
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solar mediated reduction of graphene oxide†
Manonmani
Mohandoss
ab,
Soujit Sen
Gupta
c,
Anith
Nelleri
b,
T.
Pradeep
c and
Shihabudheen M.
Maliyekkal
*a
aEnvironmental Engineering Division, School of Mechanical and Building Sciences, VIT University, Chennai Campus, Chennai – 600 127, India. E-mail: shihabudheen.m@vit.ac.in; sm.maliyekkal@gmail.com; Fax: +91-44-3993-2555; Tel: +91-44-3993-1266
bSchool of Electronics Engineering, VIT University, Chennai Campus, Chennai – 600 127, India
cDST Unit on Nanoscience and Thematic Unit of Excellence (TUE), Department of Chemistry, Indian Institute of Technology Madras, Chennai 600 036, India
First published on 4th January 2017
Abstract
This paper explores the reduction of water dispersed graphene oxide (GO) by sunlight as an environmentally friendly alternative to conventional methods of reduction of GO. The possible mechanism of the reduction process is delineated. The electrical and thermal conductivity, the degree of reduction and structural defects of sunlight reduced GO (sRGO) are studied thoroughly and compared with RGO samples produced through hydrazine (hRGO) and hydrothermal (hyRGO) reduction routes. The study reveals that the production of sRGO is feasible and its electronic properties are on a par with those of hRGO. Interestingly, sRGO showed the least structural defects, good dispersibility and higher conductivity vis-a-vis its counterparts. This cost effective and environmentally friendly method of reducing GO to RGO with enhanced electronic properties may find applications in bio-sensing and electrochemical energy storage devices.
Introduction
Graphene, one atom thick 2D sheets of sp2 hybridised carbon atoms arranged in a honeycomb lattice,1 has attracted overwhelming academic and industrial research interest due to its excellent mechanical, electrical, thermal and optical properties with promising applications.2 Diverse methods of graphene synthesis have been developed over the years expecting distinct physical and chemical properties.3 The journey started with the successful separation of graphene by scotch tape based micro-cleaving of highly oriented pyrolytic graphite (HOPG).4 The low yield and difficulty in tearing of graphene from the substrate restricted its use in laboratory scale applications for making proof-of-concept devices. This method of mechanical exfoliation may also inhibit the quality of graphene due to the possible attachment of adhesive to the graphene surface.5 Epitaxial growth on silicon carbide or metal surfaces6 and chemical vapours deposition (CVD) technique7 have emerged as alternatives to the exfoliation strategies to produce better quality graphene, especially when expecting the coverage of large substrate areas with graphene.8 Though the epitaxial method delivers high quality graphene, the stringent growth conditions and the use of costly single crystal substrates make it economically unviable for large-scale production.9 Despite the fact that the recent advancement in CVD techniques allows synthesis of graphene monolayers with large surface area, the low yield of production limits its large-scale deployment and the method is typically restricted for the precise fabrication of devices.10 Chemical conversion of graphite to graphene is attractive over other methods due to its high reduction efficiency, good dispersibility and non-requirement of sophisticated experimental conditions.11 The wet chemical exfoliation of graphite seems to be promising for the bulk production of graphene.12,13 The method typically consists of weakening of the graphite interlayer van der Waals force and disrupting it by chemical intercalation,14 thermal expansion,15 oxidation–reduction16 or by the use of surfactants.17 Hummer's method18 of chemical exfoliation is a well-established and widely practiced wet chemical technique for the bulk synthesis of water dispersible graphene oxide (GO). The process involves oxidation of graphite to GO, where large fraction of sp2 carbon atoms is converted to sp3 configurations. GO is then transformed to reduced graphene oxide (RGO) by reduction methods including chemical,19 hydrothermal,20 thermal,21 photo22,23 or combinations thereof.24 However, the need for the critical treatment conditions, use of hazardous and toxic reducing agents (e.g. hydrazine hydrate,25 hydroquinone26) and energy intensive synthesis procedures need further attention to make the production process more sustainable. Besides, the extent of removal of oxygen functionalities on GO may vary with different reduction processes. This can results in RGO with different chemical, physical and electronic properties.27 Hence, understanding the influence of reduction techniques on the properties of RGO is also important to decide its suitability for a specific application.In this paper, we demonstrate a simple and green technique for the reduction of GO to RGO as a sustainable alternative to conventional reduction techniques. The proposed method uses sunlight as the sole medium for the reduction of GO to RGO. The samples were characterized thoroughly by spectroscopic and microscopic techniques. The possible mechanism of reduction of GO to RGO is proposed. The electrical conductivity, dispersibility, degree of reduction, defect repair, and thermal conductivity of sRGO were compared systematically with RGO samples synthesised through other popular reduction techniques, which includes hydrothermal and hydrazine reduction. The sunlight mediated reduction strategy seems to be viable and can produce RGO with good electrical conductivity and less structural defects.
Materials and methods
Natural graphite powder (100 μm; 12.01 g mol−1) was obtained from S.D. Fine Chem. Pvt. Ltd., India. Sulphuric acid (H2SO4, 95–98%), potassium permanganate (KMnO4, 98.5%), potassium peroxy disulphate (K2S2O8, 98%), phosphorus pentoxide (P2O5, 95%), hydrogen peroxide (H2O2, 98%), were procured from Sisco Research Lab Chem. Pvt. Ltd., India. The dialysis tubing cellulose membrane with average diameter of 6 mm was purchased from Sigma Aldrich. All the chemicals were of analytical grade and used as received without any further purification. Unless otherwise mentioned, solutions and suspensions used in the study were prepared using deionized water (DW) of conductivity 0.056 μ℧ cm−1.Preparation of reduced graphene oxide (RGO)
GO was synthesized by modified Hummers method.28 This method involves the oxidation of graphite powder by sulphuric acid and potassium permanganate to produce graphite oxide and exfoliation of graphite oxide to GO. A detailed procedure is given in S1.1 of the ESI.† The purified dispersion of GO was stored in brown bottles (covered with aluminium foil) to cut the entry of light. The reduction of GO was carried out by exposing 0.01% dispersion of GO to sunlight for a duration of ∼16 h. The temperature of GO dispersion was measured and found to be around ∼42 ± 2 °C. Initial pH of the dispersion was measured to be 4.0 ± 0.3 and no significant change in pH was observed after the reduction process. In-order to prevent possible evaporation loss and associated change in the concentration of dispersion of GO during irradiation, the reactor was completely sealed. For kinetic studies, a series of sealed bottles containing dispersion of GO were irradiated. At periodic time intervals, a pre-designated sample bottle was withdrawn and analysed using UV-Vis and Raman spectroscopy. The incident solar light intensity at the surface of irradiance during irradiation period (from 11 a.m to 4 p.m) was measured to be in the range of 42 W m−2 to 373 W m−2. The experiment was also performed using photovoltaic solar simulator (Ecosense Insight Solar PV simulator) with halogen lamps (350–2500 nm) as the artificial source of solar radiation. The intensity of radiation from halogen lamp was adjusted similar to the natural sunlight throughout the experiment. No significant change in the rate of reduction of GO was observed under natural and simulated conditions. For comparison purpose, RGO samples were also synthesised using hydrazine (hRGO) and hydrothermal (hyRGO) heat as the reducing agents (see S1.3, S1.2 of ESI† for detailed procedure). The samples were later dialyzed against DW to remove ionic impurities.Instrumentation
Thermo-fisher Scientific (EVO 300 PC) UV/Vis spectrophotometer was used to measure UV/Vis spectra of GO and RGO samples. XPS measurements were done with Omicron ESCA Probe spectrometer with un-monochromatized Mg Kα X-rays (hν = 1253.6 eV). Processing of XPS spectra was done using Casa XPS software. Calibration was carried by alignment of the spectra with reference to the C 1s line at 284.5 ± 0.2 eV associated with graphitic carbon. The confocal Raman Microscope, CRM a300 S (WITec GmbH) coupled with AFM was used for collecting Raman spectra and AFM images. The samples were also imaged using a high-resolution transmission electron microscope (HRTEM) with a UHR pole piece equipped with an EDAX (JEOL 3011, 300 kV). The attenuated total reflectance infrared spectroscopic (ATR-IR) measurement was performed using Thermo Scientific NICOLET iS10 FT-IR Spectrometer. Indosaw four-probe set-up (SN168) was used to measure the surface resistance for the calculation of electrical conductivity. The Horibo Jobin Yvon Uvisel spectroscopic ellipsometer was used to measure the thickness of RGO sheets. Tenmars-TM207 solar power meter was used to measure the intensity of solar radiation. Dynamic light scattering Zetasizer (Horiba SZ100) was used to study the dispersibility of RGO samples by measuring their zeta potential.Results and discussion
The as-produced RGO samples were characterised in detail with various spectroscopic and microscopic techniques. Fig. 1A shows UV/Vis spectrum of GO sample synthesized through modified Hummer's method (red trace). The data show a prominent absorbance peak of GO centred at 239 nm and a shoulder peak at 302 nm, which can be ascribed to π–π* transition of C–C bonds and n–π* transition of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively. The corresponding sRGO spectrum after exposing to sunlight for a period of 16 h is also shown in Fig. 1A (blue trace). Upon exposure to sunlight, the peak corresponding to π–π* transition of C–C bonds is red shifted to 263 nm and the shoulder peak at 302 nm is completely disappeared, which indicates the restoration of sp2 carbon structure. UV/Vis spectra in Fig. 1B shows the reduction of GO to sRGO with respect to time. The shoulder peak at 302 nm is started diminishing in less than 8 h of exposure to sunlight and completely vanished after 16 h. ATR-IR spectrum in Fig. 1C shows a broad band of 3200–3400 cm−1, which is due to O–H stretching vibrations. GO shows peak centred at 1026 cm−1 (epoxy or alkoxy C–O), 1251 cm−1 (epoxy C–O), 1419 cm−1 (carboxyl C–O), 1650 cm−1 (aromatic CC), 1744 cm−1 (CO of carboxylic acid). Upon exposure of GO to sunlight, most of the peaks correspond to oxygen functionalities are disappeared. Even the peaks intensity of carboxyl group at 1419 cm−1 is reduced significantly after 16 h of exposure. The reduced sample shows two prominent peaks centred at 1571 cm−1 and 1150 cm−1 and are due to the aromatic CC and C–O stretching, respectively.29
O bonds, respectively. The corresponding sRGO spectrum after exposing to sunlight for a period of 16 h is also shown in Fig. 1A (blue trace). Upon exposure to sunlight, the peak corresponding to π–π* transition of C–C bonds is red shifted to 263 nm and the shoulder peak at 302 nm is completely disappeared, which indicates the restoration of sp2 carbon structure. UV/Vis spectra in Fig. 1B shows the reduction of GO to sRGO with respect to time. The shoulder peak at 302 nm is started diminishing in less than 8 h of exposure to sunlight and completely vanished after 16 h. ATR-IR spectrum in Fig. 1C shows a broad band of 3200–3400 cm−1, which is due to O–H stretching vibrations. GO shows peak centred at 1026 cm−1 (epoxy or alkoxy C–O), 1251 cm−1 (epoxy C–O), 1419 cm−1 (carboxyl C–O), 1650 cm−1 (aromatic CC), 1744 cm−1 (CO of carboxylic acid). Upon exposure of GO to sunlight, most of the peaks correspond to oxygen functionalities are disappeared. Even the peaks intensity of carboxyl group at 1419 cm−1 is reduced significantly after 16 h of exposure. The reduced sample shows two prominent peaks centred at 1571 cm−1 and 1150 cm−1 and are due to the aromatic CC and C–O stretching, respectively.29
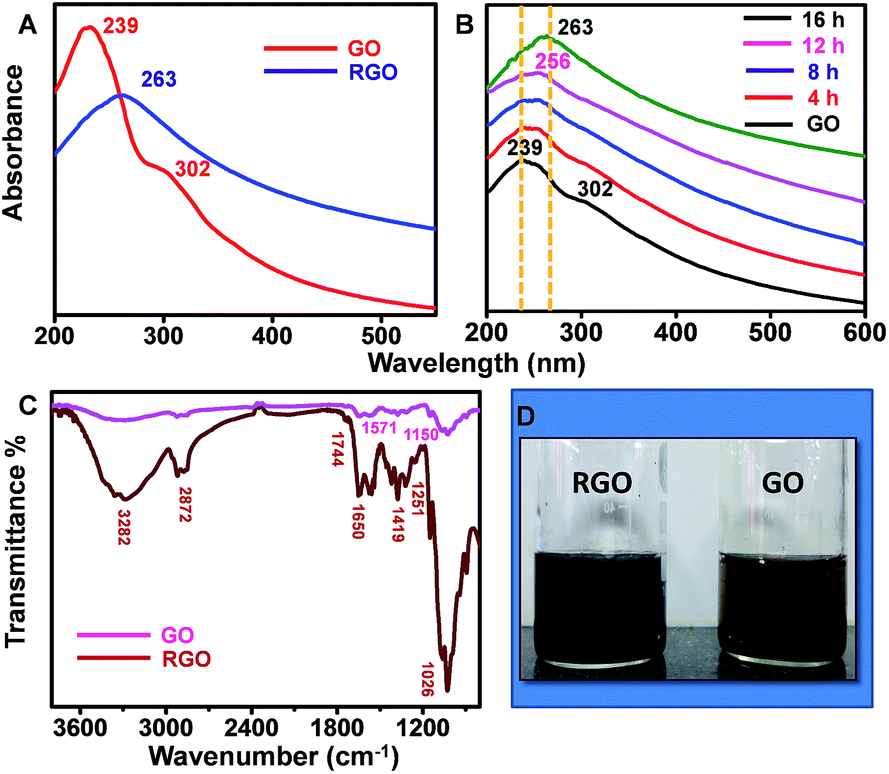 | ||
| Fig. 1 (A) UV/Vis spectrum of GO and sRGO (B) the kinetics data showing the transformation of GO to sRGO with respect to time (C) ATR-IR spectrum of GO and sRGO (D) photographic images of GO (right) and sRGO (left). Traces are shifted vertically for clarity. | ||
Raman spectroscopy was employed to study the structural changes in graphene.30 It also helps in correlating the electrical conductivity with the structural defects.31 The features like local defects and disorders at the edges are reflected in two peaks namely D band (∼1350 cm−1) and G band (∼1580 cm−1) of the Raman spectra. The D band is due to the breathing modes of sp2 atoms in the aromatic rings and the G band corresponds to the single phonon process at the Brillouin point. In GO, π bonds are replaced by σ carbon–oxygen/carbon–carbon bonds and the carbon atom hybridization change from sp2 to sp3, resulting in structural defects such as displacement of C atoms. The disappearance of the π electrons increases the band gap. The Raman spectrum in Fig. 2A shows the D and G bands of GO at 1345 cm−1 and 1601 cm−1, respectively. After reduction, the D band position is intact, but the G band is shifted to 1591 cm−1. The shift in the G band is attributed to the increase in carrier concentration and mobility after reduction.32 The peak located at ∼2683 cm−1 represents 2D band, which originates from a two phonon inter-valley double resonance mechanism. The D + D′ peak at 2923 cm−1 is due to the intra-valley scattering due to lattice defects. The shift in G band to a lower energy and a prominent increase in its 2D band confirm that GO sheets are converted to graphitic structures. The Fig. 2B compares Raman spectra of sRGO with hRGO and hyRGO. The enlarged 2D region of Raman spectra is shown in Fig. 2C. The figure clearly shows that there is a significant up-shift in the 2D band. The shift is more prominent in case of sRGO followed by hRGO. The up-shift variation of the 2D peak is due to the charge transfer with hole doping.33 For the systematic comparison of the charge transfer properties of RGO samples, ID/IG and I2D/IG ratio are measured and correlated. It is established that ID/IG ratio gives the degree of defects present in RGO samples and I2D/IG ratio indicates the recovery of sp2 CC bond in graphitic structure and hole mobility.34 Lower value of ID/IG ratio indicates lesser defects and higher value of I2D/IG ratio indicates higher charge mobility.35Table 1 summarises the ID/IG and I2D/IG values obtained for various RGO samples. On comparison of I2D/IG ratio, it is clear that sRGO has the highest conductivity and least structural defects followed by hRGO and hyRGO.
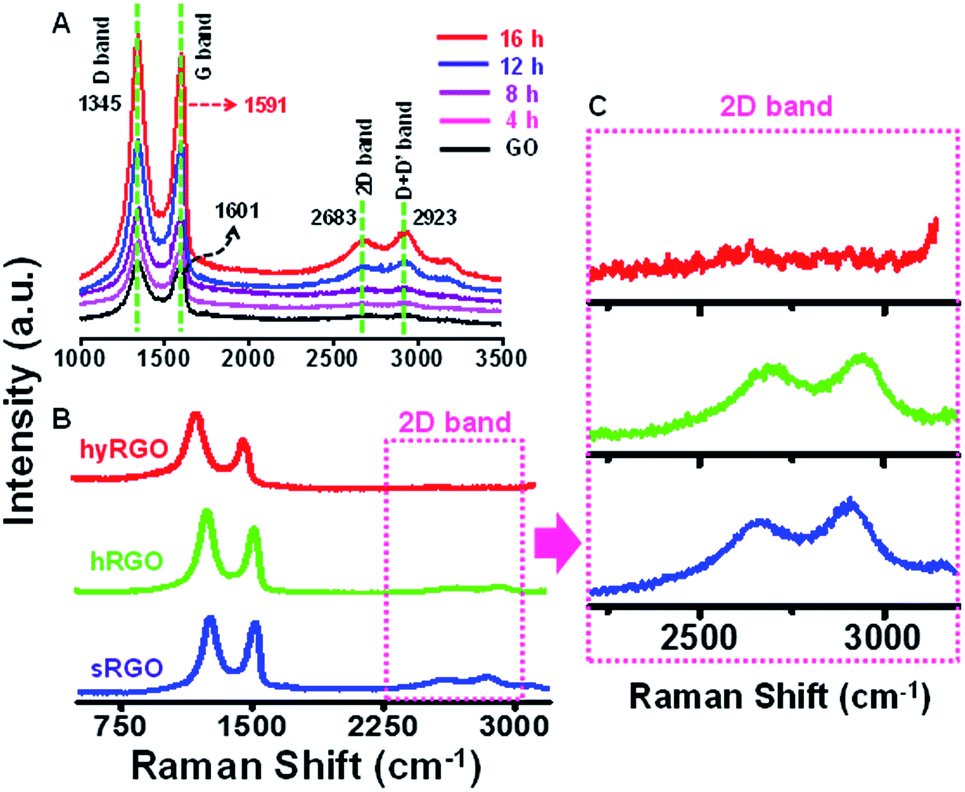 | ||
| Fig. 2 Raman spectra showing (A) the reduction of GO to sRGO with respect to time, (B) the comparison of Raman spectra of RGO samples synthesised by different reduction methods and (C) the enlarged 2D regions of various RGO samples. | ||
| Samples | I D/IG | I 2D/IG | Electrical conductivity (in S m−1) |
|---|---|---|---|
| sRGO | 1.016 | 0.136 | 166.34 |
| hRGO | 1.194 | 0.069 | 133.06 |
| hyRGO | 1.550 | 0.010 | 68.56 |
For further confirming the charge mobility behaviour, the electrical conductivities of various RGO samples were measured using four-probe method [for detailed calculations see S2 of ESI†]. The four-probes were placed collinearly and inter-electrode spacing of 2.5 mm was maintained. The current was passed through the two outer probes and the potential was measured between the two inner probes. The error due to electrical contacts are absent because the current and voltage leads are separate. The last column of Table 1 shows the electrical conductivities of various RGO samples. We can see that the conductivity data is consistent with Raman data.
Fig. 3 shows XPS spectra of carbon and oxygen content before and after photo-reduction. XPS peaks were fitted to Voigt functions having 80% Gaussian and 20% Lorentzian character, after performing Shirley background subtraction.36 The binding energy of C sp2 group of graphene lies at 284.5 eV or 284.6 eV. The other carbon–oxygen groups are shifted with respect to C sp2. The peak positions at 286.4 eV and 287.1 eV corresponds to hydroxyl (C–OH) and epoxy (C–O–C) groups, respectively. The smaller components at 288.1 eV and 289.2 eV are due to carbonyl (CO) and carboxyl (COOH) groups, respectively.37 From the relative intensity of the peaks, it is clear that oxygen groups present in GO are mainly due to epoxy, aldehyde and ketone groups located in the basal plane of GO sheets.
 | ||
| Fig. 3 (A) XPS survey spectra of GO, hRGO, hyRGO and sRGO with their respective (B) deconvoluted C 1s and (C) deconvoluted O 1s plots. | ||
Upon reduction, there is a notable decrease in the intensity of the components associated to these basal plane groups. The deconvoluted C 1s spectra of various RGO samples (hRGO, hyRGO and sRGO) show maximum intense peak at 284.5 eV that confirms the restoration of C–C bonds after the reduction. The intensity of peaks at 286.4 eV, 287.2 eV and 288.1 eV corresponding to hydroxyl, epoxy and carbonyl groups, respectively decreases after the reduction. The peak at 289.1 eV, which is due to the carboxylic group, remains after reduction. These residual peaks can be seen in hRGO and sRGO, but diminishes completely in the case of hyRGO. The data is also consistent with stability of different RGO dispersions prepared through various reduction routes. The stability of the samples in water follows the increasing order of sRGO ≈ hRGO > hyRGO. This is evident from the zeta potential values (Table 1 of ESI 3†). The lower stability (low zeta potential value) of hyRGO can be due to complete reduction of carboxylic groups. It is reported that carboxylic acid groups are difficult to reduce by chemical or photochemical route. But, heating at 100–150 °C in hydrothermal condition can reduce the group significantly.38
The O 1s spectra were de-convoluted into six components that are assigned to quinone (530.5 eV), C–OH (531.9 eV), C–O–C (532.7 eV), CO (533.5 eV), C–O (534.3 eV) and water (535 eV).39 The reduction in oxygen functionalities varies with different reduction methods.
The relative atomic contents (in %) of carbon and oxygen components were calculated from the analysis of area under C 1s, O 1s survey spectra. The values are summarised in Table 3. The relative sensitivity factors of carbon and oxygen were obtained from CASA XPS standard table.40 The calculated C/O ratio of GO and RGO samples are summarised in Table 2.
| Samples | C (%) | O (%) | C/O |
|---|---|---|---|
| GO | 67.87 | 32.13 | 2.11 |
| hRGO | 81.50 | 16.72 | 4.87 |
| hyRGO | 81.81 | 18.19 | 4.49 |
| sRGO | 78 | 22 | 3.54 |
Tables 3 and 4 show the fitted results of carbon (C 1s) and oxygen (O 1s) XPS spectra of RGO samples, respectively. The values represent the absolute amount of carbon and oxygen content before and after reduction.41Table 3 shows that C sp2 component increases after reduction, indicating the restoration of the graphitic structure. Meanwhile, majority of C–O, C–OH groups are reduced considerably after reduction.
| B.E (eV) | 284.5 | 285.2 | 286.4 | 287.1 | 288.1 | 289.1 |
| Assignment | C sp2 | C sp3 | C–OH | C–O–C | CO |
C–OOH |
| GO | 29.57 | — | 15.12 | 19.34 | 0.45 | 3.34 |
| hRGO | 50.63 | 16.65 | 9.26 | 0.96 | 2.77 | 1.16 |
| hyRGO | 51.92 | 17.61 | 8.52 | 0.84 | 2.90 | — |
| sRGO | 40.03 | 14.52 | 11.58 | 4.70 | 4.44 | 2.71 |
| B.E (eV) | 530.5 | 531.9 | 532.7 | 533.5 | 534.3 | 535 |
| Assignment | Quinone | C–OH | C–O–C | CO |
C–O | H2O |
| GO | 1.24 | 5.46 | 19.44 | 4.54 | 0.65 | — |
| hRGO | 1.68 | 4.16 | 6.21 | 3.77 | 0.92 | 0.27 |
| hyRGO | 1.94 | 4.50 | 6.37 | 3.76 | 1.26 | 0.34 |
| sRGO | 1.71 | 9.54 | 7.63 | 2.74 | — | 0.35 |
The fitted results of O 1s XPS spectra in Table 4 shows that the CO peak decreases upon reduction whereas an increase in the quinone and C–O peak is observed. This may be due to the transformation of the unstable oxygen groups to stable components during reduction process.41
Attempts were also done to assess the thermal conductivities of RGO samples. Equal concentrations of RGO samples (0.001 wt%) were dispersed in distilled water and were subjected to thermal conductivity measurements by Prok2D probe at room temperature (30 ± 0.5 °C). The results are compared against distilled water to check the possible enhancement in the thermal conductivity. It is observed that all the samples tested showed comparable enhancement in the conductivity.
The data is summarised in Table 5. The dimensions of the graphene flakes including shape, size and the stability of nanofluids plays a major role in describing the physics behind the heat conduction. Sen Gupta et al.,42 observed that, at lower concentrations of graphene dispersion, Brownian motion plays dominant role in deciding the thermal conductivity of the nanofluids. However, at higher concentrations, percolation starts dominating. In the present experiments, the shape and size of RGO sheets are comparable as we have used same GO as the starting material irrespective the reduction methods. However, the presence of functional group can vary with respect to the reducing agents used. At low concentration, the dispersibility of hRGO and sRGO are comparable and thus showed more or less similar results. The presence of less functional group in hyRGO and associated reduction in dispersibility may be the reason for the observed small reduction in the thermal conductivity of hyRGO compared to its counterparts. But one may expect a different behaviour in thermal conductivity at higher concentrations as the dominant mechanism involved is percolation and not the Brownian motion. It is worth to note that different reduction strategies result in different amounts and types of functional groups and hence likely change in thermal conductivity. But more investigations are required to establish the differences.
| Samples | Thermal conductivity (W m K−1) | Enhancement (%) |
|---|---|---|
| DI water | 0.670 | — |
| hRGO | 0.710 | 5.9 |
| sRGO | 0.709 | 5.8 |
| hyRGO | 0.706 | 5.4 |
Fig. 4A and B represent HRTEM images of RGO showing a sheet like morphology at different magnifications. The folding and the wrinkles that characterize the presence of 2D graphene sheets43 are marked with white arrows in the Fig. 4A. The HRTEM data in Fig. 4B shows the grain boundary of RGO sheet marked as [1] represents 2 layered sheet and [2] represents 10 layered sheets. Fig. 4C shows AFM topological image and the corresponding height profile of the image is shown in Fig. 4D, which is taken on the horizontal blue line shown in the figure. The height image reveals the presence of 2–4 layers of RGO corresponding to 0.7–1.4 nm height. There are regions that show height profile of a few layers of RGO sheets one above another. This may be due to stacking of layers during the drying process of the specimen used for the analysis. This is also in consistent with HRTEM data.
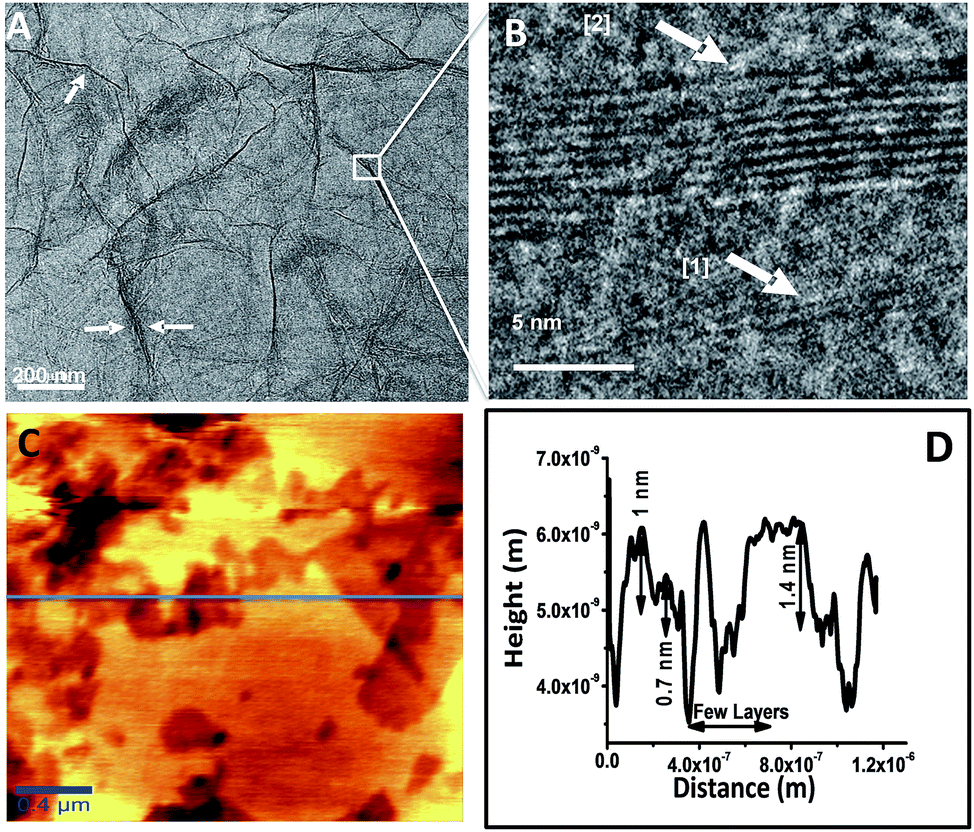 | ||
| Fig. 4 (A) HRTEM image of sRGO sheets (white arrows marked in the figure represents the wrinkles) (B) the grain boundary of RGO sheets of 2 and 10 layers magnified at the specified location (C) AFM image of sRGO and (D) height profile of sRGO sheets (along the blue line in (C)). | ||
Proposed photo-reduction mechanism of GO to RGO
The synthesis of GO involves oxygenation of graphite with the loss of π electrons of the carbon atoms. This covalent functionalization converts sp2 to sp3 configuration. GO consist of both highly conducting sp2 regions and insulating or semi-conducting sp3 regions. The disappearance of π electrons opens the bandgap which can be tuned in proportion with the oxygen functional groups. Normally, during photo-reduction, the bandgap excitation of the semiconducting region is responsible for reduction process and not the thermal effect.44 Also, the absorbance of light depends on the bandgap. In the present study, the photo-reduction of GO with bandgap energy of 3.09 eV [see S4 of ESI† for details] is carried out by exposing the aqueous dispersion of 0.01% GO (pH ∼ 4) to sunlight. The liberation of H+ and OH− ions and solvated electrons during photodecomposition of water molecules can enhances the reduction process. The possible reduction mechanism of GO to RGO is explained.On exposure of GO to sunlight, it absorbs photons of energy equal to or larger than its bandgap in-order to generate electrons in the conduction band and holes in the valence band. These electrons and holes contribute to the redox reactions on GO sheet. The single bonded hydroxyl and epoxy groups attached to the basal plane are considered as highly reactive groups and hence the reduction of such groups happens readily.
The disappearances of these groups at early hours of the exposure to sunlight are evident from the Fig. 1B. A complete removal of these groups was observed within 8 h of exposure to sunlight. The possible pathways of reduction of these groups are described by the chemical reactions (R1) and (R2). Visible formation of gas bubbles in the reactor during the first 3 h of exposure to sunlight also supports a similar reduction route. The presence of functional groups in graphene and the liberation of CO2 during the reduction of GO are expected to increase the defects and in turn reduce the conductivity of graphene.45 The release of CO2 is also confirmed by decrease in total carbon content [see S5 of ESI† for details] with an increase in the inorganic carbon.46 However, the experimental evidences have shown a reverse behaviour. Interestingly, sRGO has showed enhanced conductivity compared to RGO samples prepared through other routes. This enhancement in conductivity can be due to the liberation of H2 at the defect region as given in the eqn (R3).
| GO (sp3 region) + hγ → e− + h+ |
| 2C–OH + H2O + 3h+ + e− → C (defects) + CO2 + H2O | (R1) |
| 2C–O–C + H2O + 2h+ + 2e− → C (defects) + C–C (defects) + CO2 + H2O | (R2) |
| 2H2O + C (defect) + 2e− + 2h+ → H2 + CO2 + H2O | (R3) |
It is to be noted that there is a tendency for the electropositive carbon defect to attract the highly electronegative oxygen atom of the water molecules (Fig. 5). This process can enable the liberation of H2 at the defect sites and re-establish the conjugated π networks and provide higher conductivity. As the irradiation duration is increased, the production of H2 also increased.47
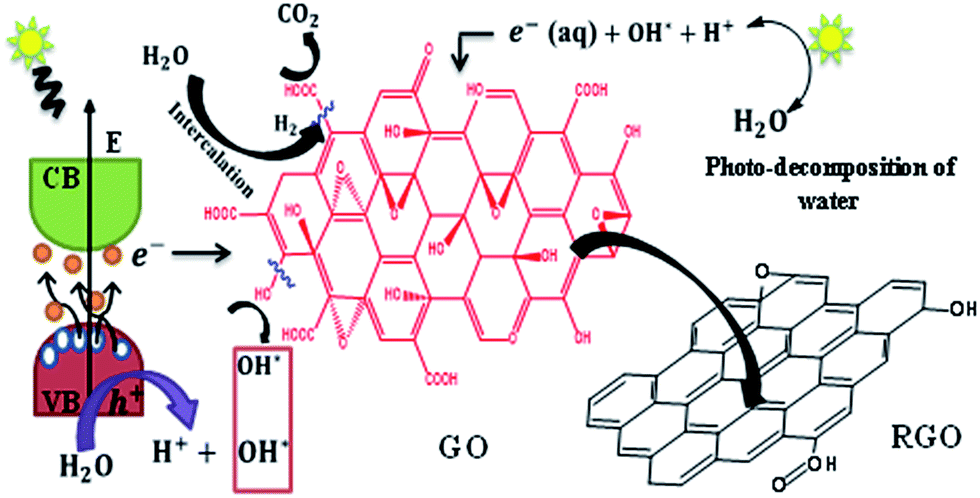 | ||
| Fig. 5 Schematic showing the formation of sRGO from GO. | ||
The possible enhanced release of H2 and corresponding re-establishment of the conjugated π networks can be the reason for the observed higher conductivity. The higher electrical conductivity of hRGO compared to other RGO samples is due to the nitrogen incorporation from the reducing agent (hydrazine monohydrate). The formation of hydrazones results in the removal of oxygen functional groups in hRGO.48
Conclusion
The reduction of GO by sunlight can be employed as an energy saving alternative for the large-scale preparation of RGO. The method is simple and sustainable compared to the conventional methods of reduction of GO. sRGO synthesized by this method exhibited higher electrical conductivity, least structural defects and good dispersibility compared to other RGO samples tested. The data shows that the proposed reduction route is feasible and a sustainable option to produce dispersible RGO with good electrical conductivity. The study also caution using GO in any device components which may undergo prolonged exposure to sunlight.Acknowledgements
Authors gratefully acknowledge the Department of Science and Technology (DST), Government of India, Research and development grant under water technology initiative (DST/WTI/2K14/228) for partially supporting the work. The authors also thank VIT University for supporting the work. Thanks are due to Mr Sudhakar, DST unit on Nanoscience and Technology, IIT Madras for helping in XPS analysis.References
- J. C. Meyer, A. K. Geim, M. Katsnelson, K. Novoselov, T. Booth and S. Roth, Nature, 2007, 446, 60–63 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- D. Sen, K. S. Novoselov, P. M. Reis and M. J. Buehler, Small, 2010, 6, 1108–1116 CrossRef CAS.
- J. Robinson, X. Weng, K. Trumbull, R. Cavalero, M. Wetherington, E. Frantz, M. LaBella, Z. Hughes, M. Fanton and D. Snyder, ACS Nano, 2009, 4, 153–158 CrossRef.
- D. Arco, L. Gomez, Y. Zhang, A. Kumar and C. Zhou, IEEE Trans. Nanotechnol., 2009, 8, 135–138 Search PubMed.
- A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2009, 9, 3087 CrossRef CAS.
- J. K. Wassei and R. B. Kaner, Mater. Today, 2010, 13, 52–59 CrossRef CAS.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- K.-H. Liao, A. Mittal, S. Bose, C. Leighton, K. A. Mkhoyan and C. W. Macosko, ACS Nano, 2011, 5, 1253–1258 CrossRef CAS PubMed.
- T. Sreeprasad, S. M. Maliyekkal, K. Lisha and T. Pradeep, J. Hazard. Mater., 2011, 186, 921–931 CrossRef CAS.
- S. H. Anderson and D. D. L. Chung, Carbon, 1984, 22, 253–263 CrossRef CAS.
- C. Botas, P. Álvarez, C. Blanco, R. Santamaría, M. Granda, M. D. Gutiérrez, F. Rodríguez-Reinoso and R. Menéndez, Carbon, 2013, 52, 476–485 CrossRef CAS.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- M. J. Fernández-Merino, J. I. Paredes, S. Villar-Rodil, L. Guardia, P. Solís-Fernández, D. Salinas-Torres, D. Cazorla-Amorós, E. Morallón, A. Martínez-Alonso and J. M. D. Tascón, Carbon, 2012, 50, 3184–3194 CrossRef.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- X. Mei, X. Meng and F. Wu, Phys. E, 2015, 68, 81–86 CrossRef CAS.
- D. Zhan, Z. Ni, W. Chen, L. Sun, Z. Luo, L. Lai, T. Yu, A. T. S. Wee and Z. Shen, Carbon, 2011, 49, 1362–1366 CrossRef CAS.
- Y. Matsumoto, M. Koinuma, S. Y. Kim, Y. Watanabe, T. Taniguchi, K. Hatakeyama, H. Tateishi and S. Ida, ACS Appl. Mater. Interfaces, 2010, 2, 3461–3466 CrossRef CAS.
- P. Kumar, K. Subrahmanyam and C. Rao, Int. J. Nanosci., 2011, 10, 559–566 CrossRef CAS.
- H. Wang, J. T. Robinson, X. Li and H. Dai, J. Am. Chem. Soc., 2009, 131, 9910–9911 CrossRef CAS.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- S. Jin, Q. Gao, X. Zeng, R. Zhang, K. Liu, X. Shao and M. Jin, Diamond Relat. Mater., 2015, 58, 54–61 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS.
- D. Konios, M. M. Stylianakis, E. Stratakis and E. Kymakis, J. Colloid Interface Sci., 2014, 430, 108–112 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- D. M. Basko, S. Piscanec and A. C. Ferrari, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 165413 CrossRef.
- S. Eigler, F. Hof, M. Enzelberger-Heim, S. Grimm, P. Müller and A. Hirsch, J. Phys. Chem. C, 2014, 118, 7698–7704 CrossRef CAS.
- Y. Su, J. Du, D. Sun, C. Liu and H. Cheng, Nano Res., 2013, 6, 842–852 CrossRef CAS.
- C.-Y. Su, Y. Xu, W. Zhang, J. Zhao, X. Tang, C.-H. Tsai and L.-J. Li, Chem. Mater., 2009, 21, 5674–5680 CrossRef CAS.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field and C. A. Ventrice, Carbon, 2009, 47, 145–152 CrossRef CAS.
- L. Stobinski, B. Lesiak, A. Malolepszy, M. Mazurkiewicz, B. Mierzwa, J. Zemek, P. Jiricek and I. Bieloshapka, J. Electron Spectrosc. Relat. Phenom., 2014, 195, 145–154 CrossRef CAS.
- Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950–2956 CrossRef CAS.
- S. Rella, A. Giuri, C. E. Corcione, M. R. Acocella, S. Colella, G. Guerra, A. Listorti, A. Rizzo and C. Malitesta, Vacuum, 2015, 119, 159–162 CrossRef CAS.
- V. Štengl, S. Bakardjieva, T. M. Grygar, J. Bludská and M. Kormunda, Chem. Cent. J., 2013, 7, 1–12 CrossRef.
- C.-M. Chen, Q. Zhang, M.-G. Yang, C.-H. Huang, Y.-G. Yang and M.-Z. Wang, Carbon, 2012, 50, 3572–3584 CrossRef CAS.
- S. Sen Gupta, V. Manoj Siva, S. Krishnan, T. S. Sreeprasad, P. K. Singh, T. Pradeep and S. K. Das, J. Appl. Phys., 2011, 110, 084302 CrossRef.
- S. M. Maliyekkal, T. Sreeprasad, D. Krishnan, S. Kouser, A. K. Mishra, U. V. Waghmare and T. Pradeep, Small, 2013, 9, 273–283 CrossRef CAS.
- B. Konkena and S. Vasudevan, J. Phys. Chem. C, 2015, 119, 6356–6362 CrossRef CAS.
- M. Agharkar, S. Kochrekar, S. Hidouri and M. A. Azeez, Mater. Res. Bull., 2014, 59, 323–328 CrossRef CAS.
- W.-C. Hou, I. Chowdhury, D. G. Goodwin Jr, W. M. Henderson, D. H. Fairbrother, D. Bouchard and R. G. Zepp, Environ. Sci. Technol., 2015, 49, 3435–3443 CrossRef CAS.
- Y. Matsumoto, M. Koinuma, S. Ida, S. Hayami, T. Taniguchi, K. Hatakeyama, H. Tateishi, Y. Watanabe and S. Amano, J. Phys. Chem. C, 2011, 115, 19280–19286 CrossRef CAS.
- P. Zhu, M. Shen, S. Xiao and D. Zhang, Phys. B, 2011, 406, 498–502 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24696f |
| This journal is © The Royal Society of Chemistry 2017 |