
Synthesis of tricyclic 3-hydroxyisoindolin-1-ones via triethylamine-catalyzed domino reactions of electron-deficient alkynes with phthalimidomalonate derivatives†
Li
Zhang
,
Beichen
Zhang
,
Teng
Jiang
,
Tao
Lu
* and
Qingfa
Zhou
*
State Key Laboratory of Natural Medicines, Department of Organic Chemistry, China Pharmaceutical University, Nanjing, 210009, P. R. China. E-mail: zhouqingfa@cpu.edu.cn
First published on 25th October 2016
Abstract
A triethylamine-catalyzed domino reaction between phthalimidomalonate derivatives and activated alkynes has been developed. This catalytic protocol is applicable to the synthesis of structurally functionalized N-fused tricyclic 3-hydroxyisoindolin-1-ones.
The development of new methods that can construct diversely nitrogen-containing heterocycles remains a preeminent goal and a great challenge in the pharmaceutical and agrochemical industries. Towards this target, domino reactions are among the most powerful processes in synthetic organic chemistry, allowing rapid generation of molecular complexity and diversity in a one-pot transformation from easily available starting materials.
Isoindolones are structural motifs that are widely present in natural products and medicinally important agents. In particular, 3-hydroxyisoindolin-1-ones bearing a hydroxyl group at the 3-position are key structural elements that appear in the core structures of a lot of naturally occurring isoindolone alkaloids and pharmaceutically relevant compounds (Fig. 1).1 For instance, chilenine (1), an isoindole-containing benzazepine, is isolated from Chilean barberries.1a Chlorphthalidone (2) has been widely used as an antihypertensive agent.1c The isoindolin-1-one derivative (3) has demonstrated antibacterial activity.1e Other interesting 3-hydroxyisoindolone derivatives have also shown inhibition of the MDM2-p53 protein–protein interaction,2a HIV integrase2b as well as cyclin-dependent kinases (CDKs).2c In addition, 3-hydroxyisoindolones are also versatile building blocks that can undergo synthetically useful transformations to other heterocycles.3 However, only a few approaches have been reported thus far to construct this novel N-fused structure, which mainly included (1) nucleophilic addition, reduction or photolysis reactions of phthalimide derivatives;4 (2) cyclization of ortho functional benzoic acid derivatives.5 Despite these presented methods for the synthesis of isoindolin-1-ones, it is in high demand to develop new and efficient protocols for the synthesis of 3-hydroxyisoindolin-1-one derivatives due to the unique status of 3-hydroxyisoindolin-1-ones in pharmaceutical chemistry and synthetic chemistry.
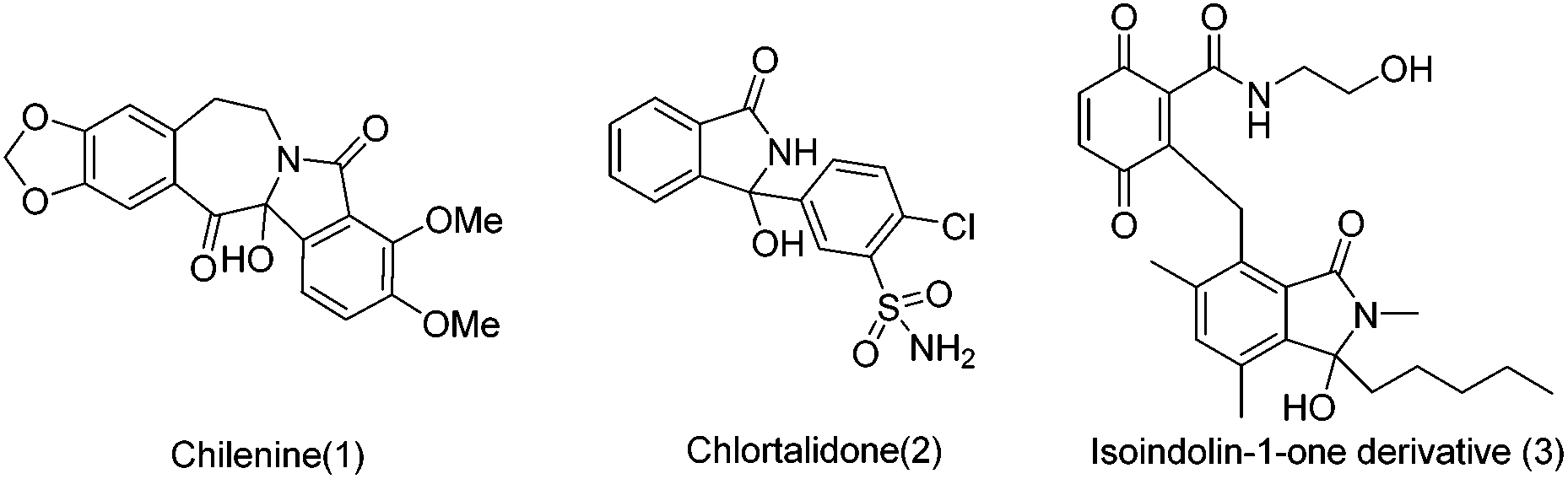 | ||
| Fig. 1 The related structures of 3-hydroxyisoindolin-1-ones. | ||
Yavari and Esmaili reported a strategy for the synthesis of tetraalkyl 2,3-dihydro-5-oxopyrrolo[2,1-a]isoindole-1,2,3,3-tetracarboxylates using dialkyl acetylenedicarboxylate, dialkyl phthalimidomalonates and a stoichiometric amount of triphenylphosphine (Scheme 1a).6 Very recently, we found that phosphine can effectively catalyse the [3 + 2] annulation of electron-deficient alkynes with phthalimide derivatives for the synthesis of 3-hydroxyisoxazolo[3,2-a]isoindol-8(3aH)-ones (Scheme 1b).7 Based on these earlier studies, we hypothesized that phthalimidomalonates could react with electron-deficient alkynes in the presence of a phosphine catalyst to yield 3-hydroxyisoindolin-1-ones. To our surprise, no product was found when the reaction was disposed with a phosphine catalyst, but the expected tricyclic 3-hydroxyisoindolin-1-one was formed under the influence of a tertiary amine catalyst (Scheme 1c). To the best of our knowledge, this is the first example of an amine-catalyzed domino reaction of activated alkynes with an amide as electrophile partners, although the reactivity of electron-deficient alkynes with aldehyde, imine or activated ketone electrophiles to construct azacyclic compounds has been studied extensively.8 Herein, we wish to report a novel example of amine-catalyzed domino reactions of phthalimidomalonates with electron-deficient alkynes for the construction of highly functionalized 3-hydroxyisoindolin-1-one derivatives.
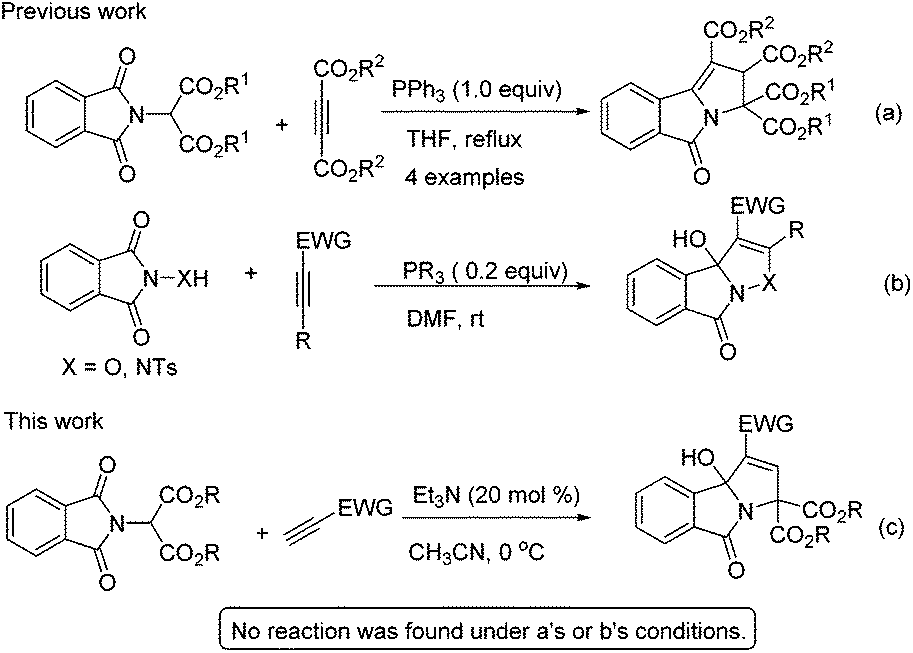 | ||
| Scheme 1 Base-mediated synthesis of isoindolones using phthalimide derivatives. | ||
As a test case for the construction of 3-hydroxyisoindolin-1-one derivatives, we subjected the nucleophile diethyl 2-(1,3-dioxoisoindolin-2-yl)malonate (1a) and benzyl propiolate (2a) to our previously reported cyclization reaction conditions: i.e., 20 mol% of triphenylphosphine in DMF at room temperature (Table 1, entry 1). No product was formed. Even when a stronger nucleophilic base, Bu3P, was used as a catalyst, no product was found (entry 2). To our delight, the 3-hydroxyisoindolin-1-one (3a) with multiple functional groups was obtained with a moderate yield, when triethylamine was applied in the reaction system (entry 3). To improve the chemical yield, various solvents were examined. Acetonitrile proved to be the most efficient reaction medium, providing the 3-hydroxyisoindolin-1-one in 51% isolated yield after 24 h (entry 4). The product 3a was obtained in 13% yield when EtOAc was used as a solvent. Other solvents, such as CH2Cl2, toluene, THF, 1,4-dioxane and ether, were infeasible (entries 5–10). Further screening of bases revealed that triethylamine was the optimal base (entries 11–15). DBU could also effectively promote cyclization and give a matched yield. The desired product was given only in 8% yield when DMAP was used as a promoter. No reaction was occurred when the reaction was carried out in the presence of pyridine. Further investigation showed that the temperature was an important parameter for the formation of 3a (entries 16–19). For example, when the reaction is run at 0 °C, 3a was obtained in 55% yield; no reaction was found when the reaction was performed at −35 °C even with a prolonged reaction time. The starting material diethyl 2-(1,3-dioxoisoindolin-2-yl)malonate (1a) was consumed quickly, but the reaction became very complex when the reaction was proceeded at 60 °C. Decreasing the loading of the catalyst Et3N to 10 mol% did affect the reaction rate and the yield to a certain extent (entry 20). No reaction was found when the reaction was performed without Et3N.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Solvent | Temp. [°C] | Time [h] | Yieldb [%] |
| a Unless noted otherwise, reaction of diethyl 2-(1,3-dioxoisoindolin-2-yl)malonate (1a) (0.4 mmol, 122 mg) and benzyl propiolate (2a) (0.8 mmol, 128 mg) was performed in 5 mL of solvent under N2. b Isolated yield based on 1a. c Only the Michael addition product was formed. | |||||
| 1 | Ph3P (20) | DMF | 25 | 24 | NR |
| 2 | Bu3P (20) | DMF | 25 | 24 | NR |
| 3 | Et3N (20) | DMF | 25 | 24 | 40 |
| 4 | Et3N(20) | CH3CN | 25 | 24 | 51 |
| 5 | Et3N (20) | CH2Cl2 | 25 | 12 | 5 |
| 6 | Et3N (20) | Toluene | 25 | 12 | 7 |
| 7 | Et3N (20) | EA | 25 | 24 | 13 |
| 8 | Et3N (20) | THF | 25 | 48 | NR |
| 9 | Et3N (20) | 1,4-Dioxane | 25 | 48 | NR |
| 10 | Et3N (20) | Ether | 25 | 48 | NR |
| 11 | NaOH (20) | CH3CN | 25 | 24 | 62c |
| 12 | PPh3 (20) | CH3CN | 25 | 24 | Trace |
| 13 | Pyridine (20) | CH3CN | 25 | 36 | NR |
| 14 | DBU (20) | CH3CN | 25 | 24 | 50 |
| 15 | DMAP (20) | CH3CN | 25 | 24 | 8 |
| 16 | Et3N (20) | CH3CN | 40 | 1.5 | 45 |
| 17 | Et3N (20) | CH3CN | 60 | 0.5 | 43 |
| 18 | Et3N (20) | CH3CN | 0 | 100 | 55 |
| 19 | Et3N (20) | CH3CN | −35 | 200 | NR |
| 20 | Et3N (10) | CH3CN | 0 | 100 | 32 |
| 21 | Et3N (40) | CH3CN | 0 | 100 | 51 |
| 22 | Et3N (0) | CH3CN | 0 | 100 | 0 |
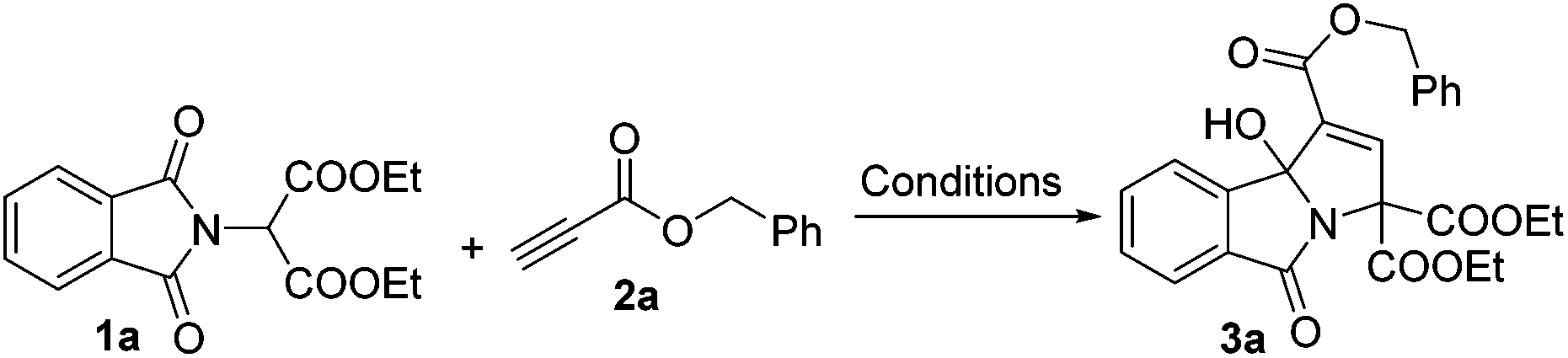
With the optimized reaction conditions in hand, a variety of 3-hydroxyisoindolin-1-one derivatives were efficiently synthesized in good chemical yields (Table 2). Generally, all terminal alkynoates were proved to be applicable to this reaction under the optimized reaction conditions. The nature of the substituent on the benzene ring of the benzyl propiolate had a great effect on the yields. For example, for a substrate with a strong electron-donating substituent methoxy group attached on the benzene ring, the yield of the corresponding product became very low, while 4-(trifluoromethyl)benzyl propiolate gave the product with a very good yield. The reaction was equally effective at producing the corresponding product in good yield when a 2-naphthyl group was introduced. Gratifyingly, the benzene ring of benzyl propiolate could be replaced with heteroaryl groups, such as furan-2-yl or thiophen-2-yl, and gave the desired products in good yields. The alkynoates bearing little alkyl units, namely ethyl and methyl, could give the products in 53 and 50% yields respectively and the alkynoate with a branched bulky alkyl group, such as t-butyl, gave the product in a very low yield. No product was formed when a β-substituted alkynoate was applied in the reaction. This protocol could be readily extended to various alkynyl ketones. For example, using the same reaction conditions and starting with but-3-yn-2-one, we isolated the corresponding 3-hydroxyisoindolin-1-one 3p in 87% yield. However, the reaction became very complex when 1-phenylprop-2-yn-1-one was used in the reaction (Scheme 2).
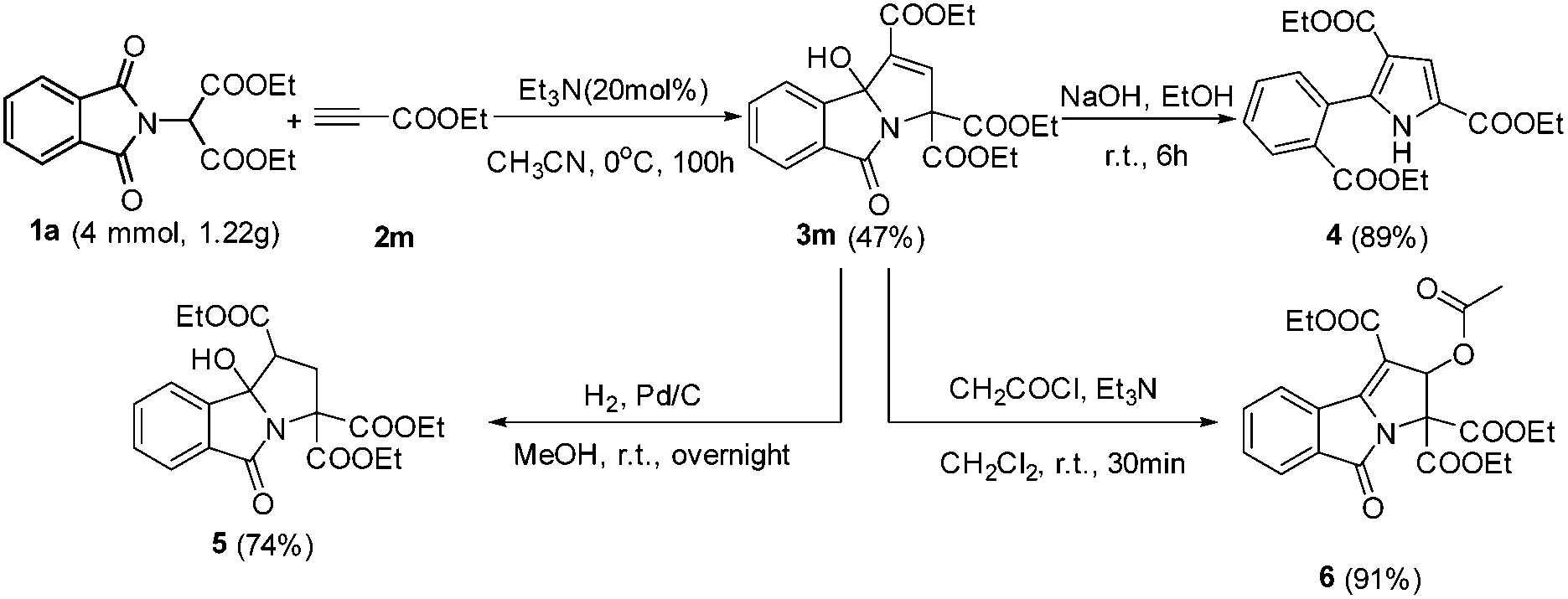 | ||
| Scheme 2 Gram-scale synthesis of 3-hydroxyisoindolin-1-one 3m and further transformations. | ||
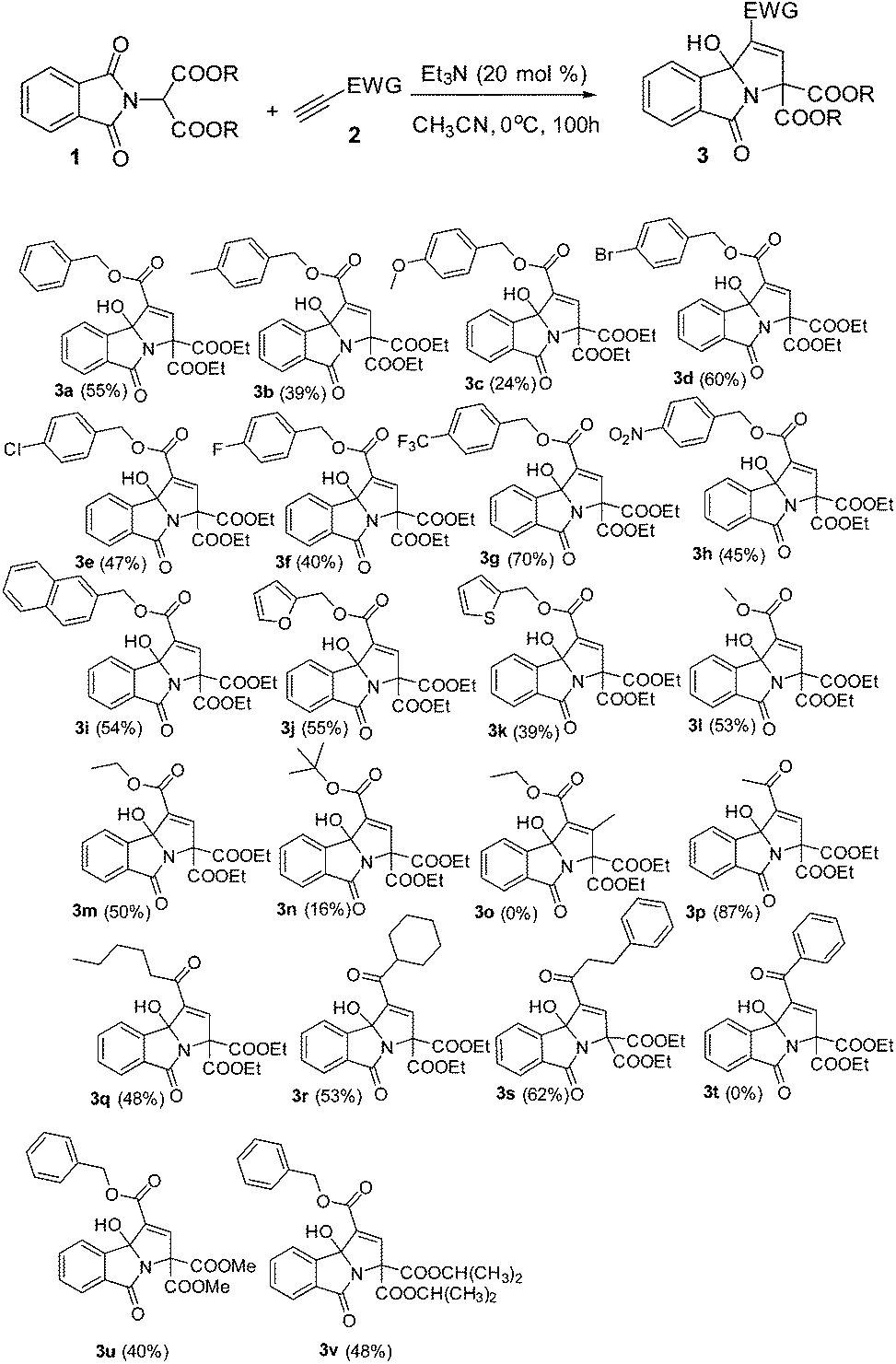
No reaction was found when N,N-diethylpropiolamide or N,N-dibenzylpropiolamide was used as a reaction partner in place of terminal alkynoates. Furthermore, different phthalimidomalonates, such as dimethyl 2-(1,3-dioxoisoindolin-2-yl)malonate and diisopropyl 2-(1,3-dioxoisoindolin-2-yl)malonate, also worked nicely with ethyl propiolate in the presence of Et3N (20 mol%), providing 3u and 3v with good yields. However, other CH-active units in place of malonate, such as ethyl 2-(1,3-dioxoisoindolin-2-yl)-3-oxobutanoate or 2-(2,4-dioxopentan-3-yl)isoindoline-1,3-dione, were used as a reaction partner, but no product was formed.
The structures of the products were undeniably confirmed by X-ray crystallographic analysis of compound 3m.9 The ORTEP diagram of 3m is shown in Fig. 2.
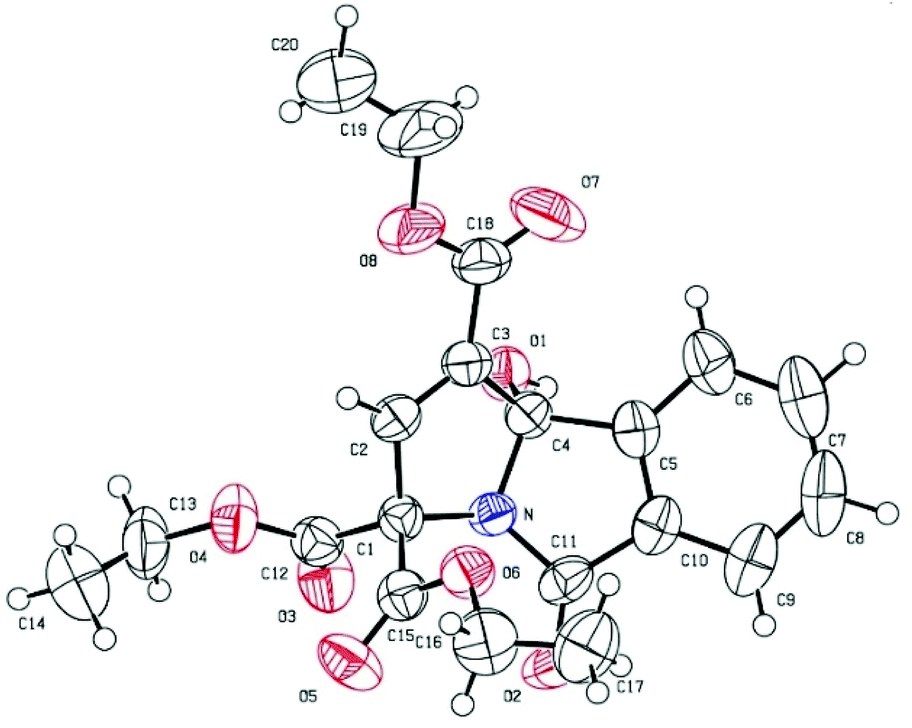 | ||
| Fig. 2 X-ray crystal structure of 3m. | ||
To demonstrate the synthetic potential of this catalytic system, the gram-scale preparation of highly functionalized 3-hydroxyisoindolin-1-one 3m was investigated. The reaction of 4 mmol of the starting material (1a) proceeded smoothly, delivering the corresponding product 3m without a significant loss of efficiency (small scale, 50%) (Scheme 3). The 3-hydroxyisoindolin-1-ones can be readily converted into various interesting compounds (Scheme 3). First, treatment of 3m with NaOH in EtOH afforded the functional 1H-pyrrole 4 in 89% yield. Next, the 3-hydroxyisoindolin-1-one 3m was treated with Pd/C under the atmosphere of H2 in MeOH at room temperature, leading to isoindolin-1-one 5 in 74% yield. Interestingly, triethyl 2-acetoxy-5-oxo-2,5-dihydro-3H-pyrrolo[2,1-a]isoindole-1,3,3-tricarboxylate 6 was obtained by treatment of 3m with AcCl and Et3N in CH2Cl2 at room temperature.
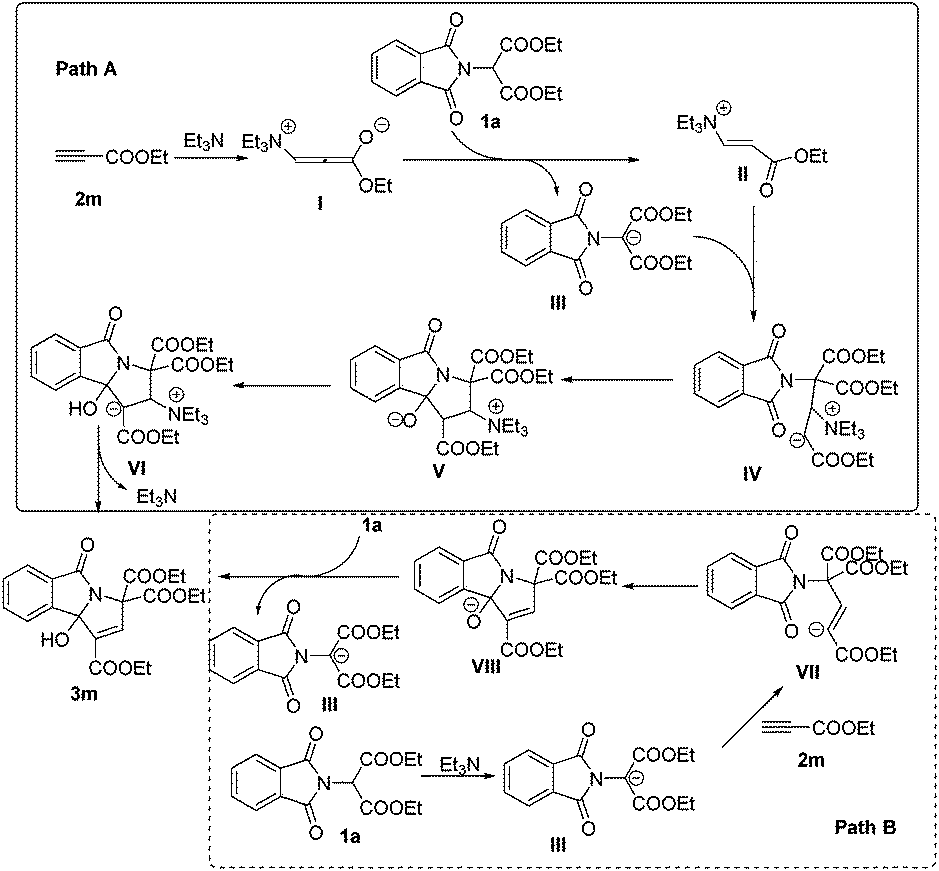 | ||
| Scheme 3 Mechanism for the formation of 3-hydroxyisoindolin-1-one derivatives 3m. | ||
At present, the reaction mechanism is not clear. However, on the basis of our experimental observations and known literature, we tentatively proposed two mechanisms for the formation of 3-hydroxyisoindolin-1-one derivatives 3 in Scheme 3. For path A, the nucleophile triggers the reaction by Michael type nucleophilic conjugate addition at the carbon–carbon triple bond of 2m to produce vinylammonium carbanion I, which can then deprotonate phthalimidomalonate 1a to generate intermediates II and III. Intermediate III then undergoes Michael addition to II to give IV. Intermediate IV might then undergo intramolecular nucleophilic attack to form intermediate V, followed by proton transfer and elimination of Et3N to produce the desired product 3m. For path B, Et3N abstracts a proton from an active methylene of 1a to form the nucleophilic carbanion III. Addition of this anion to the carbon–carbon triple bond of 2m generates intermediate VII. Intramolecular addition in VII produces intermediate VIII, which abstracts a proton from phthalimidomalonate 1a to give intermediate III and product 3m.
Conclusions
In summary, we have developed a triethylamine-catalyzed domino reaction of phthalimidomalonate derivatives with various activated alkynes under mild reaction conditions to give the 3-hydroxyisoindolin-1-one derivatives. This reaction could be performed on the gram scale and is a practical protocol. Subsequent transformations provided a concise access to various heterocycles and pharmaceutically attractive compounds.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant no. 21102179, 21572271 and 81473078), the Qing Lan Project, the project-sponsored by SRF for ROCS, SEM and National Found for Fostering Talents of Basic Science (Grant no. J1030830).References
- (a) G. Kim, P. Jung and A. T. Le, Tetrahedron Lett., 2007, 49, 2391 CrossRef; (b) M. H. A. Zarga, S. S. Sabri, S. Firdous and M. Shamma, Phytochemistry, 1987, 26, 1233 CrossRef; (c) J. G. Topliss, L. M. Konzelman, N. Sperber and F. E. Roth, J. Med. Chem., 1964, 7, 453 CrossRef CAS PubMed; (d) B. R. Davis, J. A. Cutler, C. D. Furberg, J. T. Wright, M. A. Farber and J. V. Felicetta, Ann. Intern. Med., 2002, 137, 313 CrossRef CAS PubMed; (e) A. Mikolasch, S. Hessel, M. G. Salazar, H. Neumann, K. Manda, D. Gördes, E. Schmidt, K. Thurow, E. Hammer, U. Lindequist, M. Beller and F. Schauer, Chem. Pharm. Bull., 2008, 56, 781 CrossRef CAS PubMed.
- (a) I. R. Hardcastle, S. U. Ahmed, H. Atkins, G. Farnie, B. T. Golding, R. J. Griffin, S. Guyenne, C. Hutton, P. Källblad, S. J. Kemp, M. S. Kitching, D. R. Newell, S. Norbedo, J. S. Northen, R. J. Reid, K. Sarvanan, H. M. G. Willems and J. Lunec, J. Med. Chem., 2006, 49, 6209 CrossRef CAS PubMed; (b) J. M. Chen, X. Chen, M. Fardis, H. Jin, C. U. Kim and L. N. Schacherer, World Pat, WO2004035577-A3, 2004 Search PubMed; Chem. Abstr. 2004 140 375156 Search PubMed; (c) T. Honma, N. Yoshizumi, K. Hayashi, N. Kawanishi, K. Fukasawa, T. Takaki, C. Ikeura, M. Ikuta, T. I. Suzuki, T. Hayama, S. Nishimura and H. Morishima, J. Med. Chem., 2001, 44, 4628 CrossRef CAS PubMed.
- (a) G. Blaskó, D. J. Gula and M. Shamma, J. Nat. Prod., 1982, 45, 105 CrossRef; (b) L. Li, M. Wang, X. Zhang, Y. Jiang and D. Ma, Org. Lett., 2009, 11, 1309 CrossRef CAS PubMed; (c) M. Lamblin, A. Couture, E. Deniau and P. Grandclaudon, Org. Biomol. Chem., 2007, 5, 1466 RSC; (d) S. Couty, C. Meyer and J. Cossy, Tetrahedron Lett., 2006, 47, 767 CrossRef CAS; (e) S. Couty, B. Liegault, C. Meyer and J. Cossy, Tetrahedron, 2006, 62, 3882 CrossRef CAS; (f) P. Pigeon and B. Decroix, Tetrahedron Lett., 1996, 37, 7707 CrossRef CAS; (g) Y. Koseki and T. Nagasaka, Chem. Pharm. Bull., 1995, 43, 1604 CrossRef CAS.
- (a) T. Bootwicha, D. Panichakul, C. Kuhakarn, S. Prabpai, P. Kongsaeree, P. Tuchinda, V. Reutrakul and M. Pohmakotr, J. Org. Chem., 2009, 74, 3798 CrossRef CAS PubMed; (b) T. Bousquet, J. F. Fleury, A. Daïch and P. Netchitaïlo, Tetrahedron, 2006, 62, 706 CrossRef CAS; (c) A. G. Griesbeck, K. D. Warzecha, J. M. Neudörfl and H. Görner, Synlett, 2004, 2347 CrossRef CAS; (d) M. Othman, P. Pigeon and B. Decroix, Tetrahedron, 1997, 53, 2495 CrossRef CAS; (e) C. H. Yeh, Y. C. Lin, S. Mannathan, K. Hung and C. H. Cheng, Adv. Synth. Catal., 2014, 356, 831 CrossRef CAS; (f) H. N. Nguyen, V. J. Cee, H. L. Deak, B. Du, K. P. Faber, H. Gunaydin, B. L. Hodous, S. L. Hollis, P. H. Krolikowski, P. R. Olivieri, V. F. Patel, K. Romero, L. B. Schenkel and S. D. Geuns-Meyer, J. Org. Chem., 2012, 77, 3887 CrossRef CAS PubMed.
- (a) G. Kim, P. Jung and L. A. Tuan, Tetrahedron Lett., 2008, 49, 2391 CrossRef CAS; (b) Y. Zhou, Y. Zhai, J. Li, D. Ye, H. Jiang and H. Liu, Green Chem., 2010, 12, 1397 RSC; (c) S. Sharma, E. Park, J. Park and I. S. Kim, Org. Lett., 2012, 14, 906 CrossRef CAS PubMed; (d) X. X. Zheng, C. Du, X. M. Zhao, X. Zhu, J. F. Suo, X. Q. Hao, J. L. Niu and M. P. Song, J. Org. Chem., 2016, 81, 4002 CrossRef CAS PubMed; (e) L. Yang, L. Han, B. Xu, L. Zhao, J. Zhou and H. Zhang, Asian J. Org. Chem., 2016, 5, 62 CrossRef CAS.
- I. Yavari and A. A. Esmaili, J. Chem. Res., 1998, 714 RSC.
- (a) Q. F. Zhou, X. P. Chu, F. F. Ge, Y. Wang and T. Lu, Adv. Synth. Catal., 2013, 355, 2787 CrossRef CAS; (b) Q. Chen, K. Li, T. Lu and Q. Zhou, RSC Adv., 2016, 6, 24792 RSC; (c) X. Chen, F. F. Ge, T. Lu and Q. F. Zhou, J. Org. Chem., 2015, 80, 3295 CrossRef CAS PubMed.
- (a) D. Tejedor, S. López-Tosco, G. Méndez-Abt, L. Cotos and F. García-Tellado, Acc. Chem. Res., 2016, 49, 703 CrossRef CAS PubMed; (b) G. L. Zhao and M. Shi, J. Org. Chem., 2005, 70, 9975 CrossRef CAS PubMed; (c) C. H. Chen, G. S. Yellol, P. T. Lin and C. M. Sun, Org. Lett., 2011, 13, 5120 CrossRef CAS PubMed; (d) Y. G. Wang, S. L. Cui and X. F. Lin, Org. Lett., 2006, 8, 1241 CrossRef CAS PubMed; (e) P. D. Armas, F. García-Tellado, J. J. Marrero-Tellado, D. Tejedor, M. A. Maestro and J. Gonzalez-Platas, Org. Lett., 2001, 3, 1905 CrossRef PubMed; (f) D. Tejedor, S. López-Tosco, J. González-Platas and F. García-Tellado, J. Org. Chem., 2007, 72, 5454 CrossRef CAS PubMed; (g) D. T. Aragón, G. V. López, F. García-Tellado, J. J. Marrero-Tellado, P. D. Armas and D. Terrero, J. Org. Chem., 2003, 68, 3363 CrossRef PubMed; (h) S. L. Cui, X. F. Lin and Y. G. Wang, Eur. J. Org. Chem., 2006, 5174 CrossRef CAS; (i) D. Tejedor, F. García-Tellado, J. J. Marrero-Tellado and P. D. Armas, Chem. – Eur. J., 2003, 9, 3122 CrossRef CAS PubMed; (j) D. Tejedor, D. González-Cruz, A. Santos-Expósito, J. J. Marrero-Tellado, P. D. Armas and F. García-Tellado, Chem. – Eur. J., 2005, 11, 3502 CrossRef CAS PubMed; (k) D. Tejedor, L. Cotos and F. García-Tellado, Org. Lett., 2011, 13, 4422 CrossRef CAS PubMed; (l) D. Tejedor, G. Méndez-Abt, J. González-Platas, M. A. Ramírez and F. García-Tellado, Chem. Commun., 2009, 17, 2368 RSC.
- CIF files have also been deposited with the Cambridge Crystallographic Data Centre as “supplementary publication no. CCDC 1493448 for compound 3m”.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1493448. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00544f |
| This journal is © the Partner Organisations 2017 |