
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of alkyl substitution on the supramolecular organization of thiophene- and dioxine-based oligomers
Wojciech Zajaczkowskia,
Shivananda Kammasandra Nanajundab,
Yoav Eichenb and
Wojciech Pisula*ac
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: pisula@mpip-mainz.mpg.de
bSchulich Faculty of Chemistry, Technion-Israel Institute of Technology, Technion City, 3200008 Haifa, Israel
cDepartment of Molecular Physics, Faculty of Chemistry, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
First published on 9th January 2017
Abstract
The interplay between the molecular structure, position and type of alkyl substituents on morphology and molecular packing is essential for the development of high-performance solution-processable organic semiconductors. This study focuses on the influence of the position and geometry of alkyl side chains on the supramolecular organization of thiophene- and dioxine-based oligomers. The structural investigation is performed by X-ray scattering for bulk and thin film samples. It is shown that attaching the side chains at the lateral position of the rigid oligomers mainly results in the formation of one-dimensional stacks. On the other hand, terminal alkyl substitution increases the steric hindrance between side chains and reduces the molecular interactions lowering the sample crystallinity.
Introduction
The development of new high performance organic semiconductors, which have great potential in plastic electronics applications, including light-emitting diodes, organic field-effect transistors, sensors or photovoltaic cells, has been one of the most active research fields in the last few years.1–3 The design of high performance semiconducting materials requires a thorough understanding of the interplay between molecular structure, solid-state packing, and the influence of both factors on the charge-carrier transport.The most frequently applied driving forces for the self-assembly are π-stacking interactions through conjugated aromatic units and hydrogen bonds introduced via functional groups.4 The supramolecular organization can also be controlled by the shape of the molecular core and alkyl substituents.5,6 Side-chain engineering is a powerful technique to improve the properties and solubility of semiconducting materials, but can affect the molecular organization as well.7 While the length of alkyl chains solely impacts the intermolecular distance, the geometry of the substituents, e.g. branched vs. linear, has direct effect on the self-assembly. For example, replacing linear with branched side chains generates pronounced steric hindrance between substituents, hinders π-interactions, and dramatically reduces the crystallinity and the semiconductor conductivity.8,9 When alkyl chains are introduced around a disk-shaped aromatic core, the molecules assemble into columnar stacks, which can serve as one-dimensional charge-carrier pathways.10,11 Rigid rod-like conjugated oligomers with flexible chains typically organize in two-dimensional layered structures, which are more favourable for the transport of charge-carriers compared with one-dimensional columnar structures. Substituents with different hydrophobic/hydrophilic properties can trigger an additional phase-separation in the periphery of the molecular core and generate more complex structures, including tubular fibre and helical packing.11 In the case of rigid rod-like small molecules, the side chains can be attached at both lateral and terminal positions of the aromatic core, influencing in different manner the supramolecular assembly.
Thiophene- and dioxine-based oligomers are well known organic semiconductors obtained by “click” chemistry. The great advantage of those molecules is solution processability into thin film devices and controlling their properties, such as solubility and thermal stability, by the introducing different lateral side chains.
This work focuses on the variation of the supramolecular organization of a series of thiophene- and dioxine-based oligomers by tuning not only the substitution pattern, but also the core structure. The solid-state assembly of these oligomers substituted by terminal and lateral by N-alkyl chains is compared. The structural analysis based on X-ray scattering exhibits differences in oligomer organization depending on the attachment position of the alkyl chains. The position changes at the peripheral thiophene rings lead to dramatically reduced molecular interactions and induce disorder. On the other hand, minor increase in the size of the oligomer core does not cause major changes in molecular order.
Results and discussion
Structural study
To understand the influence of the different N-alkyl substituents and their substitution position on the organization of the oligomers P1–P5 (Fig. 1) in bulk two-dimensional wide-angle X-ray scattering (2DWAXS) measurements were performed. The synthesis route of P1–P5 has been reported elsewhere.19 The samples were prepared by fibre extrusion, which induces a macroscopic orientation of the molecules by mechanical shear forces in the specimen. These extruded fibres were mounted vertically in front of the 2D detector. This setup allows an insight into the molecular arrangement within the corresponding superstructure.12 For the analysis of the data, the recorded 2DWAXS pattern can be divided into a meridional and equatorial planes on which the reflections are located. In bulk, the oligomers reveal variations in crystallinity and molecular long-range orientation, as evident from differences in number and angular intensity distribution of the reflections. These distinct changes in molecular orientation and supramolecular organization are essentially influenced by the design and substitution position of the alkyl side chains as well as to certain extend to the length of the oligomer core. For the structural analysis, the unit cell parameter a is related to the short oligomer axis and c corresponds to the long axis of molecule, while b is related to the π-stacking period.13–15 | ||
| Fig. 1 Chemical structures of oligomers (a) P1, (b) P2, (c) P3, (d) P4 and (e) P5. | ||
Fig. 2 shows the 2DWAXS patterns for P1 and P2 recorded at room temperature after thermal annealing at 120 °C for 1 hour under vacuum. The pattern for P1 (Fig. 2a) indicates a herringbone-like structure of molecules arranged in stacks that are aligned along the extruded fibre (Fig. 2c). Reflections on the equatorial plane in the small-angle range correspond to the Miller's index of 001 and d-spacing of 2.11 nm, which is in agreement with the molecular long axis (Fig. 2c). Further higher order reflections confirm the long-range order and high crystallinity of the compound. Additional 100 reflections positioned also on the equatorial plane are attributed to the distance of 1.44 nm, which is related to the short oligomer axis (Fig. 2c). Off-meridional wide-angle X-ray scattering 010 reflections as indicated in the pattern by dashed circles in Fig. 2a, correspond to the π-stacking distance of 0.35 nm and a molecular tilt angle of ∼47° towards the stack axis (see intra-stack arrangement in side view illustration in Fig. 2e). Based on the structural data, a monoclinic unit cell is derived with parameters of a = 1.25 nm, b = 0.52 nm, c = 1.99 nm and γ = 97.1° between the b and c axes. Thereby, the a axis coincidences with the alignment direction of the molecules in the fibre sample and with the axis of the stacks, the b parameter represents the intra-stack period. While the direction of b is fixed, axes a and c are arranged perpendicular to b (Fig. 3c and d). The proposed model was confirmed by Cerius2 simulations, yielding an identical position and distribution of reflections (Fig. 3a and b).
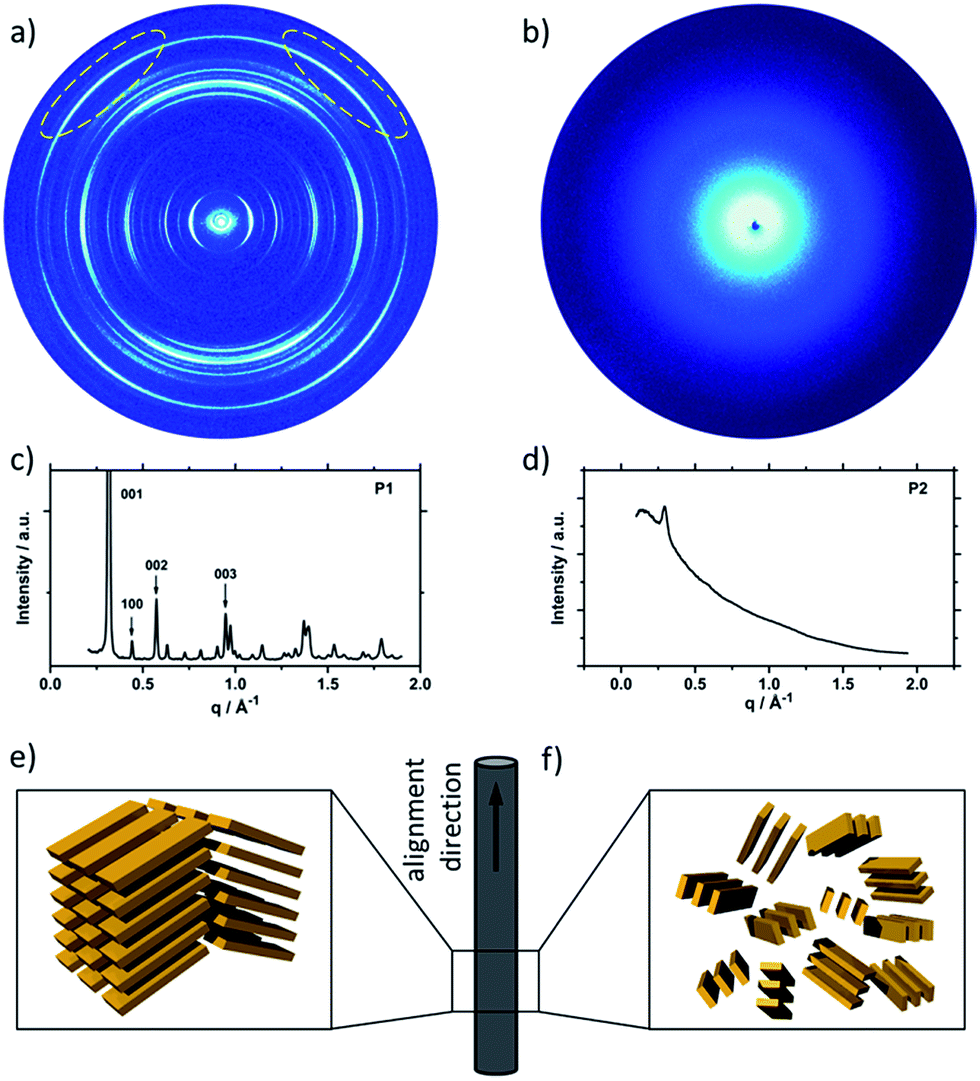 | ||
| Fig. 2 Fibre 2DWAXS patterns for (a) P1 (off-meridional reflection related to π-stacking of tilted molecules are indicated by dash circles) and (b) P2; corresponding equatorial integrations for (c) P1 and (d) P2; schematic illustrations of the supramolecular organization in the fibre for (e) P1 and (f) P2. | ||
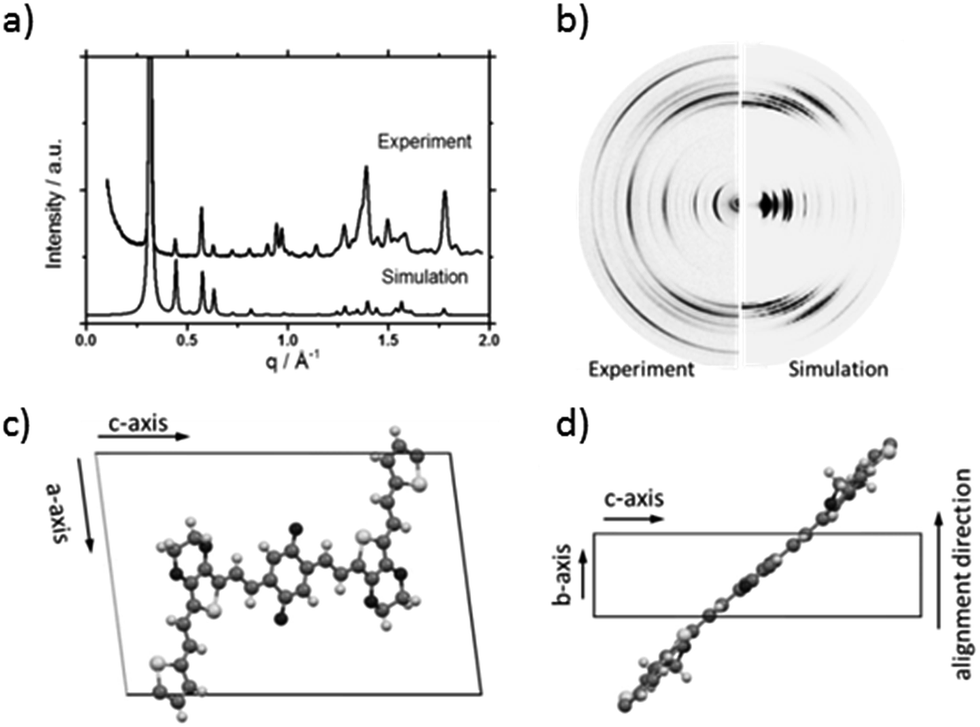 | ||
| Fig. 3 Comparison between experiment and simulation of (a) peak integration (radial integration for the whole pattern) and (b) 2DWAXS patterns for experiment (left side) and simulation (right side). (c) Top view and (d) side view of the unit cell of P1. | ||
The scattering pattern of P2 (Fig. 2b) significantly differs from P1. The 2DWAXS measurement provides only one single weak reflection which corresponds to a d-spacing of 2.12 nm. This can be assigned to the long axis of molecule and is in agreement with the reflection of 001 of P1 (d-spacing of 2.11 nm). Higher order scattering intensities and the π-stacking peak are missing indicating a high disorder of the molecules. The isotropic distribution of the reflection over the azimuthal angle χ shown in Fig. 4a, confirms the long-range disorder.16,17 The ratio between the in-plane to out-of-plane intensity indicates the macroscopic orientation of sample and has been found for the 001 reflection to be isotropic with Rin/out ∼ 1. In contrast, oligomer P1 exhibits much higher macroscopic orientation as evident from the azimuthal intensity distribution of the 001 reflection over the angle χ (Fig. 4a). It has to be emphasized that all fibre samples have been prepared under the same conditions. The maxima of this reflection at 0° and 180° are indicative for the alignment of the stacks along the fibre extrusion. The high value of Rin/out = 7.1 verifies the pronounced structural anisotropy of P1 which is also in agreement with the high crystallinity.
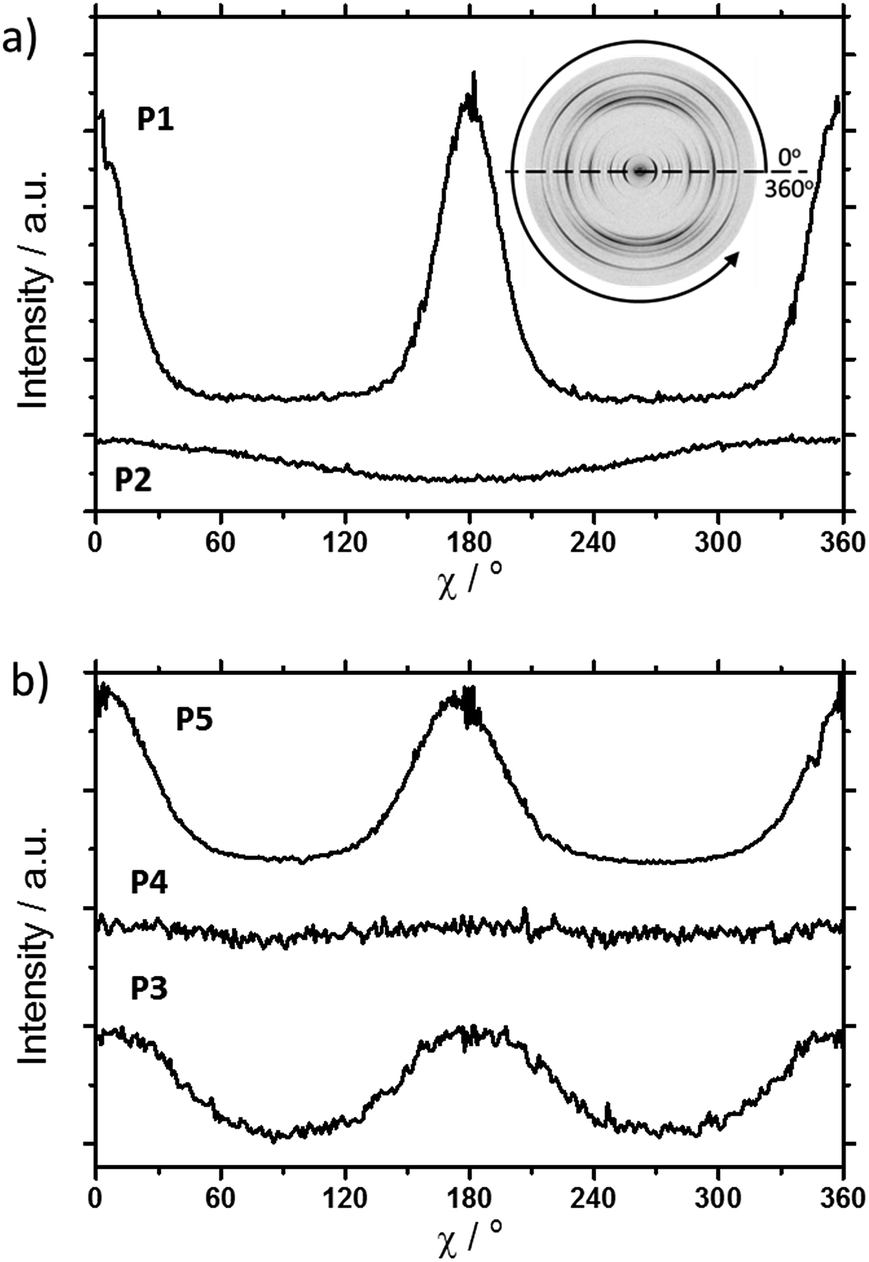 | ||
| Fig. 4 Azimuthal intensity distribution of the 001 Bragg peak as a function of χ referred to the equatorial axis (0° and 180° correspond to the equatorial plane as illustrated in the inset for P1 in (a)). Comparison between (a) P1 and P2, as well as (b) P3, P4 and P5. | ||
Interestingly, P3, substituted laterally by n-octyl side chains, is as highly ordered as P1, however, exhibits a different type of molecular orientation towards the extrusion direction of the fibre specimen (Fig. 5a). The strong intensity reflections located in the wide-angle range are assigned to the π-stacking period (Miller's index of 020) of 0.37 nm. In contrast to the meridional location of the π-stacking reflection for P1, the peak appears for P3 on the equatorial plane. Furthermore, the small-angle 001 reflection related to the d-spacing of 2.12 nm is also located on equatorial plane of the pattern and is in agreement with the long molecular axis of P3. The 200 reflections on the meridional plane in the middle-angle range correspond to the d-spacing of 1.47 nm and are attributed to the short molecular axis. As already observed for P1, the higher order reflections confirm the long-range order and high crystallinity of the compound. This distribution of reflections suggests that, in contrast to P1, in the extruded sample molecules are arranged with their long axis perpendicular towards the extrusion direction (Fig. 5a).
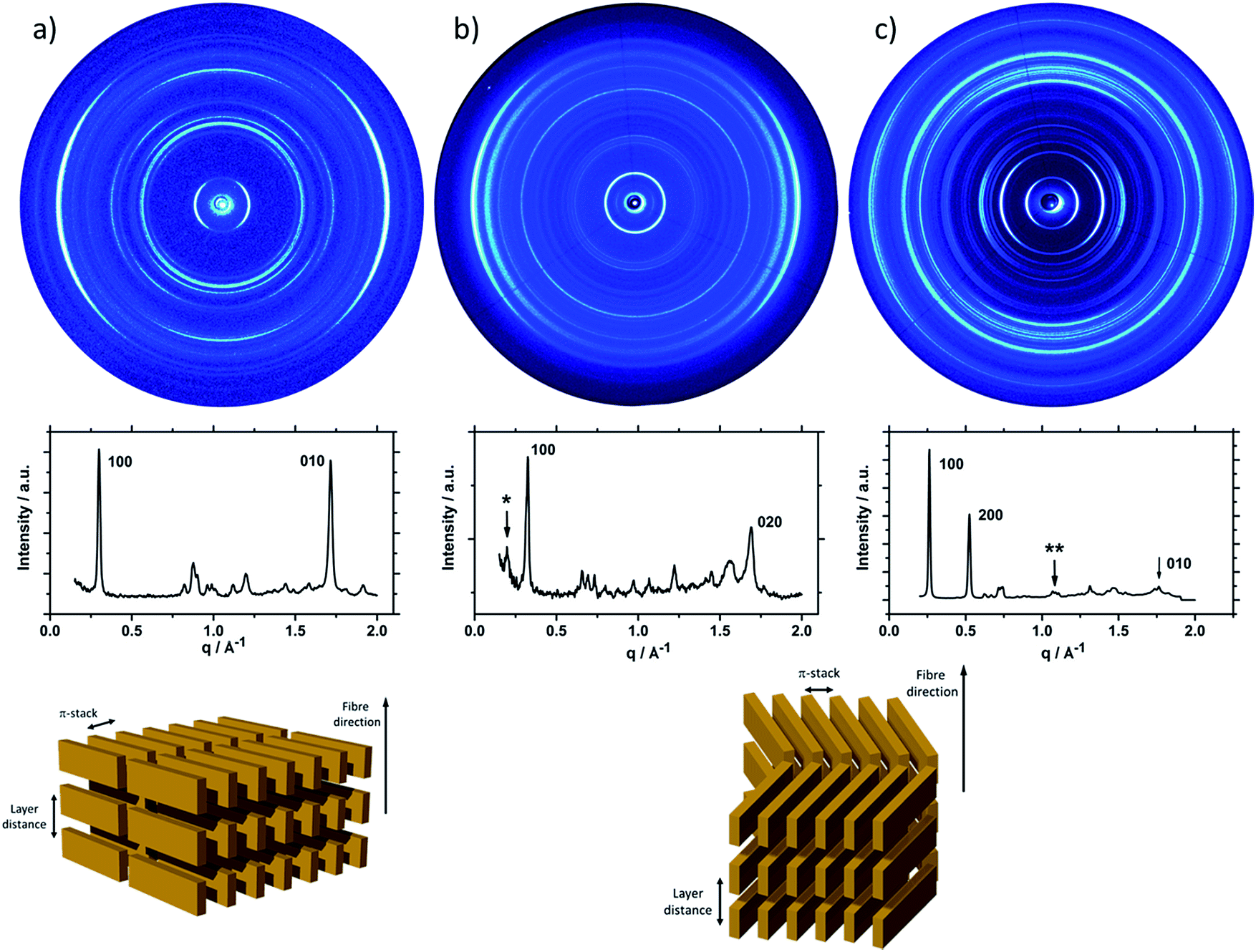 | ||
| Fig. 5 Fibre 2DWAXS patterns for (a) P3, (b) P4 and (c) P5 with corresponding equatorial integrations (asterisk for P4 indicates the reflection related to the long molecular axis) and schematic illustrations of the organization in the fibre in top view. | ||
Compound P4 shows high crystallinity similar to P1 and P3 as obvious from the large number of distinct reflections. However, the isotropic distribution of all reflections over the azimuthal integration (Fig. 5b) and a Rin/out value of only ∼1 indicate lack of preferential long-range orientation. Nevertheless, the pattern suggests the formation of a layer structure in which the molecules are packed in a tilted fashion as determined from two characteristic pairs of reflections. Firstly, in the equatorial wide-angle range two d-spacing of 0.37 nm and 4.01 nm are found that are attributed to the π-stacking and intralayer distance. Secondly, two scattering intensities appear in the small-angle range related to the layer organization. The peak labelled by the asterisk in the equatorial integration in Fig. 5b corresponds to a d-spacing of 3.08 nm and can be attributed to the molecular long axis. Furthermore, the second reflection indexed as 001 is assigned to d-spacing of 2.01 nm for the interlayer distance. Therefore, the organization is similar to P1, however, without long-range orientation of the molecules.
The replacement of the linear alkyl chains by more bulky branched ones at the lateral position does not change the organization P5 in comparison to P4. Similar to P1 and P3, compound P5 is highly crystalline as proven by the large number of distinct reflections (Fig. 5c). The intensive small-angle reflection 001 positioned on the equatorial plane corresponds to the interlayer distance of 2.40 nm. The equatorial wide-angle reflections 010 related to the π-stacking of 0.36 nm implies a perpendicular molecular alignment of P5 as found for P4. The marked peak (**) in the integration plot in Fig. 5c is related to an intermolecular distance of 0.57 nm along the layer axis indicating a tilting of 51° of the oligomers and the formation of a herringbone structure similar to P1 and P4. Thereby, the tilting of P5 occurs perpendicular to the fibre axis. In comparison to P3, P5 shows higher structural anisotropy of the long-range alignment as confirmed by strong maxima at 0° and 180° in the azimuthal integrations (Fig. 5c) and high Rin/out = 4.58 (Table 1).
| Oligomer | π-stacking distance | Rin/out |
|---|---|---|
| P1 | 0.35 nm | 7.1 |
| P2 | n.a. | ∼1 |
| P3 | 0.37 nm | 1.8 |
| P4 | 0.37 nm | 1 |
| P5 | 0.36 nm | 4.6 |
The molecular packing in thin film can significantly differ from the bulk structure. Grazing incidence wide-angle X-ray scattering (GIWAXS) provides information about the crystalline the molecular packing in both the out-of-plane (qz) and in-plane (qxy) directions of the thin films. Experiments were performed on thin film samples of the oligomers to further study the influence of alkyl side chains also in solution processed layers. These films were prepared by spin-coating P1–P5 from 4 mg mL−1 THF solution on hexamethyldisilazane (HMDS) modified SiO2 substrate with subsequent annealing at 120 °C for 1 h in vacuum before measurements. The 2D pattern for P1 (Fig. 6a) exhibits a highly ordered structure for the thin film which is similar to the crystallinity observed for the bulk sample. The meridional reflection in the small-angle range at qz = 0.303 Å−1 corresponds to the d-spacing of 2.07 nm, which is identical to the spacing for the bulk (2.11 nm) and is attributed to an interlayer distance. The peaks at qz = 0.607 Å−1 and 0.910 Å−1 are related to 002 and 003 reflections. Intensive reflections on the equatorial plane in the wide-angle range at a d-spacing of 0.35 nm correspond to the π-stacking distance. The positions of the interlayer and π-stacking reflections indicates that the molecules are almost parallel with their a–b plane to the substrate (schematic illustration in inset of Fig. 6a). This organization of P1 is in agreement with the previously reported film structure.20 The scattering pattern of P2 (Fig. 6b) is different from that of P1 and exhibits an amorphous film structure. The broad halo ring in the q range of 1.25–1.70 Å−1 is related to the amorphous structure of molecules as illustrated in the inset of Fig. 6b and is in agreement with the bulk organization. Compared to P1, the scattering pattern of P3 shows a reduced number of Bragg peaks. The lack of higher order reflections combined with the amorphous halo ring are an evidence for significantly reduced crystallinity. The peak at qz = 0.265 Å−1 located on the meridional plane is attributed to the d-spacing of 2.18 nm which can be correlated to the long molecular axis and is identical to the value for P1. The broad scattering intensity located on the off-meridional plane in the wide-angle range corresponds to the π-stacking of 0.37 nm implying a tilted arrangement of the molecules on the surface.18 The crystallinity further decreases for P4 which exhibits only a layer ordering as indicated by reflections on the meridional plane. The main 1st order peak corresponds to an interlayer distance of 3.40 nm. The π-stacking reflection is much weaker and is superimposed by the amorphous halo confirming low molecular order of P4 in the thin film.
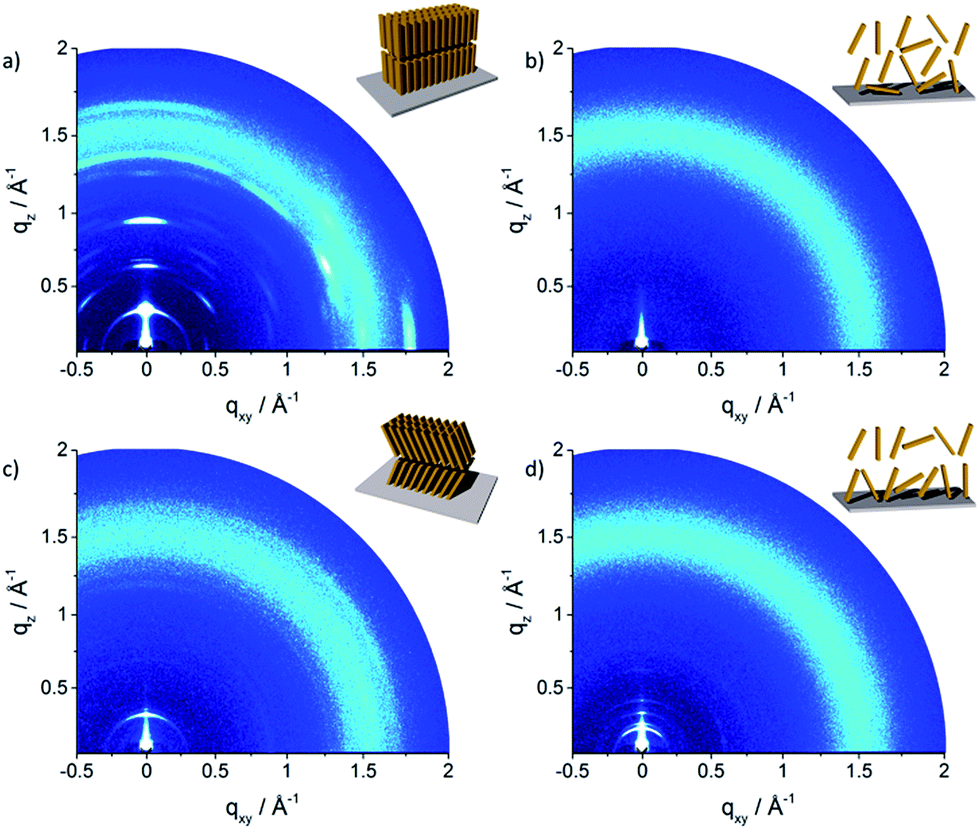 | ||
| Fig. 6 GIWAXS patterns of spin-coated oligomers: (a) P1, (b) P2, (c) P3 and (d) P4. Insets schematically illustrate the surface organization. | ||
In order to gain further insight about the influence of alkyl substituents on self-assembly of P1–P4, UV-vis for corresponding THF solutions and spin-coated thin films was performed (Fig. 7). Oligomers P1 and P2 show similar absorption bands located at middle-energy range between 450–550 nm with a small deviation between solution and thin film (Fig. 7a and b). Thin film of P1 reveals a broadened UV-vis spectra and an additional shoulder at 553 nm in comparison to the solution indicating increased intermolecular interactions. A further blue shift of the maximum absorption implies also H-aggregates in the film. These UV-vis results are in agreement to the GIWAXS data of P1 confirming a highly ordered structure with strong π-interactions. In case of P2, the UV-vis spectrum is widened and is slightly red shifted suggesting minor aggregation, which seem to be already present in solution, in the rather macroscopically disordered film. Oligomers P3 and P4 with additional phenyl rings within the extended rod-like core exhibit only small differences between solution and solid-state in the UV-vis spectra (Fig. 7c and d). The lack of pronounced low energy absorption bands in comparison to P1 confirms the disordered structure of P3 and P4. The decrease in order is related to the additional phenyl rings which probably reduce the molecular planarity and hinder intermolecular interactions.
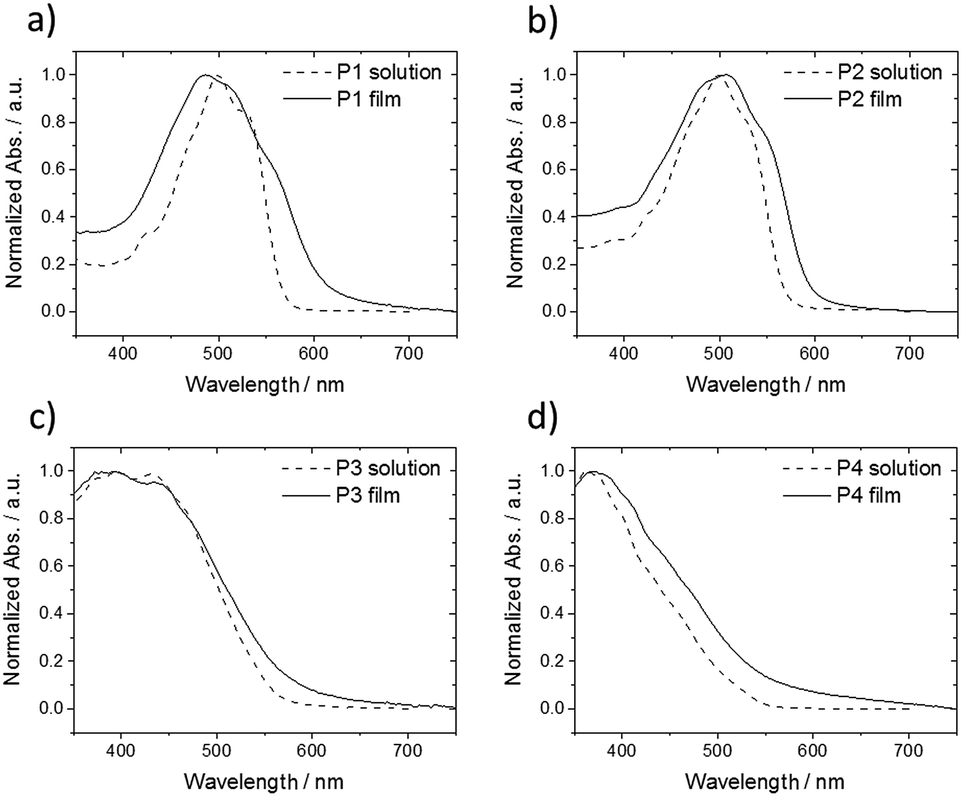 | ||
| Fig. 7 Normalized UV-vis spectra of (a) P1, (b) P2, (c) P3 and (d) P4 in THF solution (dashed lines) and spin-coated thin film (solid lines). | ||
For a series of oligomers, it has been shown that by varying the alkyl substitution pattern the organization of thiophene- and dioxine-based oligomers can be well controlled. The structure formation in bulk and thin film is a process dominated by the steric hindrance of the side chains as confirmed for the presented oligomers series. Thereby, the structure is changed from herringbone (oligomers P1 and P5) and layer packing (oligomer P3) to disordered (oligomer P2). Additionally, the long-range orientation of the oligomers is switched between parallel, perpendicular and isotropic in the extruded fibre samples.
Conclusions
Oligomer P1 bearing the terminal hexyloxy chains attached to the C-5 position of the terminal thiophene shows high crystallinity in both bulk and solution-processed thin films, with a herringbone packing. The lateral substitution of the aromatic rigid rod by 2-methyloxy-hexyloxy chains induces a perpendicular arrangement of P1 towards the shearing direction during fibre extrusion. Due to the higher steric demand of these chains in comparison to the n-octyloxy chains a more homogenous alkyl mantel is formed around the oligomer core, reducing the molecular aspect ratio. Generally, disc-like molecules with a low aspect ratio assemble in stacks which align along the orientation direction similar to the superstructure of P1. Implementation of an additional phenyl ring in P4 and P5 extends the rod-like core, leading to high crystallinity due to better molecular interactions and induces partly a parallel orientation of the molecules to the fibre axis. Interestingly, changing the position of the substituents from C5 for P1 to C3 on both thiophene rings in P2 leads to dramatically reduced molecular interaction and higher disorder. This substitution pattern increases the steric hindrance between the lateral and terminal attached side chains. The result is an amorphous structure in bulk and thin film. In thin film, P2 remains amorphous, while the organization of the other oligomers differs in comparison to bulk. The crystalline herringbone structure of P1 changes into a non-tilted surface ordering. On the other hand, P3 and P4 show significantly higher crystallinity in bulk than in thin film. This might be related to the fast solvent evaporation during spin-coating leading to a rather disordered non-equilibrium state. Additionally, for P3 the arrangement switches to a quasi-herringbone structure with tilted molecules on the surface.Experimental
Sample preparation
The synthesis of the compounds has been described in ref. 19. The fibre samples were prepared by using a custom-build mini extruder. The extruder consists of three parts: die – with an opening (ø = 0.5 mm) through which the extrusion process is carried out, barrel and piston. During extrusion process, an external force applied to the piston exerts pressure on sample located in the barrel allowing the samples extrusion. The extrusion process was conducted at room temperature. The thin films of P1, P2, P3 and P4 were spin-coated from 4 mg mL−1 THF solution on hexamethyldisilazane (HMDS) modified SiO2 substrate and annealed at 120 °C for 1 h in vacuum before measurements.Two-dimensional wide-angle X-ray scattering (2DWAXS) and grazing incidence wide-angle X-ray scattering (GIWAXS)
X-ray scattering was performed using a custom setup consisting on a Siemens Kristalloflex X-ray source with copper anode X-ray tube operated at 35 kV/30 mA and Osmic confocal MaxFlux optics monochromator. The diffraction beam was collimated with two pinholes (1.2 and 1.0 mm – Owis, Germany) and anti-scattering pinhole (1.1 mm – Owis, Germany). The 2DWAXS patterns were recorded on a MAR345 image plate detector (Marresearch, Germany). Scattering data are expressed as a function of the scattering vector: q = 4π/λ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(Θ), where Θ is a half the scattering angle and λ = 0.154 nm is the wavelength of the incident radiation. Here qxy (qz) is a component of the scattering vector perpendicular (parallel) to the fibre direction. All X-ray scattering measurements were performed under vacuum (∼1 mbar) to reduce air scattering.
sin(Θ), where Θ is a half the scattering angle and λ = 0.154 nm is the wavelength of the incident radiation. Here qxy (qz) is a component of the scattering vector perpendicular (parallel) to the fibre direction. All X-ray scattering measurements were performed under vacuum (∼1 mbar) to reduce air scattering.
GIWAXS experiments were performed by using the same beam parameter as for the 2DWAXS measurements. The films were irradiated just below the critical angle for total reflection with respect to the incoming X-ray beam (∼0.12°).
All X-ray data were processing and analysis using the software package Data squeeze (http://www.datasqueezesoftware.com).
UV-vis measurements
Solution and thin film UV-vis absorption spectra were recorded at room temperature on a Perkin-Elmer Lambda 900 spectrophotometer in THF (HPLC grade) in a conventional quartz cell (light pass 10 mm, Hellma Analytics). Thin films for UV-vis measurements were prepared by spin-coating a THF solution on glass substrate and annealed at 120 °C for 1 h before measurements.Acknowledgements
We are grateful to Klaus Müllen for stimulating discussions.Notes and references
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- M. Jung, Y. Yoon, J. H. Park, W. Cha, A. Kim, J. Kang, S. Gautam, D. Seo, J. H. Cho, H. Kim, J. Y. Choi, K. H. Chae, K. Kwak, H. J. Son, M. J. Ko, H. Kim, D. K. Lee, J. Y. Kim, D. H. Choi and B. Kim, ACS Nano, 2014, 8, 5988–6003 CrossRef CAS PubMed.
- L. Fang, Y. Zhou, Y. X. Yao, Y. Diao, W. Y. Lee, A. L. Appleton, R. Allen, J. Reinspach, S. C. B. Mannsfeld and Z. A. Bao, Chem. Mater., 2013, 25, 4874–4880 CrossRef CAS.
- S. Himmelberger, D. T. Duong, J. E. Northrup, J. Rivnay, F. P. V. Koch, B. S. Beckingham, N. Stingelin, R. A. Segalman, S. C. B. Mannsfeld and A. Salleo, Adv. Funct. Mater., 2015, 25, 2616–2624 CrossRef CAS.
- S. Chen, B. Sun, W. Hong, H. Aziz, Y. Meng and Y. Li, J. Mater. Chem. C, 2014, 2, 2183–2190 RSC.
- A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. J. Fréchet, J. Am. Chem. Soc., 2012, 134, 2180–2185 CrossRef CAS PubMed.
- A. M. van de Craats, L. D. A. Siebbeles, I. Bleyl, D. Haarer, Y. A. Berlin, A. A. Zharikov and J. M. Warman, J. Mater. Chem. B, 1998, 102, 9625–9634 CAS.
- X. Feng, V. Marcon, W. Pisula, M. R. Hansen, J. Kirkpatrick, F. Grozema, D. Andrienko, K. Kremer and K. Müllen, Nat. Mater., 2009, 8, 421–426 CrossRef CAS PubMed.
- W. Pisula, X. Feng and K. Müllen, Adv. Mater., 2010, 22, 3634–3649 CrossRef CAS PubMed.
- T. Schuettfort, L. Thomsen and C. R. McNeill, J. Am. Chem. Soc., 2013, 135, 1092–1101 CrossRef CAS PubMed.
- A. J. Kronemeijer, E. Gili, M. Shahid, J. Rivnay, A. Salleo, M. Heeney and H. Sirringhaus, Adv. Mater., 2012, 24, 1558–1565 CrossRef CAS PubMed.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- Q. Yuan, S. C. B. Mannsfeld, M. L. Tang, M. Roberts, M. F. Toney, D. M. DeLongchamp and Z. Bao, Chem. Mater., 2008, 20, 2763–2772 CrossRef CAS.
- X. Zhang, L. J. Richter, D. M. DeLongchamp, R. J. Kline, M. R. Hammond, I. McCulloch, M. Heeney, R. S. Ashraf, J. N. Smith, T. D. Anthopoulos, B. Schroeder, Y. H. Geerts, D. A. Fischer and M. F. Toney, J. Am. Chem. Soc., 2011, 133, 15073–15084 CrossRef CAS PubMed.
- L. H. Jimison, A. Salleo, M. L. Chabinyc, D. P. Bernstein and M. F. Toney, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 125319 CrossRef.
- K. N. Shivananda, I. Cohen, E. Borzin, Y. Gerchikov, M. Firstenberg, O. Solomeshch, N. Tessler and Y. Eichen, Adv. Funct. Mater., 2012, 22, 1489–1501 CrossRef CAS.
- P. Kumar, K. N. Shivananda, W. Zajaczkowski, W. Pisula, Y. Eichen and N. Tessler, Adv. Funct. Mater., 2014, 24, 2530–2536 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |