolymerization process upon oxidation and reduction (Fig. 1a). Upon oxidation, the thiol groups of the DMcT molecules form disulfide bonds and PDMcT chains. After the reduction of the PDMcT chains, the cleavage of disulfide bonds will result in the depolymerization of PDMcT and the DMcT monomers are released.To detect the release of DMcT, purified PANI/PDMcT particles were first washed several times with EA until no obvious signal of DMcT in the supernatant was observed at ∼345 nm by UV-vis spectroscopy. A known amount of EA with the addition of the reducing agent DTT at various concentrations was used to trigger the release. The release of DMcT was then achieved by the reduction of PANI and by the depolymerization of PDMcT upon the addition of DTT. The maximum equilibrated absorption of the released DMcT was set to 100% of the release.
As shown in Fig. 4, the release of DMcT from both PANI/PDMcT-1 and PANI/PDMcT-2 particles was observed upon reduction but no obvious release was observed when no DTT was applied. In both the cases, an increased amount of DTT accelerated the release process (Fig. 4). However, the release kinetics of DMcT from the two particles with different morphology were different, as indicated by the release profiles. PANI/PDMcT-1 capsules with the yolk/shell structure exhibited faster release when the DTT to DMcT molar ratio was set to 1:1. The release of DMcT from PANI/PDMcT-1 after 6 h reached 37%, and then 54% after 24 h, whereas only 14% of release was observed for PANI/PDMcT-2 after 6 h and 30% after 24 h.
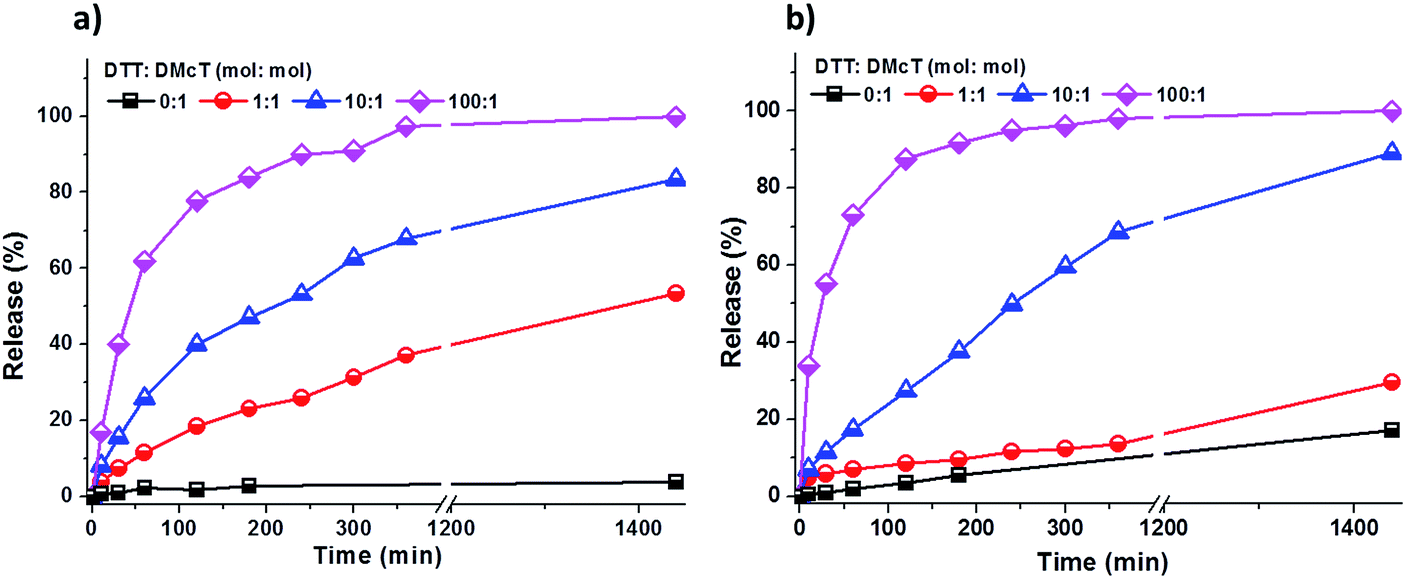 | ||
| Fig. 4 Release of DMcT from (a) PANI/PDMcT-1 and (b) PANI/PDMcT-2 under different reducing conditions. | ||
The morphology of PANI/PDMcT particles was also detected, as shown by the SEM and TEM images in Fig. 5. It was found that after the release of DMcT, PANI/PDMcT yolk/shell capsules still exhibited yolk/shell structure, whereas the solid yolk inside the shell exhibited a hollow structure. This was attributed to the depolymerization of PDMcT and the subsequent release of DMcT (Fig. 1b and 5a). The PANI/PDMcT-2 also exhibited different morphology after the release experiment. As shown in Fig. 5b and c, the particles displayed a rough surface and a smaller size of 180 ± 42 nm after the reduction as compared to the PANI/PDMcT-2 particles before release (290 ± 100 nm), which can also be explained by the depolymerization of PDMcT and the removal of DMcT (Fig. 1b).
 | ||
| Fig. 5 TEM (a and b) and SEM (c) images of PANI/PDMcT-1 (a) and PANI/PDMcT-2 (b and c) after the release of DMcT. The insets are the images with high magnification. | ||
The different release behaviors of PANI/PDMcT-1 and 2 can be explained by (a) difference in the average diffusion pathway and (b) the surface area of the PDMcT moiety. As indicated by the TEM images in Fig. 2d and e, it was assumed that the yolk–shell morphology of PANI/PDMcT-1 exhibited a shorter average diffusion pathway but a lower surface area than that of the multi-hollow structure of PANI/PDMcT-2. When a lower amount of the reducing agent DTT was used (DTT:DMcT, 1:1), the longer average diffusion pathway of PANI/PDMcT-2 contributed to the slower release of DMcT compared to that of PANI/PDMcT-1 (Fig. 4). However, the release of DMcT was accelerated as the amount of DTT increased, particularly for PANI/PDMcT-2 because of the higher surface area of PDMcT.
A qualitative profile fit of the release kinetics, as shown in Fig. S8,† further reveals the release mechanisms of PANI/PDMcT-1 and 2 particles. When the reducing agent with a DTT:DMcT molar ratio of 100:1 was used, the release of DMcT was more like Fickian diffusion, a first order t1/2-time dependent model,51,52 and was driven by the concentration gradient of DMcT, indicating that the degradation of PDMcT and relaxation (reduction) of the PANI matrix was very fast under the intense reducing conditions. For the release of DMcT under conditions with a lower concentration of the reducing agent, the Korsmeyer–Peppas model53,54 was used to fit the release profiles. Diffusion exponents 0.43 < n < 0.85 were found for all the cases, indicating the release mechanism as an anomalous non-Fickian transport that involves both Fickian diffusion and a polymer degradation procedure.51
Conclusions
In conclusion, by varying the type of oxidant, PANI/PDMcT particles with yolk/shell, multi-hollow or monolithic particle structure were synthesized via a one-pot miniemulsion polymerization process. DMcT, which is a corrosion inhibitor, was encoded as a monomer unit in the polymer PDMcT during the synthesis and its release can be controlled by controlling the redox stimuli and the nanoarchitectures of the PANI/PDMcT particles. Based on the reversible polymerization/depolymerization property of PDMcT upon reduction, these PANI/PDMcT composites particles are promising candidates for electrodes in Li–S batteries or self-healing system in anticorrosion applications, or even biomedical applications because PANI has already been used in tissue engineering,55,56 bio-actuators,57 and biosensors,58 and DMcT is used for fungicidals59 or as a vulcanizing agent60 with low-cytotoxicity.Acknowledgements
L.-P. Lv gratefully acknowledges the National Natural Science Foundation of China (51603119), Young Eastern Scholar (QD2016027) at Shanghai Institutions of Higher Learning, and the Shanghai Sailing Program (16YF1404200) for financial support.Notes and references
- M. Yadav and D. Sharma, Indian J. Chem. Technol., 2010, 17, 95–101 CAS.
- T. T. Qin, J. Li, H. Q. Luo, M. Li and N. B. Li, Corros. Sci., 2011, 53, 1072–1078 CrossRef CAS.
- W. Chen, H. Q. Luo and N. B. Li, Corros. Sci., 2011, 53, 3356–3365 CrossRef CAS.
- J. Gao, M. A. Lowe, S. Conte, S. E. Burkhardt and H. D. Abruña, Chem.–Eur. J., 2012, 18, 8521–8526 CrossRef CAS PubMed.
- T.-Y. Chi, H. Li, X.-W. Li, H. Bao and G.-C. Wang, Electrochim. Acta, 2013, 96, 206–213 CrossRef CAS.
- N. Oyama, T. Tatsuma, T. Sato and T. Sotomura, Nature, 1995, 373, 598–600 CrossRef CAS.
- A. P. Esser-Kahn, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2010, 132, 10266–10268 CrossRef CAS PubMed.
- D. Crespy, L.-P. Lv and K. Landfester, Nanoscale Horiz., 2016, 1, 268–271 RSC.
- G. Wang, L. Jin, J. Ye and X. Li, Mater. Chem. Phys., 2010, 122, 224–229 CrossRef CAS.
- G. M. Spinks, A. J. Dominis, G. G. Wallace and D. E. Tallman, J. Solid State Electrochem., 2002, 6, 85–100 CrossRef CAS.
- D. E. Tallman, G. Spinks, A. Dominis and G. G. Wallace, J. Solid State Electrochem., 2002, 6, 73–84 CrossRef CAS.
- A. Mirmohseni and A. Oladegaragoze, Synth. Met., 2000, 114, 105–108 CrossRef CAS.
- A. Vimalanandan, L.-P. Lv, T. H. Tran, K. Landfester, D. Crespy and M. Rohwerder, Adv. Mater., 2013, 25, 6980–6984 CrossRef CAS PubMed.
- M. Kendig and P. Kinlen, J. Electrochem. Soc., 2007, 154, C195–C201 CrossRef CAS.
- J. Li, H. Zhan and Y. Zhou, Electrochem. Commun., 2003, 5, 555–560 CrossRef CAS.
- D. Shu, J. Zhang, C. He, Y. Meng, H. Chen, Y. Zhang and M. Zheng, J. Appl. Electrochem., 2006, 36, 1427–1431 CrossRef CAS.
- P. J. Kinlen, US Pat. 7601280B2, 2009.
- L.-P. Lv, Y. Zhao, K. Landfester and D. Crespy, Polym. Chem., 2015, 6, 5596–5601 RSC.
- R. H. Staff, M. Gallei, M. Mazurowski, M. Rehahn, R. Berger, K. Landfester and D. Crespy, ACS Nano, 2012, 6, 9042–9049 CrossRef CAS PubMed.
- H. Wen, C. Dong, H. Dong, A. Shen, W. Xia, X. Cai, Y. Song, X. Li, Y. Li and D. Shi, Small, 2012, 8, 760–769 CrossRef CAS PubMed.
- X. Wan, D. Wang and S. Liu, Langmuir, 2010, 26, 15574–15579 CrossRef CAS PubMed.
- W. Chen, P. Zhong, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2013, 169, 171–179 CrossRef CAS PubMed.
- Y. Zhao, R. Berger, K. Landfester and D. Crespy, Small, 2015, 11, 2995–2999 CrossRef CAS PubMed.
- R. H. Staff, D. Schaeffel, A. Turshatov, D. Donadio, H. J. Butt, K. Landfester, K. Koynov and D. Crespy, Small, 2013, 9, 3514–3522 CrossRef CAS PubMed.
- J. Fickert, C. Wohnhaas, A. Turshatov, K. Landfester and D. Crespy, Macromolecules, 2013, 46, 573–579 CrossRef CAS.
- L.-P. Lv, Y. Zhao, N. Vilbrandt, M. Gallei, A. Vimalanandan, M. Rohwerder, K. Landfester and D. Crespy, J. Am. Chem. Soc., 2013, 135, 14198–14205 CrossRef CAS PubMed.
- L. Sauguet, M. Moutiez, Y. Li, P. Belin, J. Seguin, M.-H. Le Du, R. Thai, C. Masson, M. Fonvielle, J.-L. Pernodet, J.-B. Charbonnier and M. Gondry, Nucleic Acids Res., 2011, 39, 4475–4489 CrossRef CAS PubMed.
- R. L. Lundblad, Techniques in Protein Modification, 1995 Search PubMed.
- Z. Sun, Y. Geng, J. Li, X. Wang, X. Jing and F. Wang, J. Appl. Polym. Sci., 1999, 72, 1077–1084 CrossRef CAS.
- J. Yan, L. Ma, M. Gan, X. Li, Z. Li, J. Tang, Y. Tu and H. Hu, Macromol. Res., 2014, 22, 853–858 CrossRef CAS.
- D. K. Moon, K. Osakada, T. Maruyama and T. Yamamoto, Makromol. Chem., 1992, 193, 1723–1728 CrossRef CAS.
- J. Liu, S. Z. Qiao, J. S. Chen, X. W. Lou, X. Xing and G. Q. Lu, Chem. Commun., 2011, 47, 12578–12591 RSC.
- M. Ji, B. Liu, X. Yang and J. Wang, Polymer, 2009, 50, 5970–5979 CrossRef CAS.
- P. Du and P. Liu, Langmuir, 2014, 30, 3060–3068 CrossRef CAS PubMed.
- P. Du, H. Yang, J. Zeng and P. Liu, J. Mater. Chem. B, 2013, 1, 5298–5308 RSC.
- M. Zhang, Y. Lan, D. Wang, R. Yan, S. Wang, L. Yang and W. Zhang, Macromolecules, 2011, 44, 842–847 CrossRef CAS.
- W.-M. Wan and C.-Y. Pan, Macromolecules, 2010, 43, 2672–2675 CrossRef CAS.
- S. J. Visco and L. C. De Jonghe, Handbook of Solid State Batteries and Capacitors, 1995, pp. 515–540 Search PubMed.
- (a) F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed; (b) K. Zhang and X. Y. Wu, Biomaterials, 2004, 25, 5281–5291 CrossRef CAS PubMed; (c) M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC; (d) T. Chen, W. Wu, H. Xiao, Y. Chen, M. Chen and J. Li, ACS Macro Lett., 2016, 5, 55–58 CrossRef CAS.
- D. Roy, J. N. Camber and B. S Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- M. Huo, J. Yuang, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- N. Casado, G. Hernandez, H. Sardon and D. Mecerreyes, Prog. Polym. Sci., 2016, 52, 107–135 CrossRef CAS.
- R. Liu, X. Zhao, T. Wu and P. Y. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef CAS PubMed.
- Y. Yan, A. P. R. Johnston, S. J. Dodds, M. M. J. Khamphuis, C. Ferguson, R. G. Parton, E. C. Nice, J. K. Heath and F. Caruso, ACS Nano, 2010, 4, 2928–2936 CrossRef CAS PubMed.
- Z. Luo, K. Y. Cai, Y. Hu, L. Zhao, P. Liu, L. Duan and W. H. Yang, Angew. Chem., Int. Ed., 2011, 50, 640–643 CrossRef CAS PubMed.
- Z. Luo, K. Y. Cai, Y. Hu, J. H. Li, X. W. Ding, B. L. Zhang, D. W. Xu, W. H. Wang and P. Liu, Adv. Mater., 2012, 24, 431–435 CrossRef CAS PubMed.
- P. Nguyen, P. Gomez-Elipe and I. Manners, Chem. Rev., 1999, 99, 1515–1548 CrossRef CAS PubMed.
- Y. J. Ma, W. F. Dong, M. A. Hempenius, H. Mohwald and G. J. Vancso, Nat. Mater., 2006, 5, 724–729 CrossRef CAS PubMed.
- D. Janczewski, J. Song, E. Csanyi, L. Kiss, P. Blazso, R. L. Katona, M. A. Deli, G. Gros, J. Xu and G. J. Vancso, J. Mater. Chem., 2012, 22, 6429–6435 RSC.
- B. V. K. J. Schmidt, J. Elbert, D. Scheid, C. J. Hawker, D. Klinger and M. Gallei, ACS Macro Lett., 2015, 4, 731–735 CrossRef CAS.
- J. Siepmann and N. A. Peppas, Int. J. Pharm., 2011, 418, 6–12 CrossRef CAS PubMed.
- S. K. Murase, L. P. Lv, A. Kaltbeitzel, K. Landfester, L. J. del Valle, R. Katsarava, J. Puiggali and D. Crespy, RSC Adv., 2015, 5, 55006–55014 RSC.
- G. Schliecker, C. Schmidt, S. Fuchs, A. Ehinger, J. Sandow and T. Kissel, J. Controlled Release, 2004, 94, 25–37 CrossRef CAS PubMed.
- P. Costa and J. M. S. Lobo, Eur. J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS PubMed.
- D. Cheng, H. Xia and H. S. Chan, J. Nanosci. Nanotechnol., 2005, 5, 466–473 CrossRef CAS PubMed.
- M. Li, Y. Guo, Y. Wei, A. G. MacDiarmid and P. I. Lelkes, Biomaterials, 2006, 27, 2705–2715 CrossRef CAS PubMed.
- G. M. Spinks, V. Mottaghitalab, M. Bahrami-Samani, P. G. Whitten and G. G. Wallace, Adv. Mater., 2006, 18, 637–640 CrossRef CAS.
- C. Dhand, M. Das, M. Datta and B. D. Malhotra, Biosens. Bioelectron., 2011, 26, 2811–2821 CrossRef CAS PubMed.
- Tokyo Organic Chem Inds Ltd., Fungicidal thiadiazoles, Patent No. JP71023639-B, IPC: A01N-000/00; C07D-000/00, 1971.
- I. Yarzabal, T. Aubert and P. Lugez, Use of mixture comprising bis(2,5-dimercapto-1,3,4-thiadiazole) and organic base, as vulcanizing agent for unsaturated polymers, preferably polymers of unsaturated halogen and chlorine-containing polymers, Patent No. FR2975996-A1, IPC: C08K-005/46, 2012.
Footnotes |
| † Electronic supplementary information (ESI) available: UV-vis, FT-IR, 1H NMR, TGA data and spectra. See DOI: 10.1039/c6ra24796b |
| ‡ Present address: Department of Chemistry, Izmir Institute of Technology, Urla, 35430, Izmir, Turkey. |
| This journal is © The Royal Society of Chemistry 2017 |