
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal- and organophotocatalyst-free perfluoroalkylation reaction of amino(hetero)aromatics initiated by the complex [(TMEDA)I·I3] and visible light†
Damian E. Yerien,
Romina Conde,
Sebastian Barata-Vallejo,
Bruno Camps,
Beatriz Lantaño and
Al Postigo *
*
Universidad de Buenos Aires, Facultad de Farmacia y Bioquímica, Departamento de Química Orgánica, Junín 954, CP 1113-Buenos Aires, Argentina. E-mail: apostigo@ffyb.uba.ar
First published on 3rd January 2017
Abstract
Radical initiation for the perfluoroalkylation reaction of amino(hetero)aromatics has been accomplished employing the complex [(TMEDA)I·I3] and visible light. This methodology circumvents the use of metal(organo)catalysts and biologically-relevant substrates are easily substituted with RF moieties employing a mild and environmentally benign radical strategy starting from readily-available perfluoroalkyl iodides RFI.
Introduction
Aminoaromatic compounds such as aniline derivatives can be perfluoroalkylated with ease1,2 by radical reactions due to the electrophilic nature of the fluoroalkyl radical3,4 and the high electron density of the aromatic ring. Even more so, fluoroalkylation can be attempted under visible light through the homolytic cleavage of the RF–I bond upon irradiation (ca. 201 kJ mol−1, 590 nm), albeit, in poor yields. Reports on visible-light photocatalytic perfluoroalkylations utilizing photoorganocatalysts4,6 have provided alternative routes to better-yielding perfluoroalkyl-substituted compounds (Scheme 1).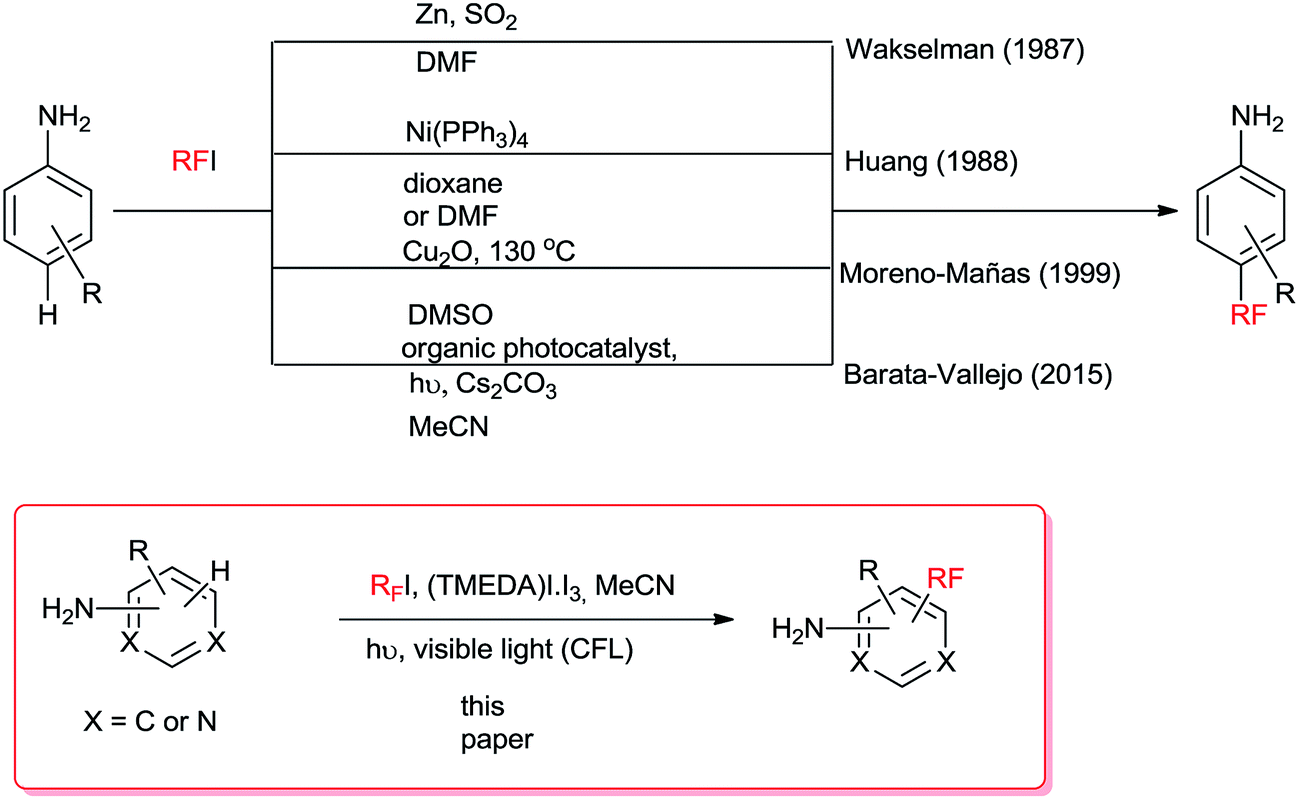 | ||
| Scheme 1 Methods for radical perfluoroalkylation of aniline derivatives. | ||
Motivated by the results of Noel and collaborators5 for the RS–H photosubstitution of cysteine derivatives employing TMEDA, RF–I and a transition metal photoorganocatalyst (POC) in MeCN, we replaced our reported strategy for the radical perfluoroalkylation reactions of anilines that employed Cs2CO3/POC5 for TMEDA and used a commercial fluorescent light CFL in the absence of organophotocatalyst to attempt the visible-light perfluoroalkylation of amino aromatic compounds in the presence of TMEDA. In this work, we will show that radical initiation can take place from visible light-irradiation of TMEDA/I2 complex and that organophotocatalysts are not necessary for improved substitution yields.
Results and discussion
At start of our studies it was not clear the species responsible for the visible-light absorption, in the absence of photoorganocatalyst POC Rose Bengal5 (RB), since product formation seemed to improve substantially upon illumination (cf. entries 2 & 5, Table 1), and none of the reagents alone (i.e.: n-C4F9I, aniline, or TMEDA) showed enough optical density at the irradiation wavelengths.| Entry | Product yield (%) | Isomer ratio (o![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :p) :p) |
|---|---|---|
| a In the presence of Rose bengal RB, 0.1 equiv.b λirradiation = 550–680 nm (λmax = 585 nm). BDE C4F9I = 201 kJ mol−1 corresponds to λmax = 590 nm.c λirradiation = 370 nm (black fluorescent light bulb, 40 watt).d Dark reaction.e In the absence of TMEDA.f I2 was removed from the reagent n-C4F9I by neutral alumina column chromatography.g 0.5 equiv. TBAI + 0.5 equiv. I2.h Only para-product observed.i I2 was thoroughly removed from the reagent C4F9I by neutral alumina column chromatography and then additional 0.5 mM of I2 to the reaction mixture was added.j 3 equiv of iodine added.k λirradiation = 410–490 nm (λmax = 425 nm). | ||
| 1 | 76a | 1:1 |
| 2 | 62 | 0.9:1.1 |
| 3 | —b | — |
| 4 | 64c | 0.9:1.2 |
| 5 | 11d | 1:3 |
| 6 | 3e | h |
| 7 | 23f | 0.8:1.2 |
| 8 | 36g,f | 1:1.5 |
| 9 | 65i | 1:1 |
| 10 | —e,f,k | — |
| 11 | —e,f | — |
When aniline was subjected to the visible-light irradiation in the presence of n-C4F9I and TMEDA in MeCN as solvent, in the absence of photocatalyst, 62% yield of substitution product was obtained (entry 2, Table 1). By irradiating at 370 nm (with a black fluorescent light lamp), a 64% yield of substitution product was obtained (entry 4, Table 1). The dark reaction (entry 5, Table 1) afforded only 11% yield of product, whereas the absence of TMEDA reduced the yield significantly (entry 6, Table 1).
When we inspected the UV-vis spectrum of the solution mixture of aniline and n-C4F9I to be irradiated at the working concentrations, we noticed an absorption band at λmax = 458 nm, corresponding to the absorption of iodine in MeCN (Fig. S1, ESI†), with enough optical density to initiate the radical reaction through the production of iodine atoms.8,9 Concerned about this absorption, we excluded iodine from the reagent n-C4F9I, passing the neat liquid through a neutral alumina column, and subjected the mixture of aniline, iodine-free n-C4F9I, and TMEDA to the photoreaction under visible-light (CFL) in MeCN as solvent. Under these latter reaction conditions, we obtained only 23% yield of substitution product, indicating that an iodine-containing species is intervening in the initiation process (entry 7, Table 1). However, when we purposely added tetra butyl ammonium iodide (TBAI) and iodine (entry 8, Table 1) to a solution of purified n-C4F9I (I2-free), a low yield of substitution product was again obtained (36%), indicating that I3− (λmax = 291 nm and 364 nm in MeCN) was not responsible for the photoinitiation. Conducting the irradiation of aniline with a medium pressure Hg lamp (employing a NaNO2/CuSO4 filter for allowing a light band path range of 410–490 nm – where I2 absorbs), in the presence of I2-free n-C4F9I and 3 additional equiv. of I2 in the absence of TMEDA, no product was obtained (entry 10, Table 1), indicating that iodine alone is not responsible for the initiation. When the same reaction was conducted with I2-free n-C4F9I, 3 additional equiv. of I2, but this time with TMEDA under visible light irradiation (CFL), a 65% yield of substitution product was obtained (entry 9, Table 1).
A complex between TMEDA and I2 (i.e.: [(TMEDA)I·I3]) has been informed by Adam and colleagues,10 with λmax = 360 nm in MeCN, (ε = 4 × 104 M−1 cm−1, a complex formation constant ca. 4.7 × 107 M−1 and ECT = 4.35 eV). Due to the traces of I2 present in the neat starting n-C4F9I, we deem proper to investigate whether complexation of these trace amounts of I2 present in n-C4F9I with TMEDA could trigger the radical initiation process. Fig. S2 and S3,† show the UV-vis spectra of mixtures of TMEDA and I2, corroborating the remarkable increase in the absorptivity at 360 nm, responsible for the absorption of light. This charge transfer complex would be responsible, upon visible light absorption (λmax = 360 nm from a commercial fluorescent light CFL or λmax = 370 nm from a black fluorescent light bulb), of the initiation process, producing excited iodine atoms, as in eqn (1).9
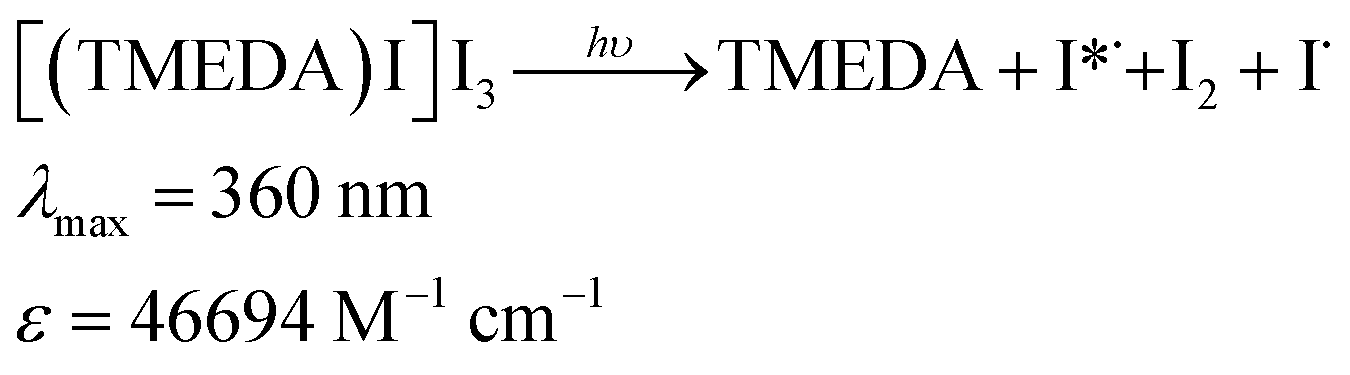 | (1) |
The iodine atoms thus produced (eqn (1)) could then abstract I atoms from perfluoroalkyl (and alkyl) iodides such as n-C4F9I,10 thus initiating the chain reaction with the production of C4F9˙ radicals (eqn (2)).
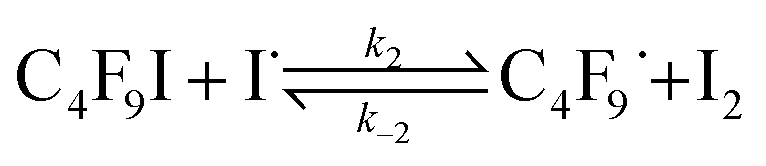 | (2) |
Rate constant k2 is a very exothermic process with very low activation energy10 provided abstraction of iodine is carried out by excited iodine atoms,11–13 which has been demonstrated to be the case when irradiation wavelengths lower than 500 nm (ref. 11) are employed.14
In order to corroborate that the complex [(TMEDA)I·I3] does produce I˙ atoms upon 370 nm irradiation, we attempted the iodine atom-induced cis/trans radical isomerization of oleic fatty acid methyl ester.16 However, elaidic acid methyl ester was not obtained, indicating that the complex [(TMEDA)I·I3] does not produce iodine atoms upon irradiation at 370 nm, as in eqn (1), responsible for the cis/trans isomerization process.
The dark reaction (TMEDA, n-C4F9I and substrate) excluding iodine, does not yield any substitution product, purporting that also the dark component accounting for product formation (entry 11, Table 1) arises from a thermal ET initiation within the complex [(TMEDA)I·I3] when present.
UV-vis spectra of mixtures of n-C4F9I and TMEDA do not show a distinct change in absorption or extinction coefficients. The TMEDA·2-CF3I complex has been postulated by Ritter15,17 and colleagues, based on calculations, X-ray crystallographic analysis, and spectroscopic data (1H, 19F, 13C NMR). A complex between a nitrogen base and RF–I has very recently been postulated to generate RF radicals upon visible light irradiation.18–20 We have also observed the same trends for the formation of a complex TMEDA·2C4F9I based on NMR data (19F, 13C NMR), where spectral changes are observed when going from free reagents to the complexed mixture, as indicated in Table S1.†
The 19F NMR upfield shifts observed in the I–CF2–C3F7 are indicative of a debilitated I–C bond in n-C4F9I when TMEDA is present, being the largest shift when a stoichiometry of the complex TMEDA:2C4F9I is reached (Table S1†). Also, from Table S1,† there seems to be the presence of complexes between perfluoroalkyl iodides and aminoaromatic compounds,16 the existence of which result in chemical shift changes of the signal from ICF2–RF in the 19F NMR spectra when n-C4F9I is in the presence of the amino substrate as compared with the innate chemical shift value of ICF2–RF signal, interpreted as halogen bonding to the nitrogen atom of the substrate.17 The –CF2I signals in the 19F NMR spectra of n-C4F9I in mixtures with amino aromatics (2,4,6-triaminopyrimidine, 2,4-dihydroxy-6-methylpyrimidine, 2,4-diamino-6-hydroxypyrimidine, respectively) are reported in Table S1,† where the similar upfield chemical shifts of the ICF2–RF can be observed. Analysis of the data presented in Table S1,† also reveals that the substituent amino group in amino aromatic compounds exerts a more favored halogen-bonding interaction with I–C4F9 than the ring-nitrogen does, based on the magnitude of the upfield shifts. Also, this effect is increased when TMEDA is present in the mixture. This is consistent with electron lone pair availability on the nitrogen atom. We therefore could postulate a more facile iodine atom abstraction from TMEDA:2n-C4F9I complex, as depicted in eqn (3).
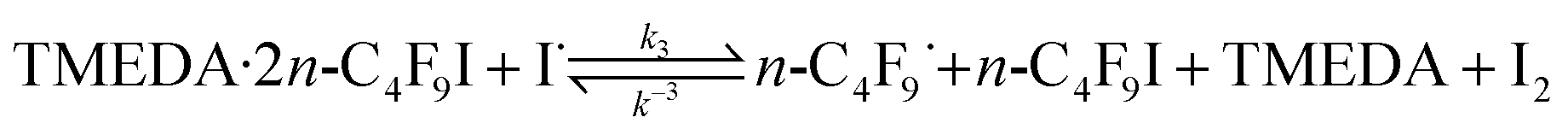 | (3) |
With these reaction conditions in hand, we undertook the perfluoroalkylation reactions of a series of aniline derivatives employing the [(TMEDA)I·I3] complex (ca. 0.25 mM) as initiator. The reaction of 2-anisidine afforded 57% yield of substitution products 1 (4-perfluorobutyl-2-methoxy aniline and 2-perfluorobutyl-2-methoxy aniline, 1:5) (Table 2), leading predominantly to the para isomer. 2,5-Dimethoxy aniline afforded 63% yield of product 2 (i.e.: 4-perfluorobutyl-2,5-dimethoxyaniline). By using other perfluoroalkyl iodides, such as I–(CF2)4–I, I–(CF2)6–I, and n-C10F21I, the corresponding substituted products 3–5 were obtained in 18, 76, and 10% yields, respectively, as indicated in Table 2. 2,3-Dimethylaniline afforded 35% yield of substitution products 6 (o:p isomer ratio = 1:2); whereas the yields for the substituted products derived from 2,3-dimethylaniline with n-C8F17I, I–(CF2)6–I, and I–(CF2)4–I, and n-C10F21I (i.e.: products 6–10, respectively), are 79, 71, 53, and 6% respectively, as indicated in Table 2. The less activated anilines, i.e.: 2,5-difluoroaniline, 2,5-dibromoaniline, 2-chloro-6-methylaniline, and 2,5-dichloroaniline, afforded products 11–14 in 9%, 15%, 15% and 16% yields respectively, as shown in Table 2. The low yields isolated from these products, i.e.: 11–14, are attributed to volatility during the work-up/purification process.
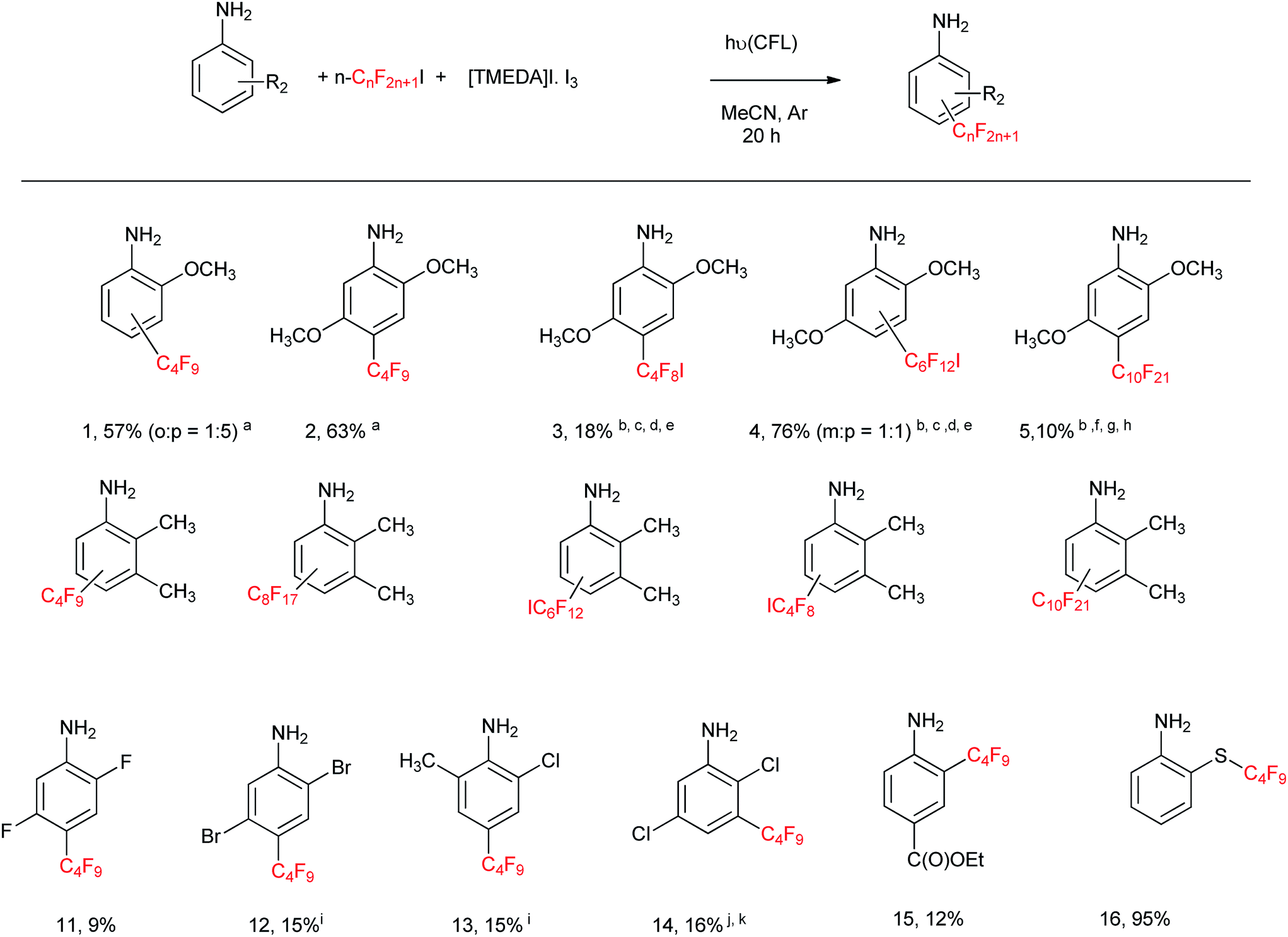
Benzocaine,4 a local anesthetic, affords 12% yield of the C4F9 substitution product 15 (Table 2). Starting from 2-mercaptoaniline, under the reaction conditions of Table 2, 2-((perfluorobutyl)thio)aniline 16 is obtained in almost quantitative yield (>95%) in 2 hour reaction.
We have attempted a large scale reaction of 2-anisidine (see Experimental) and obtained a 36% yield of purified 1 (1.474 g), indicating that a scale-up process is applicable with this methodology.
Taking into account the presence of dissolved iodine in commercial RFI, the use of TMEDA can aid in the initiation process through the [(TMEDA)I·I3] complex formation. This is likely the case in many studies7,21,22 where a combination of RFI, TMEDA, and visible light is employed.
The reactions of 2-anisidine in the presence of DBTN (di-tert-butylnitroxide, a well-known radical scavenger) as well as in the presence of 1,4-dinitrobenzene afford no substitution products, indicating the presence of radicals. The reaction of 2-anisidine in the presence of p-cresol (0.4 equiv.) affords only 20% yield of products 1.
From Table 3, the C4F9-substitution yields of a series of aminopyrimidines and aminopyridines is illustrated. 2,4,6-Triaminopyrimidine affords 5-perfluorobutyl-2,4,6-triaminopyrimidine 17 in 48% yield. 2,5-Diamino-6-hydroxy-pyrimidine affords 5-perfluorobutyl-2,4-diamino-6-hydroxypyrimidine 18 in 64% yield. 2-Aminopyridine affords a mixture of 5-perfluorobutyl-2-aminopyridine 19 in 34% yield and 3-perfluorobutyl-2-aminopyridine 20 in 23% yield. 4-Aminopyridine affords 3-perfluorobutyl-4-aminopyridine 21 in 25% yield, whereas 2,3-diaminopyridine affords 4-perfluorobutyl-2,3-diaminopyridine 22 in 12% yield. 5-Methyl-2-aminopyridine and 5-bromo-2-aminopyridine afford 4-perfluorobutyl-5-methyl-2-aminopyridine 23 and 3-perfluorobutyl-5-bromo-2-aminopyridine 24 in 47 and 50% yields, respectively. The low substitution yields encountered for compounds 19–22 is due to volatility, as these products evaporate in the rotary evaporator during the extraction and purification processes.
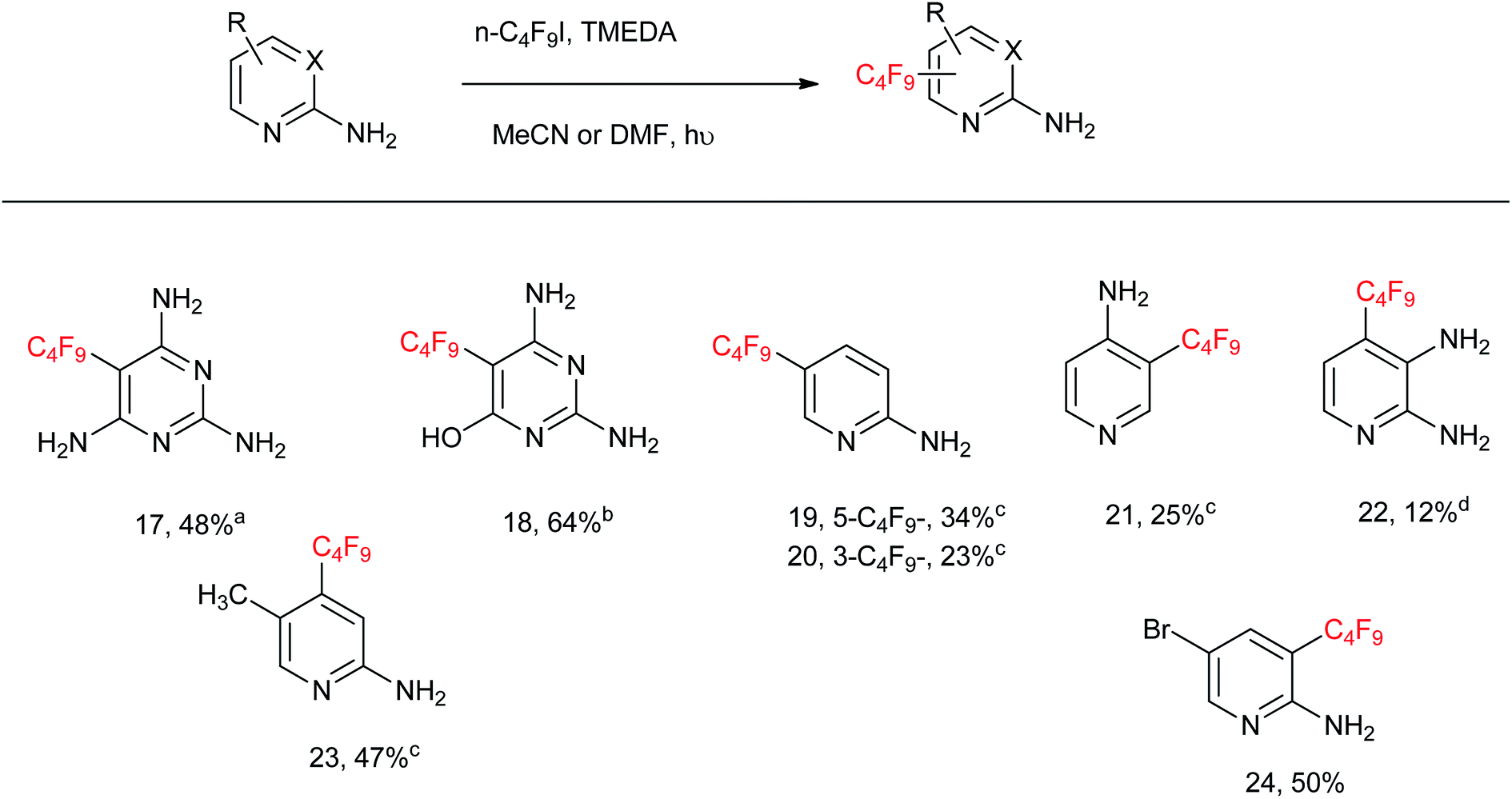
A large-scale reaction of 5-bromo-2-aminopyridine with n-C4F9I was attempted under the protocol proposed (see Experimental), and a 45% yield of purified product 24 (2.111 g) was isolated.
A proposed reaction mechanism is illustrated in Scheme 2. The complex formed by I2 and TMEDA (i.e.: [(TMEDA)I·I3], absorbs light from either a commercial fluorescent light bulb CFL, or black light fluorescent bulb, λmax = 370 nm) populating an excited state, which collapses into the excited iodine radicals (and ground state iodine radicals and I2).11,12 Then, an iodine atom abstraction from n-C4F9I takes place (as in eqn (3)), generating n-C4F9˙ radicals, which enter the electron-catalyzed cycle (Scheme 2) and substitute the amino-substrate, to generate a radical intermediate such as B, which by ET to n-C4F9I produces more n-C4F9˙ radicals and the final product C. Interestingly, [(TMEDA)I·I3] complex could be in situ re-generated through recombination of two molecules of iodine, one arising from the very decomposition of the complex, the other from n-C4F9I by iodine atom abstraction. This photocatalytic initiation accounts for the increased yield of product upon visible light illumination.
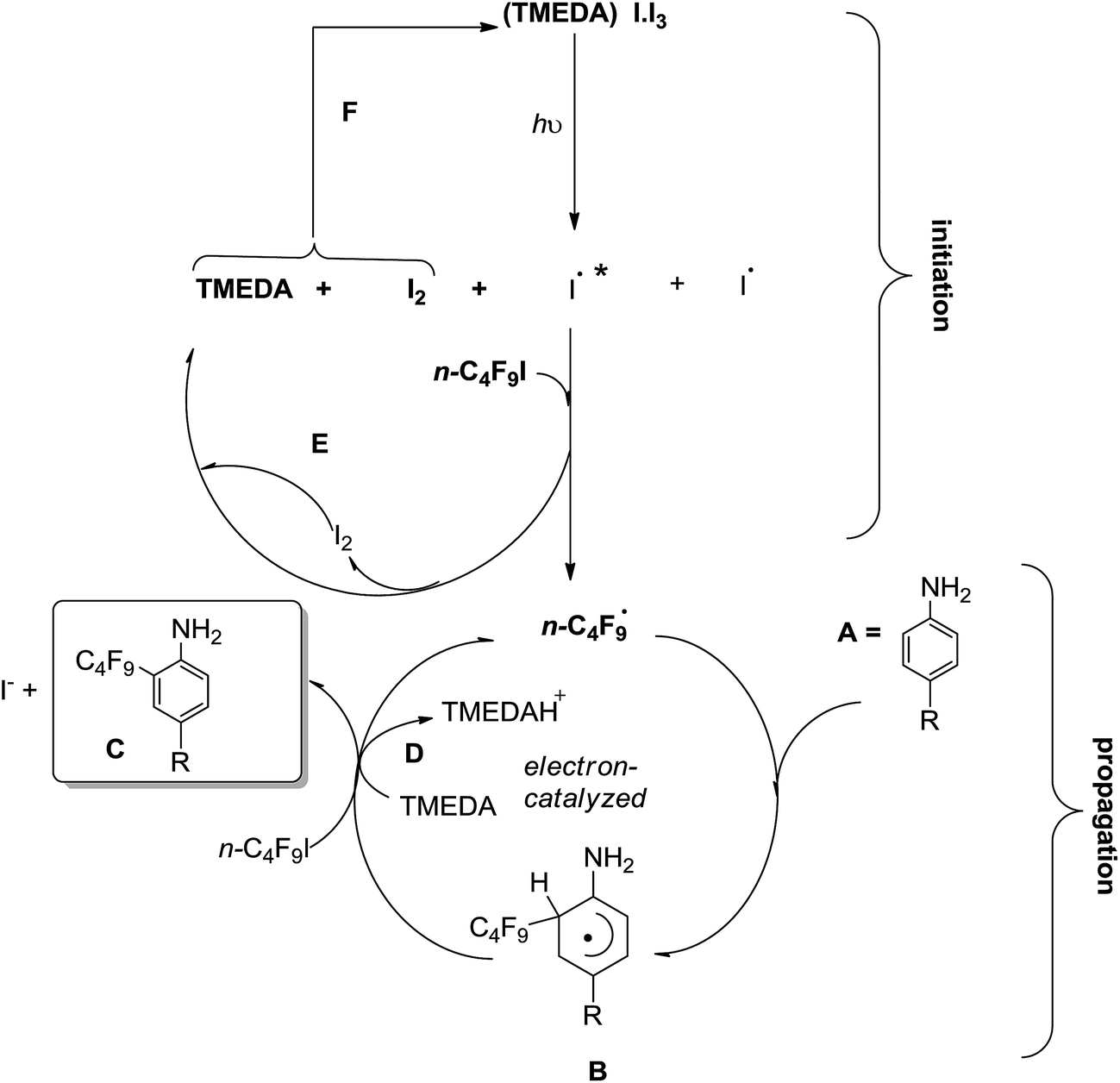 | ||
| Scheme 2 Proposed electron-catalyzed HAS of amino-substrates in the presence of TMEDA. | ||
Radical adduct B (Scheme 2) acts as a reductant to n-C4F9I, generating n-C4F9˙ radicals and a cyclohexadienyl-type substituted cation (not shown) which gets deprotonated by TMEDA to afford C. To supply more evidence in favor of the mechanism proposed in Scheme 2, we monitored the absorbance of the complex [(TMEDA)I·I3] through irradiation time, and did not observe any decrease in absorbance, purporting that the complex concentration is not depleted with irradiation time in the presence of n-C4F9I through regeneration of cycles E and F (Scheme 2).
Alternatively, we could also postulate that vertical excitation of the [(TMEDA)I·I3] complex would produce an internal ET giving iodine atom radical I˙, triiodide I3−, and the radical cation of TMEDA, which would promptly deprotonate giving the radical of TMEDA, according to Scheme 3. TMEDA radical G could act as an electron reductant to n-C4F9I, generating n-C4F9˙ radicals and the iminium ion H (Scheme 3). We did not observe any product derived from decomposition of TMEDA, through attempts of trapping any carbonyl intermediate with 2,4-dinitrophenylhydrazine/HPLC, indicating that ET from TMEDA radical G (Scheme 3) is not a major contributor to product formation.
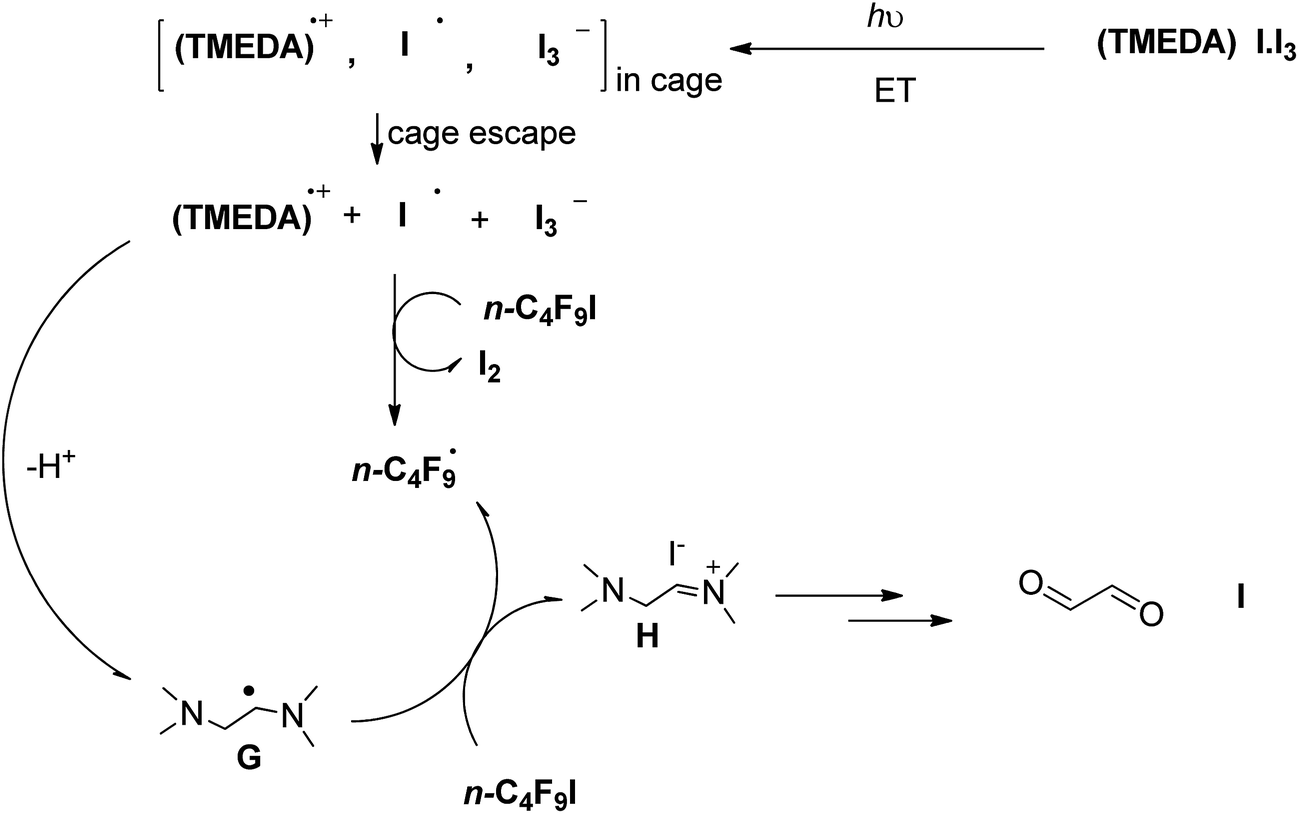 | ||
| Scheme 3 Alternative proposed mechanism where the excited complex [(TMEDA)I·I3] undergoes inner-sphere ET. | ||
The role of TMEDA is relevant in the complexation with iodine, and the deprotonation (path D, Scheme 2). In the presence of catalyst (POC), Noel and colleagues23 found that TMEDA acts as an electron donor to the catalyst, involved in a reductive quenching pathway. Being the mechanism still under investigation, a third possibility should be considered: the outer-sphere ET from [(TMEDA)I·I3] to n-C4F9I, generating n-C4F9 radicals (and iodide anion) in conjunction with the radical cation of TMEDA, which could act as an oxidant to intermediate B in Scheme 2 to re-generate thermoneutral TMEDA. Iodine could also intervene in the oxidation of intermediate B. These mechanistic alternatives are being studied at the moment.
Conclusions
A simple and metal-/photoorganocatalyst-free strategy has been presented for the perfluoroalkylation of aminoaromatics employing a complex [(TMEDA)I·I3] that upon visible light illumination produces I atoms that generate RF˙ radicals from RF–I sources. These RF˙ radicals are capable of substituting biologically-relevant targets such as local anesthetic benzocaine, etc. This convenient, high-yielding and metal/photoorganocatalyst-free strategy outperforms traditional near-UV photocatalytic methods while operating under visible light and in the presence of only an additive (TMEDA).Caution has to be exercised when employing RFI and TMEDA, since traces of iodine in the reagents (i.e.: RFI) can form a stable complex with TMEDA which could be responsible for the radical initiation process upon visible light irradiation. Moreover, given the high extinction coefficient of the complex [(TMEDA)I·I3] at the visible light wavelengths (>365 nm), iodine atom production is a key element in the radical process.
Experimental
General procedure for the ((TMEDA·I)·I3)-initiated reactions
In a 4 mL screw-cap vial provided with a micro stir bar, TMEDA (1.8 mmol, (TMEDA)I·I3 complex, ca. 0.25 mM), substrate (0.6 mmol, aniline derivative or aminoheteroaromatic compound), or photocatalyst (PC) Rose bengal where needed (0.05 equiv.) and 3 mL of acetonitrile were introduced. The mixture was de-oxygenated with a stream of Ar for 15 min and n-C4F9I or other RFI (3 equiv.) was introduced by microliter syringe, and the vial sealed. The closed reaction vessel was placed in front of a 60 watt household fluorescent light bulb (or 20 watt fluorescent black light, λmax = 370 nm) and illuminated, under constant vigorous stirring, for 24 h or otherwise noted. After the reaction time was completed, the mixture was extracted thrice with CH2Cl2/water/brine. The organic layers were gathered and dried over anhydrous Na2SO4, filtered and evaporated under vacuo. The crude reaction mixture was purified by silica-gel (60 mesh) column chromatography, with the eluants indicated in the TLC conditions (vide infra, spectral data). When a PC was used, the polarity of the dye did not introduce any particular difficulty in the separation and purification protocol, as the several CH2Cl2 extractions eliminated the PC. The eluants employed are referred to in the TLC conditions of each compound.Large scale reactions
In a 60 mL Schlenk-type tube 2-anisidine (12 mmol), TMEDA (36 mmol), and a stir bar were placed and a volume of 60 mL of MeCN was introduced by syringe through a septum. The mixture was de-oxygenated with a stream of dry Ar for 30 min, and 36 mmol of n-C4F9I were added to the mixture. The set-up was placed on a stir plate, and vigorously stirred throughout the reaction time. The vessel was placed in front (1 cm) of a 60 watt CFL, and irradiated for 40 hours. The crude reaction mixture was purified by silica-gel (60 mesh) column chromatography, with the eluants indicated in the TLC conditions (vide infra, spectral data). The large-scale reaction afforded 36% yield (1.474 g) of combined purified products 1.In a 60 mL Schlenk-type tube 5-bromo-2-aminopyridine (12 mmol), TMEDA (36 mmol), and a stir bar were placed. A volume of 60 mL of MeCN was introduced by 50 mL syringe. The mixture was de-oxygenated with a stream of dry Ar for 30 min, and 36 mmol of n-C4F9I were added to the mixture by syringe through a septum. The set-up was placed on a stir plate, and vigorously stirred throughout the reaction time. The vessel was placed in front (1 cm) of a 60 watt CFL, and irradiated for 40 hours. The crude reaction mixture was purified by silica-gel (60 mesh) column chromatography, with the eluants indicated in the TLC conditions (vide infra, spectral data). The large-scale reaction afforded 45% yield (2.111 g) of purified product 24.
Characterization of compounds
All compounds are unknown chemicals, unless otherwise noted, and are reported as % yields obtained by NMR integration (from 1H and 19F NMR integration) of the crude reaction mixtures. Isolated purified masses of compounds are expressed in gram units. Characterizations employ 1H, 13C, 19F 1D-NMR techniques, and 2D NMR spectroscopic techniques (HSQC, HMBC, COSY experiments, and DEPT-135), and HRMS measurements (see ESI†).:1 v/v): Rf = 0.5; 1H NMR (600 MHz, CDCl3) δ: 7.01 (d, J = 10 Hz, 1H), 6.92 (s, 1H), 6.73 (d, J = 10 Hz, 1H), 4.15 (b s, 2H), 3.90 (s, 3H). 13C NMR (150 MHz, CDCl3) δ: 146.5, 139.7, 120.4 (t, CF2), 118.7 (m, CF2), 118.1 (t, CF3), 116.7 (m), 113.5, 112.5, 109.7, 108.4, 55.6. 19F NMR (564.603 MHz CDCl3) δ: −81.11 (t, 3F), −109.32 (t, 2F), −122.78 (m, 2F), −125.68 (m, 2F). HRMS-EI+ (M + 1): calcd. For C11H9F9NO: 342.05349; found, 342.05388.:iso-octane 7:3 v/v): Rf = 0.69; 1H NMR (600 MHz, CDCl3) δ: 6.82 (s, 1H), 6.35 (s, 1H), 4.15 (b s, 2H), 3.83 (s, 3H), 3.76 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 154.1, 141.2, 140.6, 116.8 (CF2), 110.9, 104.9, 99.8, 56.7, 56.3. 19F NMR (564.63 MHz, CDCl3) δ: −81.00 (t, 3F), −106.20 (t, 2F), −122.25 (m, 2F), −126.06 (m, 2F). HRMS-EI+ (M + 1): calcd. For C12H10F9NO2: 372.05678; found, 372.05670.:n-hexane 8:2 v/v): Rf = 0.62; 1H NMR (600 MHz, CDCl3) δ: 6.82 (s, 1H), 6.35 (s, 1H), 4.13 (b s, 2H), 3.83 (s, 3H), 3.76 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 154.1, 141.0, 140.6, 111.0, 100.1, 99.9, 56.8, 56.3. 19F NMR (564.63 MHz, CDCl3) δ: −57.55 (t, 2F), −106.07 (t, 2F), −112.78 (m, 2F), −120.43 (m, 2F). HRMS-EI+ (M + 1): calcd. For C12H10F8INO2: 479.96285; found, 479.96280.:n-hexane 8:2 v/v): Rf = 0.52; 1H NMR (600 MHz, CDCl3) δ: 6.85 (s, 1H), 6.14 (s, 1H), 3.99 (b s, 2H), 3.70 (s, 3H), 3.63 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 152.3, 142.7, 139.9, 112.7, 108.5, 98.1, 56.7, 56.6. 19F NMR (564.63 MHz, CDCl3) δ: −58.70 (t, 2F), −107.16 (t, 2F), −113.07 (m, 2F), −119.97 (m, 2F), −120.88 (m, 2F), −121.74 (m, 2F). HRMS-EI+ (M + 1): calcd. For C14H10F12INO2: 579.95646; found, 579.95635.:n-hexane 8:2 v/v): Rf = 0.55; 1H NMR (600 MHz, CDCl3) δ: 6.45 (s, 1H), 6.31 (s, 1H), 3.99 (b s, 2H), 3.66 (s, 3H), 3.57 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 153.1, 142.6, 140.6, 112.8, 111.2, 105.3, 56.2, 55.7. 19F NMR (564.63 MHz, CDCl3) δ: −58.39 (t, 2F), −107.16 (t, 2F), −113.07 (m, 2F), −120.13 (m, 2F), −120.88 (m, 2F), −121.48 (m, 2F). HRMS-EI+ (M + 1): calcd. For C14H10F12INO2: 579.95646; found, 579.95640.:n-hexane 4:6 v/v): Rf = 0.70; 1H NMR (600 MHz, CDCl3) δ: 6.82 (s, 1H), 6.35 (s, 1H), 4.13 (b s, 2H), 3.83 (s, 3H), 3.77 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 154.1, 141.1, 140.6, 110.9, 102.1, 99.8, 56.8, 56.3. 19F NMR (564.63 MHz, CDCl3) δ: −80.74 (t, 3F), −105.98 (m, 2F), −121.33 (m, 2F), −121.78 (m, 10F), −122.67 (m, 2F), −126.08 (m, 2F). HRMS-EI+ (M + 1): calcd. For C18H10F21NO2: 672.03762; found, 672.03741.:iso-octane 7:3 v/v): Rf = 0.57; 1H NMR (600 MHz, CDCl3) δ: 7.21 (d, J = 8.6 Hz, 1H), 6.61 (d, J = 8.6 Hz, 1H), 3.88 (b s, 2H), 2.33 (s, 3H), 2.10 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 147.6, 137.6, 127.0, 121.9, 117.0, 112.3, 17.0, 13.1. 19F NMR (564.63 MHz, CDCl3) δ: −80.98 (t, 3F), −102.89 (t, 2F), −121.33 (m, 2F), −125.79 (m, 2F). HRMS-EI+ (M + 1): calcd. For C12H10F9N 340.06695; found, 340.06690.:n-hexane 1:1 v/v): Rf = 0.67; 1H NMR (600 MHz, CDCl3) δ: 7.21 (d, J = 8.6 Hz, 1H), 6.61 (d, J = 8.6 Hz, 1H), 3.88 (b s, 2H), 2.33 (s, 3H), 2.10 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 147.6, 137.7, 127.0, 121.9, 117.2, 112.3, 17.0, 13.1. 19F NMR (564.63 MHz, CDCl3) δ: −80.81 (t, 3F), −102.72 (t, 2F), −120.40 (m, 2F), −121.49 (m, 2F), −121.87 (m, 4F), −122.73 (m, 2F), −126.13 (m, 2F). HRMS-EI+ (M + 1): calcd. For C16H10F17N: 540.05418; found, 540.05420.:n-hexane 6:4 v/v): Rf = 0.66; 1H NMR (600 MHz, CDCl3) δ: 7.21 (d, J = 8.6 Hz, 1H), 6.60 (d, J = 8.6 Hz, 1H), 3.87 (b s, 2H), 2.33 (s, 3H), 2.10 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 147.6, 137.7, 127.1, 121.9, 117.3, 112.3, 17.1, 13.1. 19F NMR (564.63 MHz, CDCl3) δ: −58.58 (t, 2F), −102.70 (t, 2F), −112.99 (m, 2F), −120.36 (m, 2F), −120.87 (m, 2F), −121.48 (m, 2F). HRMS-EI+ (M + 1): calcd. For C14H10F12IN: 547.96663; found, 547.96651.:n-hexane 8:2 v/v): Rf = 0.77; 1H NMR (600 MHz, CDCl3) δ: 7.20 (d, J = 8.6 Hz, 1H), 6.60 (d, J = 8.6 Hz, 1H), 3.85 (b s, 2H), 2.32 (s, 3H), 2.10 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 147.6, 137.6, 127.1, 121.8, 117.1, 112.3, 17.0, 13.1. 19F NMR (564.63 MHz, CDCl3) δ: −57.77 (t, 2F), −102.74 (t, 2F), −112.56 (m, 2F), −119.42 (m, 2F). HRMS-EI+ (M + 1): calcd. For C12H10F8IN: 447.97302; found, 447.96999.:n-hexane 1:1 v/v): Rf = 0.88; 1H NMR (600 MHz, CDCl3) δ: 7.10 (d, J = 8.0 Hz, 1H), 6.66 (d, J = 8.0 Hz, 1H), 3.49 (b s, 2H), 2.31 (s, 3H), 2.09 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 143.9, 141.2, 125.9, 122.0, 119.6, 109.8, 20.9, 12.6. 19F NMR (564.63 MHz, CDCl3) δ: −59.21 (t, 2F), −81.03 (t, 3F), −113.20 (m, 2F), −121.02 (m, 2F), −121.98 (m, 8F), −122.89 (m, 2F), −126.32 (m, 2F). HRMS-EI+ (M + 1): calcd. For C18H10F21N: 640.04779; found, 640.04771.:n-hexane 1:1 v/v): Rf = 0.60; 1H NMR (600 MHz, CDCl3) δ: 7.21 (d, J = 8.6 Hz, 1H), 6.61 (d, J = 8.6 Hz, 1H), 3.88 (b s, 2H), 2.33 (s, 3H), 2.10 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 147.6, 137.7, 127.1, 121.9, 117.2, 112.3, 17.0, 13.1. 19F NMR (564.63 MHz, CDCl3) δ: −80.73 (t, 3F), −102.69 (t, 2F), −120.37 (m, 2F), −121.79 (m, 10F), −122.67 (m, 2F), −126.08 (m, 2F). HRMS-EI+ (M + 1): calcd. For C18H10F21N: 640.04779; found, 640.04758.:iso-octane 6:4 v/v): Rf = 0.58; 1H NMR (600 MHz, CDCl3) δ: 7.11 (dd, J = 4.6 Hz, J = 11 Hz, 1H), 6.53 (dd, J = 4.5 Hz, J = 11.6 Hz, 1H), 4.20 (b s, 2H). 13C NMR (151 MHz, CDCl3) δ: 156.6, 147.5, 139.9, 115.0 (m), 108.2, 103.8 (dd). 19F NMR (564.63 MHz, CDCl3) δ: −81.01 (t, 3F), −108.30 (m, 2F), −116.62 (td, 1F), −123.00 (m, 2F), −125.93 (m, 2F), −140.51 (d, 1F). HRMS-EI+ (M + 1): calcd. For C10H4F11N 348.01681; found, 348.01690.:petroleum ether 4:6 v/v): Rf = 0.60; 1H NMR (600 MHz, CDCl3) δ: 7.58 (s, 1H), 7.05 (s, 1H), 4.49 (b s, 2H). 13C NMR (151 MHz, CDCl3) δ: 147.9, 134.2, 120.7, 120.5, 117.6, 106.9. 19F NMR (564.63 MHz, CDCl3) δ: −80.95 (t, 3F), −105.73 (t, 2F), −120.58 (m, 2F), −125.81 (m, 2F). HRMS-EI+ (M + 1): calcd. For C10H4F9Br2N: 467.85668; found, 467.85669.:iso-octane 4:6 v/v): Rf = 0.56; 1H NMR (600 MHz, CDCl3) δ: 7.36 (s, 1H), 7.15 (s, 1H), 4.36 (b s, 2H), 2.24 (s, 3H). 13C NMR (151 MHz, CDCl3) δ = 144.5, 127.2, 126.0, 123.0, 118.6, 117.9, 18.1. 19F NMR (564.63 MHz, CDCl3) δ: −81.09 (t, 3F), −109.72 (t, 2F), −122.71 (m, 2F), −125.64 (m, 2F). HRMS-EI+ (M + 1): calcd. For C11H7F9ClN: 360.01233; found, 360.01241.:iso-octane 3:7 v/v): Rf = 0.73; 1H NMR (600 MHz, CDCl3) δ: 7.45 (d, J = 2.3 Hz, 1H), 7.24 (d, J = 2.3 Hz, 1H), 4.75 (b s, 2H). 13C NMR (151 MHz, CDCl3) δ: 142.6, 141.2, 132.9, 127.6, 121.9. 19F NMR (564.63 MHz, CDCl3) δ: −80.89 (t, 3F), −109.07 (t, 2F), −122.49 (m, 2F), −125.78 (m, 2F). HRMS-EI+ (M + 1): calcd. For C10H4Cl2F9N: 379.95771; found, 379.95784.:1 v/v): Rf = 0.6; 1H NMR (600 MHz, CDCl3) δ: 8.03 (d, J = 2 Hz, 1H), 7.94 (dd, J = 10 Hz, J = 5 Hz, 1H), 6.70 (d, J = 10 Hz, 1H), 4.66 (b s, 2H), 4.33 (q, J = 7 Hz, 2H), 1.37 (t, J = 7 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ: 165.6, 149.5, 134.2, 131.6 (t, CF2), 119.8, 118.4, 117.4, 116.9, 110.0 (t, CF2), 60.8, 14.3. 19F NMR (564.603 MHz CDCl3) δ: −81.00 (t, 3F), −109.06 (t, 2F), −122.74 (m, 2F), −125.83 (m, 2F). HRMS-EI+ (M + 1): calcd. For C13H11F9NO2: 384.06406; found, 384.06535.:iso-octane 1:1 v/v): Rf = 0.63; 1H NMR (600 MHz, CDCl3) δ: 7.47 (d, J = 7.8 Hz, 1H), 7.28 (t, J = 7.3 Hz, J = 8.2 Hz, 1H), 6.77 (d, J = 8.2 Hz, 1H), 6.74 (t, J = 7.3 Hz, J = 7.8 Hz, 1H), 4.45 (b s, 2H). 13C NMR (151 MHz, CDCl3) δ: 151.1, 140.0, 133.4, 118.8, 115.8, 104.5. 19F NMR (564.63 MHz, CDCl3) δ: −81.05 (t, 3F), −87.05 (t, 2F), −120.37 (m, 2F), −125.54 (m, 2F). HRMS-EI+ (M + 1): calcd. For C10H6F9NS: 344.00772; found, 344.00801.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from tautomer), 167.3 (from oxo tautomer), 163.1 (from oxo tautomer), 160.6, 160.3, 157.5, 156.3, 155.0 (from oxo tautomer). 19F NMR (564.63 MHz, DMSO-d6) δ: −79.10 (t, 3F) (from oxo tautomer), −80.45 (t, 3F), −102.34 (m, 2F) (from oxo tautomer), −111.83 (m, 2F), −121.54 (m, 4F), −122.38 (m, 2F), −125.35 (m, 2F). HRMS-EI+ (M + 1): calcd. For C8H5F9N4O: 345.03196; found 345.03205.NH), 3.75 (s, NH). 13C NMR (151 MHz, CDCl3) δ: 148.6, 135.6, 132.1, 120.9, 115.1. 19F NMR (564.63 MHz, CDCl3) δ: −80.99 (t, 3F), −113.22 (m, 2F), −122.75 (m, 2F), −125.76 (m, 2F). HRMS-EI+ (M + 1): calcd. For C9H6F9N3: 328.04180; found 328.04210.
O from tautomer), 167.3 (from oxo tautomer), 163.1 (from oxo tautomer), 160.6, 160.3, 157.5, 156.3, 155.0 (from oxo tautomer). 19F NMR (564.63 MHz, DMSO-d6) δ: −79.10 (t, 3F) (from oxo tautomer), −80.45 (t, 3F), −102.34 (m, 2F) (from oxo tautomer), −111.83 (m, 2F), −121.54 (m, 4F), −122.38 (m, 2F), −125.35 (m, 2F). HRMS-EI+ (M + 1): calcd. For C8H5F9N4O: 345.03196; found 345.03205.NH), 3.75 (s, NH). 13C NMR (151 MHz, CDCl3) δ: 148.6, 135.6, 132.1, 120.9, 115.1. 19F NMR (564.63 MHz, CDCl3) δ: −80.99 (t, 3F), −113.22 (m, 2F), −122.75 (m, 2F), −125.76 (m, 2F). HRMS-EI+ (M + 1): calcd. For C9H6F9N3: 328.04180; found 328.04210.Acknowledgements
Thanks are given to financial agencies: Universidad de Buenos Aires and Conicet. SBV and AP are research members of Conicet. DEY is a holder of a doctoral scholarship from Conicet. We also thank Quimey Mozetic Klik and Dr Lucas Fabian for acquiring NMR data.References
- M. Tordeux, B. Langlow and C. Wakselman, J. Chem. Soc., Perkin Trans. 1, 1990, 2293 RSC.
- M. Moreno-Mañas, R. Pleixats and S. Villarroya, Synlett, 1999, 1999, 1996–1998 CrossRef.
- S. Barata-Vallejo and A. Postigo, J. Org. Chem., 2010, 75, 6141–6148 CrossRef CAS PubMed.
- E. Baciocchi and E. Muraglia, Tetrahedron Lett., 1993, 34, 5015–5018 CrossRef CAS.
- (a) C. Bottecchia, X. Wei, K. P. L. Kuijpers, V. Hessel and T. Noël, J. Org. Chem., 2016, 81, 7301–7307 CrossRef CAS PubMed; (b) S. Barata-Vallejo, D. E. Yerien and A. Postigo, Eur. J. Org. Chem., 2015, 7869–7875 CrossRef CAS.
- H. Huo, X. Huang, X. Shen, K. Harms and E. Meggers, Synlett, 2015, 27, 749–753 CrossRef.
- C. Bottecchia, X. Wei, K. P. L. Kuijpers, V. Hessel and T. Noel, J. Org. Chem., 2016, 81, 7301–7307 CrossRef CAS PubMed.
- P. Finkbeiner and B. J. Nachtsheim, Synthesis, 2013, 45, 979–999 CrossRef CAS.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
- A. M. A. Adam, M. S. Refat, T. Sharshar and Z. K. Heiba, Spectrochim. Acta, Part A, 2012, 95, 458–477 CrossRef CAS PubMed.
- J. M. Gardner, M. Abrahamsson, B. H. Farnum and G. J. Meyer, J. Am. Chem. Soc., 2009, 131, 16206–16214 CrossRef CAS PubMed.
- B. Y. R. J. Donovan and D. Husain, Trans. Faraday Soc., 1968, 64, 3192–3199 RSC.
- A. B. Callear and J. F. Wilson, Trans. Faraday Soc., 1967, 63, 1983 RSC.
- A. B. Callear and J. F. Wilson, Trans. Faraday Soc., 1967, 63, 1358 RSC.
- D. M. Golden and S. W. Benson, Chem. Rev., 1969, 69, 125 CrossRef CAS.
- C. Chatgilialoglu, C. Ferreri, M. Melchiorre, A. Sansone and A. Torreggiani, Chem. Rev., 2014, 114, 255–284 CrossRef CAS PubMed.
- F. Sladojevich, E. McNeill, J. Börgel, S. L. Zheng and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3712–3716 CrossRef CAS PubMed.
- X. Sun, W. Wang, Y. Li, J. Ma and S. Yu, Org. Lett., 2016, 18, 4638–4641 CrossRef CAS PubMed.
- W. C. Barrett, J. P. Degnore, Y. Keng and Z. Zhang, J. Fluorine Chem., 1999, 35, 34543–34546 Search PubMed.
- Q. Y. Chen and Z. T. Li, J. Chem. Soc., Perkin Trans. 1, 1992, 1443–1445 RSC.
- N. Straathof, D. Osch, A. Schouten, X. Wang, J. Schouten, V. Hessel and T. Noël, J. Flow Chem., 2015, 4, 12–17 CrossRef.
- B. Zhang and A. Studer, Org. Lett., 2014, 16, 3990–3993 CrossRef CAS PubMed.
- Y. Su, K. Kuijpers, N. Koenig, M. Shang, V. Hessel and T. Noel, Chem.–Eur. J., 2016, 22, 12295–12300 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24786e |
| This journal is © The Royal Society of Chemistry 2017 |