
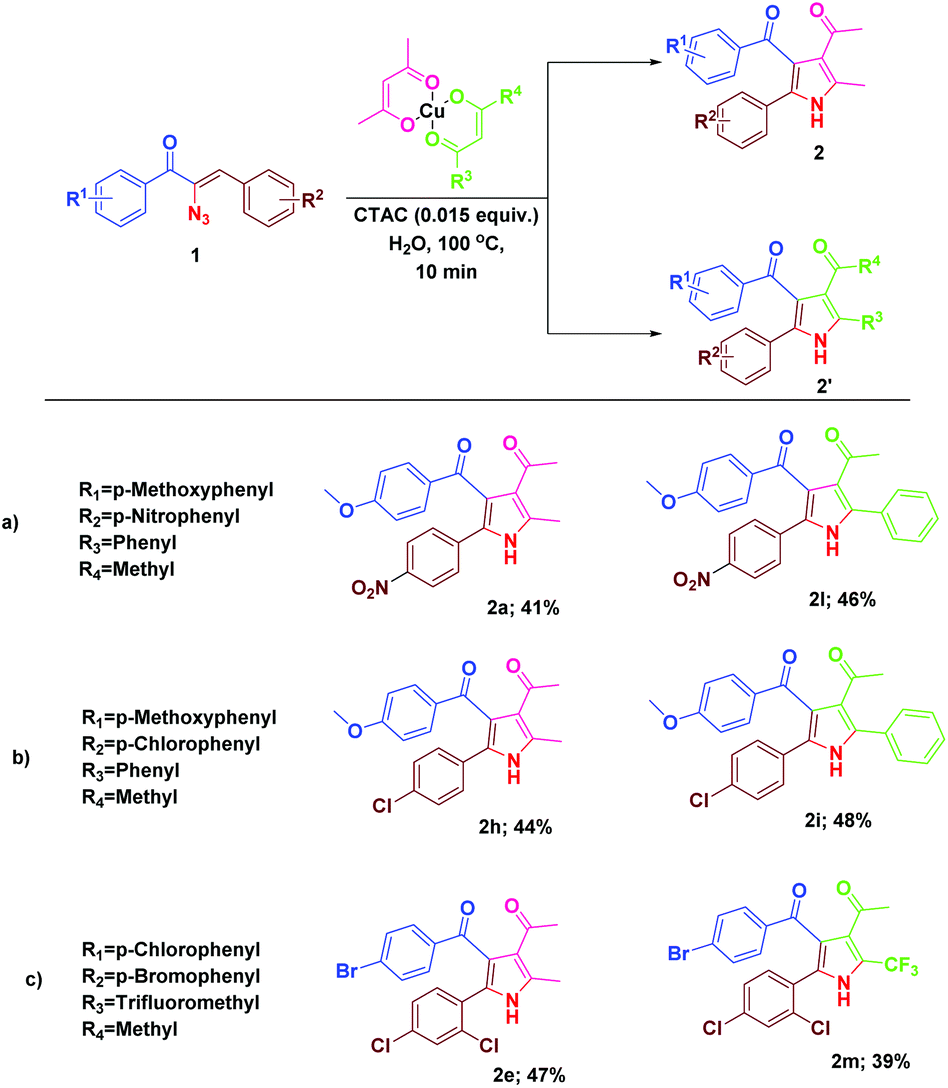
Divergent reactivity of α-azidochalcones with metal β-diketonates: tunable synthesis of substituted pyrroles and indoles†
Kandasamy
Rajaguru
a,
Arumugam
Mariappan
a,
Shanmugam
Muthusubramanian
*a and
Nattamai
Bhuvanesh
b
aDepartment of Organic Chemistry, School of Chemistry, Madurai Kamaraj University, Madurai - 625 021, India. E-mail: muthumanian2001@yahoo.com
bX-ray Diffraction Laboratory, Department of Chemistry, Texas A & M University, College Station, Texas 77842, USA
First published on 25th October 2016
Abstract
A divergent reactivity of α-azidochalcones with metal β-diketonates for the synthesis of substituted pyrroles and indoles is described. Metal β-diketonates have been applied as bifunctional reactive partners. With a copper complex, the synthesis of substituted pyrroles in micellar media via 2H-azirine intermediates has been achieved under mild conditions, while with Fe(acac)2 as a catalyst, the reaction proceeds smoothly to yield indoles regioselectively.
Introduction
The advancement of synthetic methods for synthesizing the privileged heterocyclic frameworks has acquired substantial consideration by synthetic chemists.1 Aza heterocycles are indispensable frameworks in many natural products with an extensive array of bioactivity. Particularly pyrrole and its benzo-fused analogue indole are widespread structural entities in biologically active natural and unnatural components, as well as in other functional materials (Fig. 1).2 The contradictory synthetic strategy seems to be common and expedient in the construction of aza heterocycles, demanding an understanding of the reactivity of intermediates chemoselectively.3 Many protocols involve direct C–N bond formation by the loss of H2, N2, or H2O to construct complex nitrogen heterocycles.4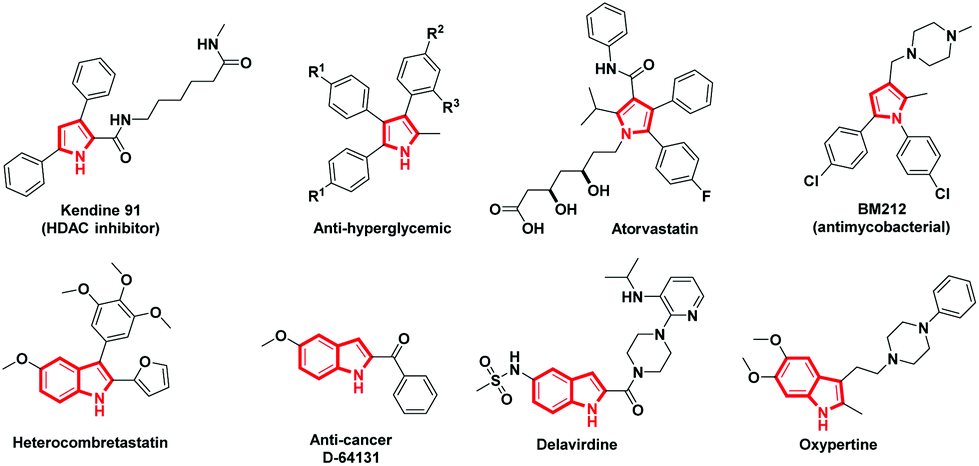 | ||
| Fig. 1 Examples of biologically active pyrrole and indole moieties. | ||
In recent times, vinyl azides are being focused largely for their inimitable properties which permit them to act as a nucleophile, electrophile, or radical acceptor for the synthesis of diverse heterocycles.5 It is common that vinyl azides undergo photolysis or thermolysis to yield highly strained three-membered 2H-azirines, which can act as building blocks in organic synthesis.6 α-Azidochalcones in particular retain distinctive chemical reactivity and aid as resourceful synthons for the synthesis of numerous aza heterocycles.7 The synthesis of indoles and carbazoles can be achieved by the intramolecular C–H amination by photolysis or thermal decomposition of azides. Still, these methods suffer from harsh conditions with an inadequate scope.8 Metal β-diketonates are gaining substantial attention as they can be used as catalysts in organic synthetic transformations and have enormous material applications.9
Results and discussion
Earlier we have reported the indium trichloride catalyzed synthesis of substituted pyrrole in water.7b In continuation of our exploitation of α-azidochalcones as precursors for aza heterocycles, the regioselective syntheses of pyrroles have been reinvestigated under milder reaction conditions compared to the reported methods.10 Apart from that, the regioselective synthesis of indoles10 has also been accomplished (Scheme 1). α-Azidochalcones can be synthesized from the corresponding chalcones in two steps following the literature method.7b,e,11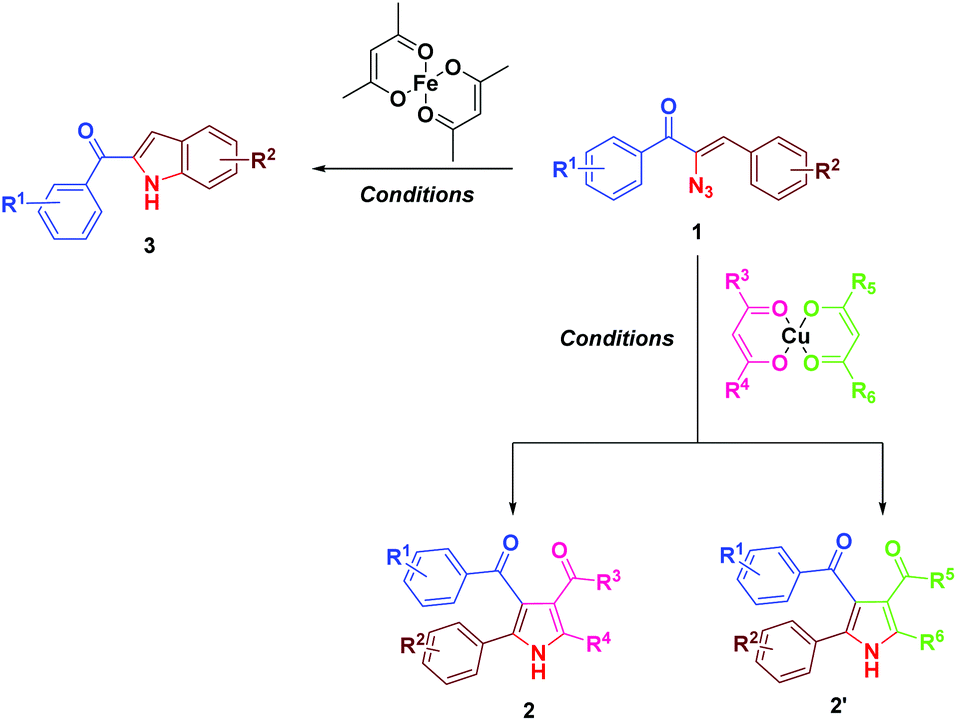 | ||
| Scheme 1 Synthesis of aza heterocycles 2 and 3. | ||
As a green reaction medium, water has several advantages as it is copiously accessible in nature, virtually safe and environment friendly among all the existing solvents.12 However, the solubility of most of the organic reactants in water is an issue that affects the execution of reactions in aqueous media. The introduction of aqueous surfactants by the materialization of micelles enriches the reactivity of water mediated reactions.13
During the course of our investigation for the construction of aza heterocycles from α-azidochalcones, we have intended to synthesise indoles from α-azidochalcones through the intramolecular C–H amination strategy in water using copper(II) acetylacetonate as a catalyst. Accordingly, we have initiated our experiment with α-azidochalcone 1 (R1 = 4-Cl, R2 = 2,4-di-OCH3) and Cu(acac)2 (0.1 equiv.) at 100 °C in a micellar medium employing cetyl trimethyl ammonium chloride (CTAC) (0.015 equiv.) as a surfactant for 12 h, but there was no product formation. The reaction was continued by increasing the catalytic loading upto 0.4 equiv. at 100 °C and decreasing the reaction time to 10 minutes. After completion of the reaction, a solid was obtained, when the reaction mixture was treated with a saturated solution of ammonium chloride and subsequently recrystallized from ethanol.14 Upon characterization by using NMR and mass spectral data, the product formed was found to be the substituted pyrrole 2a and not the expected indole 3a. The acetylacetonate component from Cu(acac)2 has been incorporated with an acceptable yield of 2a (Table 1, entries 2 and 3). A relatively good yield of pyrrole 2a (94%) was obtained by increasing the amount of Cu(acac)2 to 0.5 equiv. under the same reaction conditions (Table 1, entry 4).
|
|
||||
|---|---|---|---|---|
| S. no. | Catalyst (equiv.) | Conditions | Yielda % | |
| 2a | 3a | |||
| a Isolated yield. b Reaction under a nitrogen atmosphere. c Surfactant: 0.015 equiv. | ||||
| 1b | Cu(acac)2 (0.1) | H2O, CTAC,c 100 °C, 12 h | — | — |
| 2 | Cu(acac)2 (0.2) | H2O, CTAC,c 100 °C, 10 min | 31 | — |
| 3 | Cu(acac)2 (0.4) | H2O, CTAC,c 100 °C, 10 min | 66 | — |
| 4 | Cu(acac)2 (0.5) | H2O, CTAC,c 100 °C, 10 min | 94 | — |
| 5 | Ni(acac)2 (0.5) | H2O, CTAC,c 100 °C, 1 h | 66 | — |
| 6 | Fe(acac)2 (0.5) | H2O, CTAC,c 100 °C, 1 h | 73 | — |
| 7b | Cu(acac)2 (0.5) | CH3CN, 80 °C, 8 h | 52 | — |
| 8 | Cu(acac)2 (0.5) | DMF, 100 °C, 3 h | 43 | — |
| 9 | Cu(acac)2 (0.5) | Dioxane, 80 °C, 3 h | 36 | — |
| 10 | Cu(acac)2 (0.5) | n-BuOH, 80 °C, 8 h | 68 | — |
| 11 | Ni(acac)2 (0.5) | CH3CN, 80 °C, 8 h | 43 | — |
| 12b | Fe(acac)2 (0.5) | CH3CN, 80 °C, 8 h | 64 | 23 |
| 13b | Fe(acac)2 (0.2) | Toluene, 100 °C, 12 h | 12 | 73 |
| 14b | Fe(acac)2 (0.01) | DCE, 80 °C, 7 h | — | 53 |
| 15b | Fe(acac)2 (0.02) | DCE, 80 °C, 4 h | — | 87 |
| 16b | Fe(acac)3 (0.2) | DCE, 80 °C, 3 h | 33 | — |
| 17b | Pd(acac)2 (0.2) | DCE, 80 °C, 16 h | — | — |
| 18b | Ni(acac)2 (0.2) | DCE, 80 °C, 16 h | — | — |
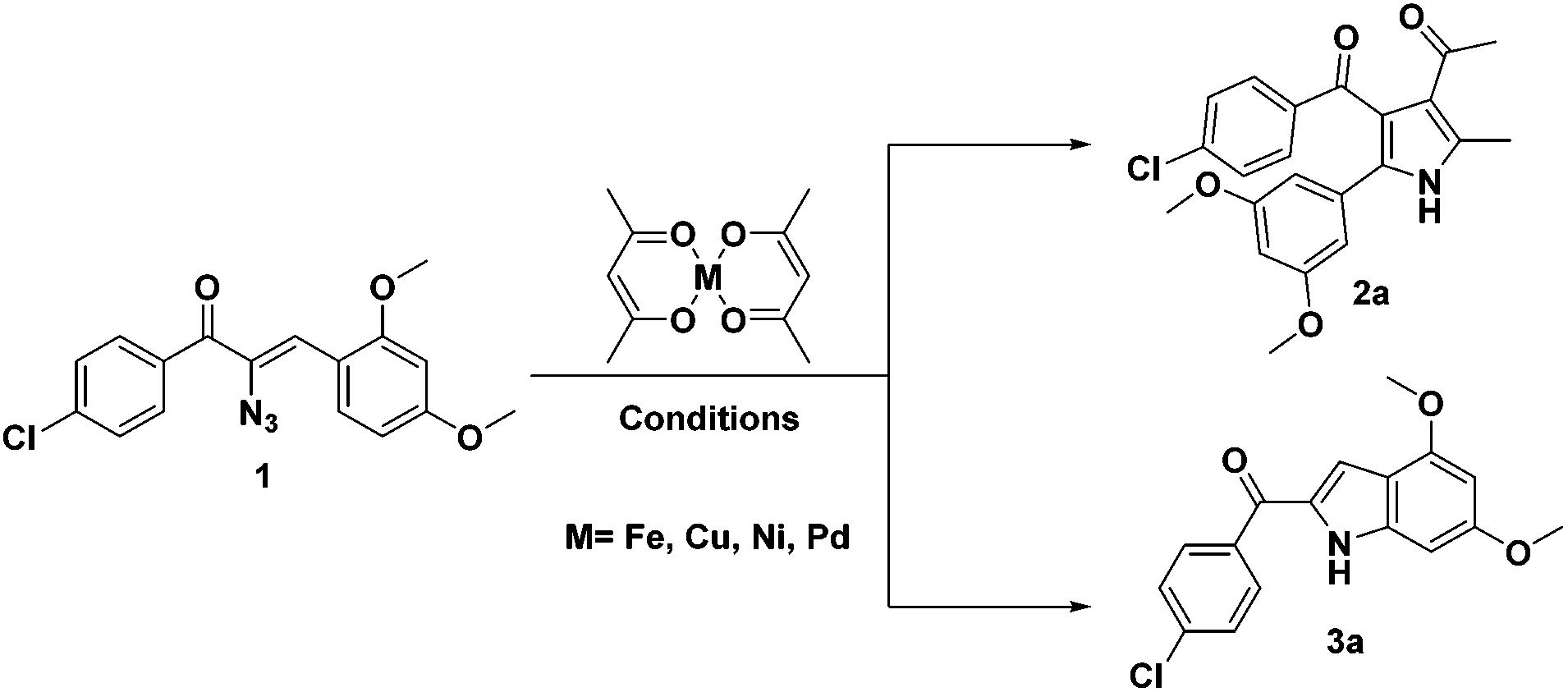
The investigation was further carried out by changing a number of experimental variables such as solvents, catalysts and temperature, but there is no improvement in the yield of 2a (Table 1, entries 5–11). When the experiment was carried out with 0.5 equiv. of Fe(acac)2 in acetonitrile at 80 °C for 8 h, the formation of pyrrole 2a (64%) was accompanied by a minor amount of substituted indole 3a (23%) (Table 1 entry 12). Attempts to improve the yield of 3a over 2a were made and when the reaction was carried out in toluene at 100 °C for 12 h with a reduced amount of Fe(acac)2 (0.2 equiv.), the formation of 3a was appreciable (73%) with a poor yield of 2a (12%) (Table 1, entry 13). Exclusive formation of 3a was possible in dichloroethane at 80 °C in 4 h with a very small amount of Fe(acac)2 (Table 1, entry 15). Nickel and palladium diketonates did not participate in the reaction, either as the catalyst or as the substrate (Table 1, entries 17 and 18).
With the optimal reactivity of α-azidochalcones with copper β-diketonates, the scope of the reaction was extended towards the synthesis of substituted pyrroles 2a–2o (Table 2). The reaction proficiency was not greatly affected by the electron donating or withdrawing substituents at different positions of the aryl rings of α-azidochalcones. Under the optimized conditions, the reaction is much compatible with α-azidocinnamate as well (Table 2).
| a Reaction conditions: α-azidochalcone (0.10 mmol), metal-β-diketonate (0.5 mmol) and CTAC (0.015 equiv.) in water (5 mL) heated at 100 °C for 10 min under open air. |
|---|
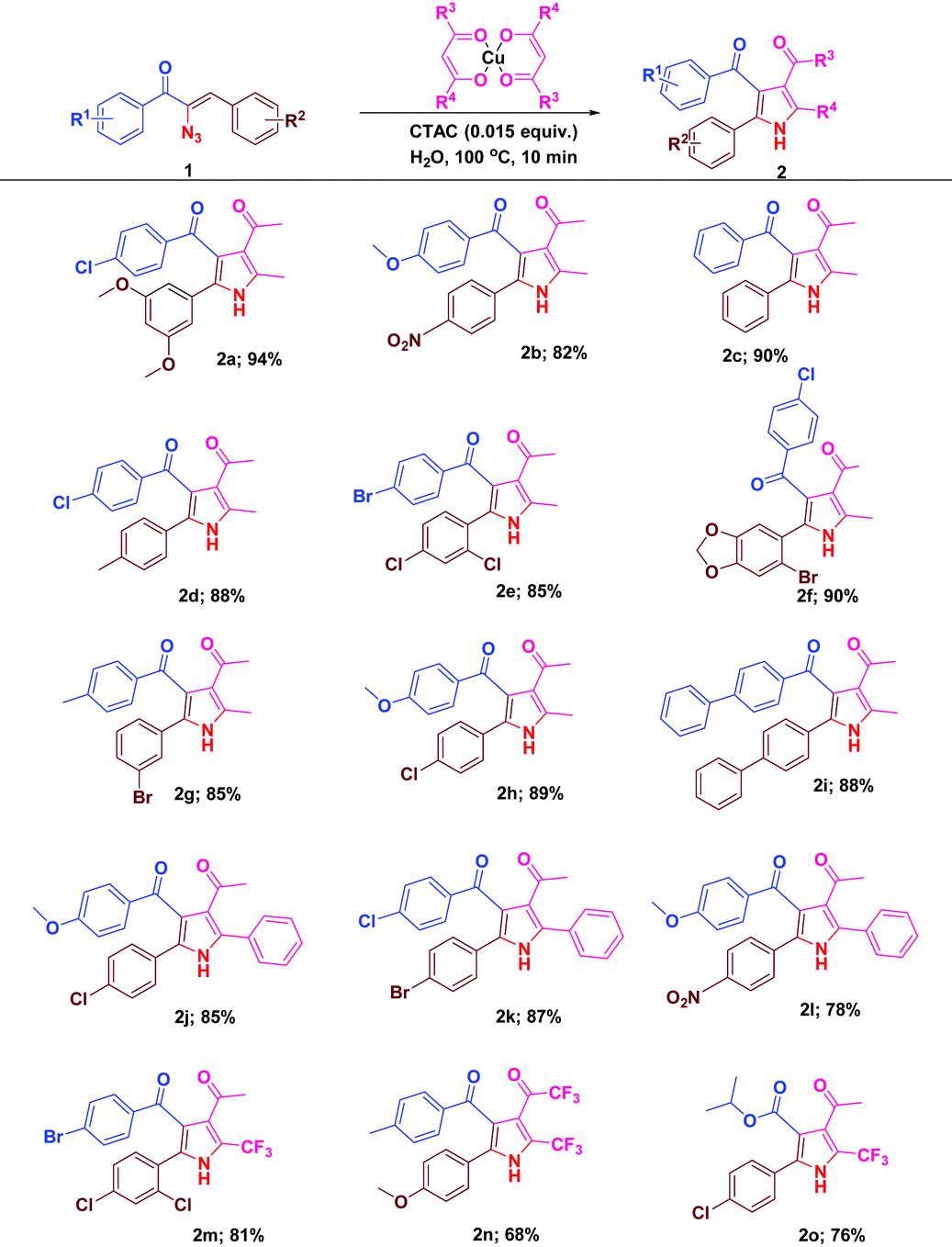
|
We were then prompted to explore the generality of this pyrrole formation using mixed ligand complexes with different β-diketone ligands, which could be easily prepared in a straightforward manner.15 The reaction of α-azidochalcones and the mixed ligand complexes of β-diketonates under the optimized reaction conditions went on successfully yielding mixtures of substituted pyrroles in appreciable yields (Table 3).
| a Reaction conditions: α-azidochalcone (0.10 mmol), mixed ligand complexes of metal-β-diketonate (0.5 mmol) and CTAC (15 mol%) in water (5 mL) heated at 100 °C for 10 min under open air. |
|---|
|
The mechanism for the formation of substituted pyrroles 2 is given in Scheme 2. Initially, thermolysis of α-azidochalcone results in denitrogenative decomposition to form a cyclic imine, a highly strained three membered 2H-azirine, 4. The solvolysis of metal β-diketonate followed by the addition of intermediate 4 results in the aziridine adduct 5, which then undergoes intramolecular nucleophilic addition on the carbonyl group with a nitrogen lone pair, yielding 2,5-dihydro-1H-pyrrole intermediate 6. Further isomerization yields the substituted pyrrole 2 ultimately.10c
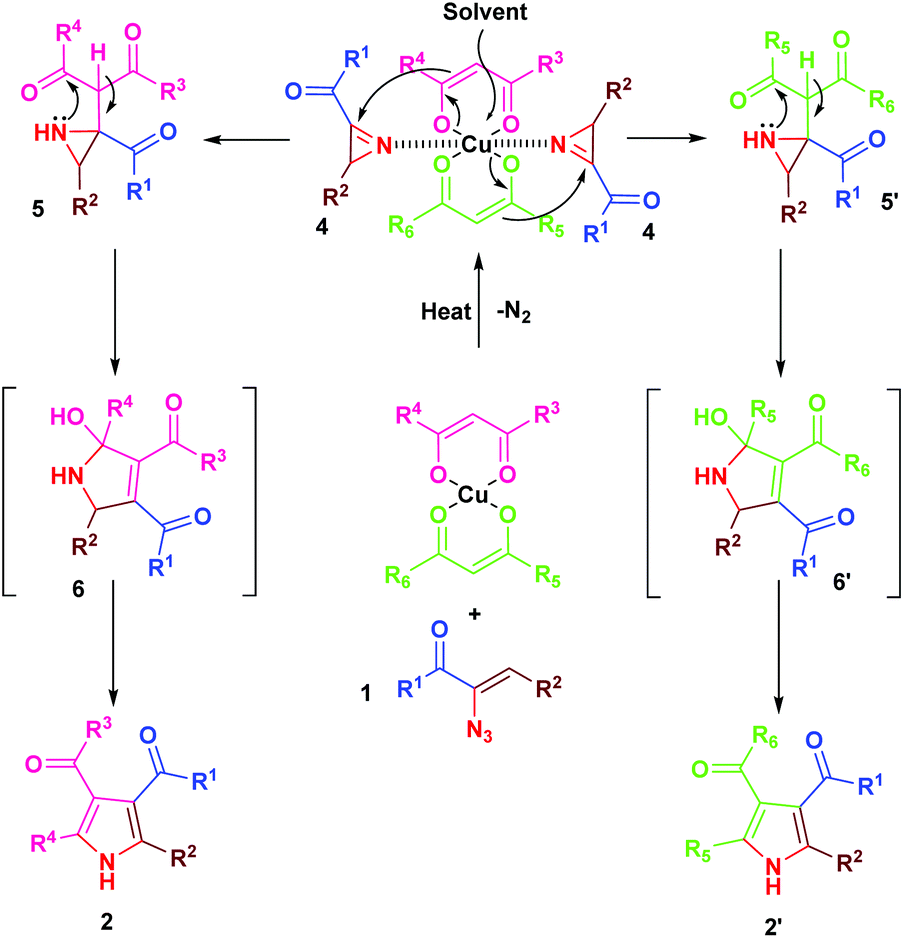 | ||
| Scheme 2 Mechanism for the formation of pyrroles 2. | ||
Regarding the mode of cyclization involving the unsymmetrical β-diketone-aziridine adduct 5, a lone pair of electrons on the nitrogen prefers the more electrophilic carbonyl carbon and leads to the respective substituted pyrrole regioselectively (Scheme 3). This is well supported by the single crystal X-ray structures of 2b and 2j (Fig. 2) with the expected regioisomers in each case.16
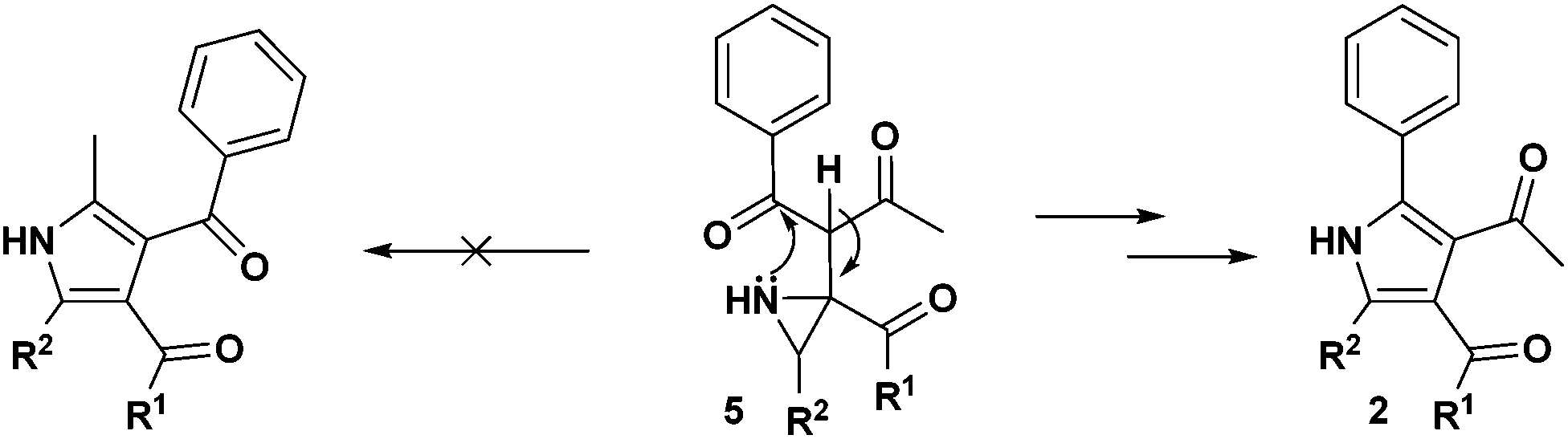 | ||
| Scheme 3 Regiochemical preferences in cyclization for pyrrole 2. | ||
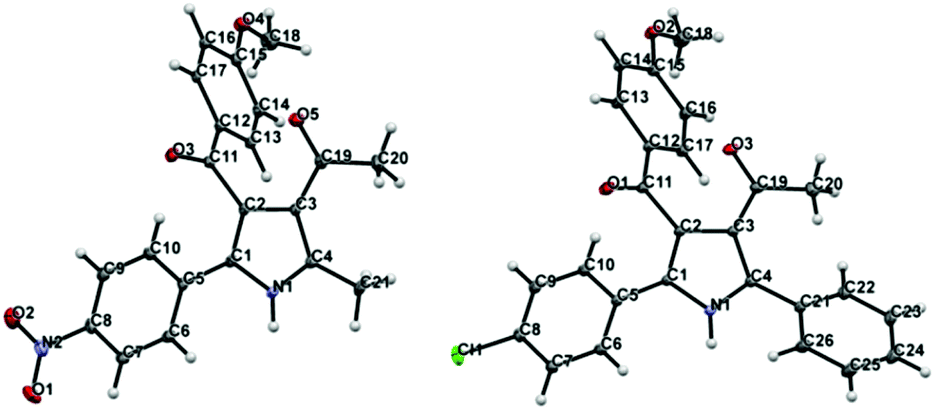 | ||
| Fig. 2 X-ray crystal structures of 2b and 2j. Thermal ellipsoids are shown at 50% probability level. | ||
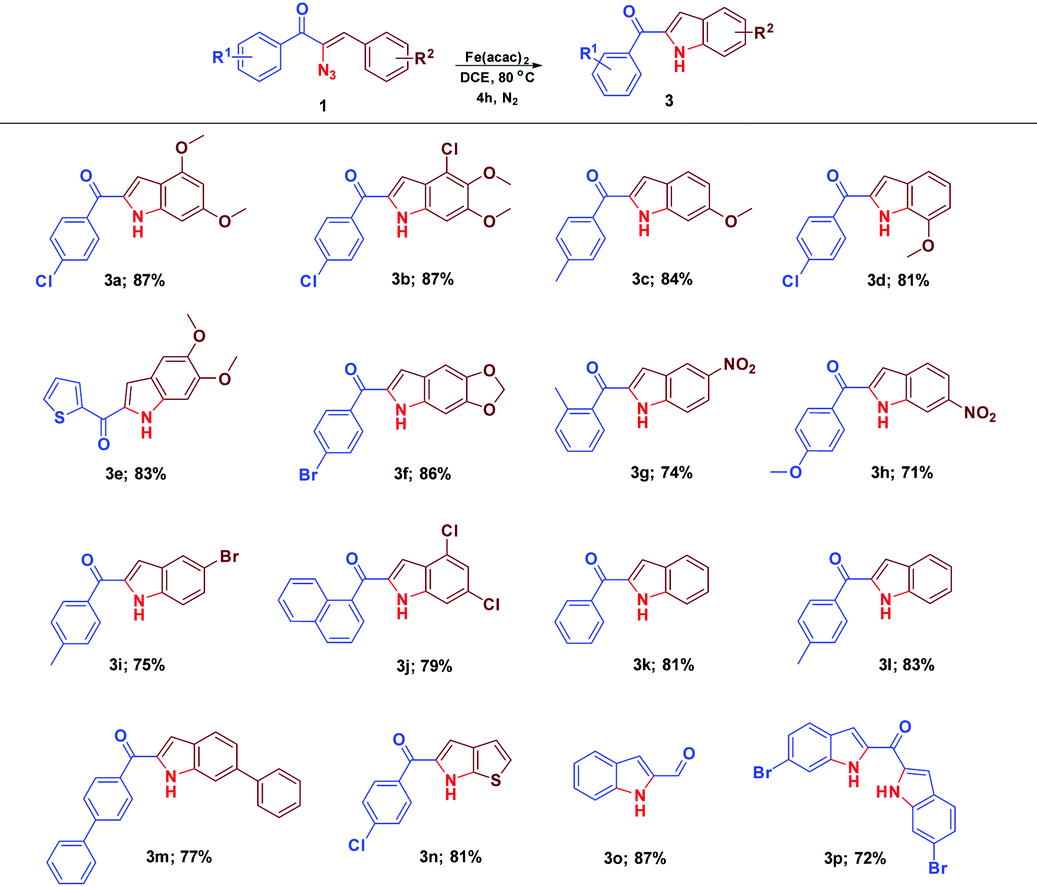
The scope and the chemistry of the Fe(acac)2 promoted C–H amination strategy for the synthesis of substituted indoles from various α-azidochalcones have been studied with further interest. The reaction progressed fairly with substrates comprising various substituents on the aryl ring (–R2) and the synthesized library of compounds is summarized in Table 4. Fascinatingly, this optimized C–H amination works well with the heteroaromatic system (3n) as well. The reaction takes place even with α-azido cinnamaldehyde instead of α-azidochalcone resulting in indole-2-carbaldehyde (3o) in good yield. An attempt to accomplish the transformation with the diazido substrate under the optimized conditions is successful yielding 3p.
| a Reaction conditions: α-azidochalcone (0.10 mmol) and Fe(acac)2 (0.02 equiv.; 0.04 equiv. for 3p) in DCE (3 mL) heated at 80 °C for 4 h under a nitrogen atmosphere. |
|---|
|
Initially, thermolysis of α-azidochalcone would have produced strained 2H-azirine intermediate 4, which forms iron azirine complex 7 with Fe(II) and subsequent cleavage of the C–N bond would afford iron vinyl nitrene complex 8. A five centered 6-π electrocyclization17via intermediate 9 results in indole 3 (Scheme 4).18
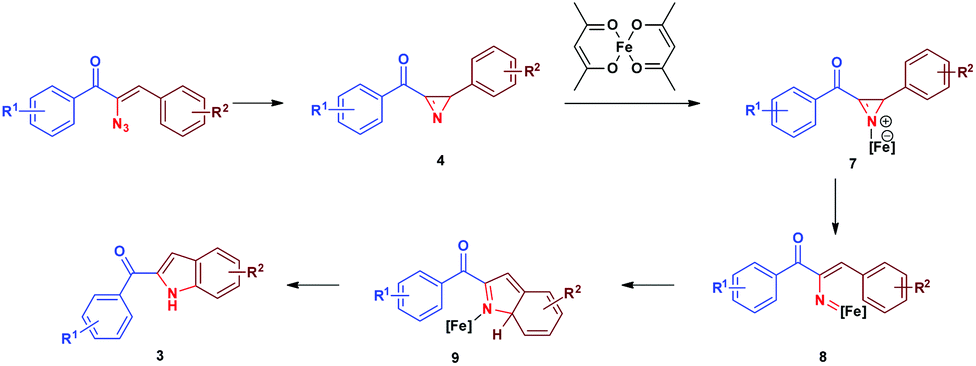 | ||
| Scheme 4 Mechanism for the formation of indole 3. | ||
The cyclization occurs at the styryl ring rather than the aroyl ring, probably due to the deactivation of the aroyl ring towards a five centered 6-π electrocyclization step (Scheme 5). This hypothesis has been proved as evidenced by the single crystal X-ray analysis of 3c (Fig. 3)19 and also by the fact that the reaction goes well with α-azido cinnamaldehyde giving an indole derivative. Though a catalyst free conversion has been reported,10g the milder reaction conditions of the present method of generating 3 are noteworthy.
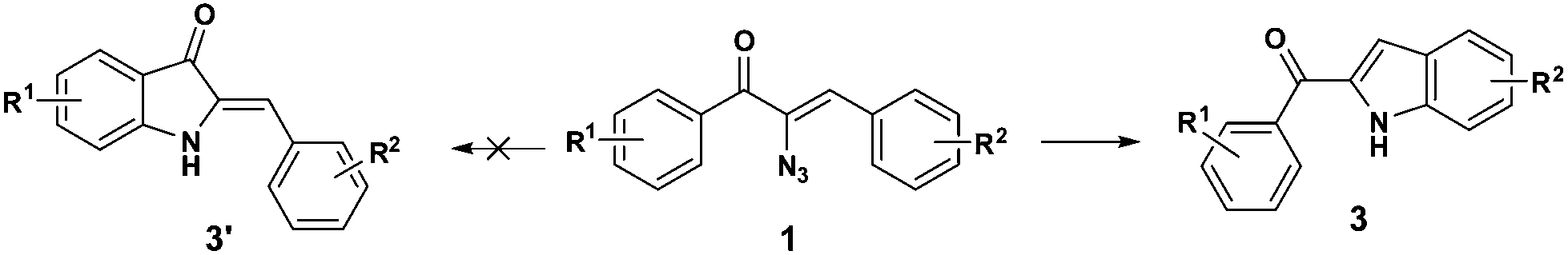 | ||
| Scheme 5 Regioselectivity in C–H amination for indole formation 3. | ||
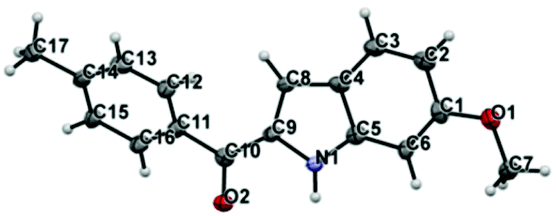 | ||
| Fig. 3 X-ray crystal structure of 3c. Thermal ellipsoids are shown at 50% probability level. | ||
When water is employed as the solvent the free ligand gets released in the equilibrium enabling the stoichiometric involvement of the complex in the reaction. But when DCE is employed the liberation of the free ligand for the pyrrole formation is restricted, where the Lewis acidic behaviour of iron dominates the course of the reaction. Thus the solvent polarity seems to be vital in dictating the course of the reaction. In the present protocol, the yields of both substituted pyrroles and indoles were poor at higher temperature. The poor atom economy associated with the formation of 2 is also a matter of concern.
Conclusions
In summary, the reactivity of α-azidochalcones with metal β-diketonates towards the synthesis of substituted pyrroles in micellar media via the 2H-azirine intermediate has been achieved. Regioselective Fe(II) catalyzed C–H amination of α-azidochalcones leading to substituted indoles has also been described. Thus the diverged reactivity of α-azidochalcones with metal β-diketonates as bifunctional reactive partners by variation of the reaction conditions has been demonstrated.Acknowledgements
The authors thank DST, New Delhi for assistance under the IRHPA program for the NMR facility.Notes and references
- (a) H.-J. Knçlker and K. R. Reddy, Chem. Rev., 2002, 102, 4303 CrossRef; (b) M. D'Ischia, A. Napolitano and A. Pezzella, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 3, p. 353 Search PubMed; (c) H. Fan, J. Peng, M. T. Hamann and J.-F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS PubMed; (d) A. J. K. Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef PubMed; (e) A. W. Schmidt, K. R. Reddy and H.-J. Knçlker, Chem. Rev., 2012, 112, 3193 CrossRef CAS PubMed.
- (a) D. Black, in Science of Synthesis, ed. G. Maas, Thieme, Stuttgart, 2000, vol. 9, p. 441 Search PubMed; (b) J. A. Joule, in Science of Synthesis, ed. E. J. Thomas, Thieme, Stuttgart, 2000, vol. 10, p. 361 Search PubMed; (c) T. Gallagher, in Science of Synthesis, ed. E. J. Thomas, Thieme, Stuttgart, 2000, vol. 10, p. 693 Search PubMed; (d) G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045 RSC; (e) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed; (f) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS PubMed; (g) S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, 215 CrossRef PubMed; (h) M. Platon, R. Amardeil, L. Djakovitch and J. C. Hierso, Chem. Soc. Rev., 2012, 41, 3929 RSC; (i) M. Inman and C. Moody, J. Chem. Sci., 2013, 4, 29 RSC.
- (a) S. L. Schreiber, Science, 2000, 287, 1964 CrossRef CAS PubMed; (b) F. G. Kuruvilla, A. F. Shamji, S. M. Sternson, P. J. Hergenrother and S. L. Schreiber, Nature, 2002, 416, 653 CrossRef CAS PubMed; (c) D. D. Dixon, J. W. Lockner, Q. Zhou and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 8432 CrossRef CAS PubMed; (d) H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222 CrossRef CAS PubMed.
- (a) M. Lhassani, O. Chavignon, J.-M. Chezal, J.-C. Teulade, J.-P. Chapat, R. Snoeck, G. Andrei, J. Balzarini, E. De Clercq and A. Gueiffier, Eur. J. Med. Chem., 1999, 34, 271 CrossRef CAS; (b) K. C. Rupert, J. R. Henry, J. H. Dodd, S. A. Wadsworth, D. E. Cavender, G. C. Olini, B. Fahmy and J. J. Siekierka, Bioorg. Med. Chem. Lett., 2003, 13, 347 CrossRef CAS PubMed; (c) C. E. Gueiffier and A. Gueiffier, Mini-Rev. Med. Chem., 2007, 7, 888 CrossRef.
- (a) W. T. Chen, M. Hu, J. W. Wu, H. B. Zou and Y. P. Yu, Org. Lett., 2010, 12, 3863 CrossRef CAS PubMed; (b) Y. Wang, K. K. Toh, E. P. J. Ng and S. Chiba, J. Am. Chem. Soc., 2011, 133, 6411 CrossRef CAS PubMed; (c) B. Hu, Z. Wang, N. Ai, J. Zheng, X. Liu, S. Shan and Z. Wang, Org. Lett., 2011, 13, 6362 CrossRef CAS PubMed; (d) N. Jung and S. Brase, Angew. Chem., Int. Ed., 2012, 51, 12169 CrossRef CAS PubMed; (e) B. K. Dinda, A. K. Jana and D. Mal, Chem. Commun., 2012, 48, 3999 RSC; (f) G. Zhang, H. Ni, W. Chen, J. Shao, H. Liu and Y. Yu, Org. Lett., 2013, 15, 5967 CrossRef CAS PubMed; (g) Y. F. Wang, G. H. Lonca and S. Chiba, Angew. Chem., Int. Ed., 2014, 53, 1067 CrossRef CAS PubMed; (h) S. Liu, W. Chen, J. Luo and Y. Yu, Chem. Commun., 2014, 50, 8539 RSC; (i) S. Liu, J. Shao, X. Guo, J. Luo, M. Zhao, G. Zhang and Y. Yu, Tetrahedron, 2014, 70, 1418 CrossRef CAS; (j) B. Hu and S. G. Di Magno, Org. Biomol. Chem., 2015, 13, 3844 RSC.
- (a) F. W. Fowler and A. Hassner, J. Am. Chem. Soc., 1968, 90, 2875 CrossRef CAS; (b) A. Kakehi, S. Ito, T. Manabe, H. Amano and Y. Shimaoka, J. Org. Chem., 1976, 41, 2739 CrossRef CAS; (c) F. Palacios, A. M. O. de Retana, E. M. de Marigorta and J. M. de los Santos, Eur. J. Org. Chem., 2001, 2401 CrossRef CAS; (d) T. Ooi, M. Takahashi, K. Doda and K. Maruoka, J. Am. Chem. Soc., 2002, 124, 7640 CrossRef CAS PubMed; (e) T. M. V. D. Pinhoe Melo, A. L. Cardoso, C. S. B. Gomes and A. M. d'A Rocha Gonsalves, Tetrahedron Lett., 2003, 44, 6313 CrossRef CAS; (f) D. A. Candito and M. Lautens, Org. Lett., 2010, 12, 3312 CrossRef CAS PubMed; (g) S. Jana, M. D. Clements, B. K. Sharp and N. Zheng, Org. Lett., 2010, 12, 3736 CrossRef CAS PubMed; (h) A. F. Khlebnikov, M. V. Golovkina, M. S. Novikov and D. S. Yufit, Org. Lett., 2012, 14, 3768 CrossRef CAS PubMed; (i) A. F. Khlebnikov and M. S. Novikov, Tetrahedron, 2013, 69, 3363 CrossRef CAS; (j) A. Prechter, G. Henrion, P. Faudot dit Bel and F. Gagosz, Angew. Chem., Int. Ed., 2014, 53, 4959 CrossRef CAS PubMed; (k) K. Rajaguru, A. Mariappan, R. Suresh, P. Manivannan and S. Muthusubramanian, Beilstein J. Org. Chem., 2015, 11, 2021 CrossRef PubMed.
- (a) R. Suresh, S. Muthusubramanian, M. Boominathan and G. Manickam, Tetrahedron Lett., 2013, 54, 2315 CrossRef CAS; (b) R. Suresh, S. Muthusubramanian, M. Nagaraj and G. Manickam, Tetrahedron Lett., 2013, 54, 1779 CrossRef CAS; (c) R. Suresh, S. Muthusubramanian, N. Paul, N. Kalidhasan and V. Shanmugaiah, Med. Chem. Res., 2014, 23, 4367 CrossRef CAS; (d) K. Rajaguru, R. Suresh, A. Mariappan, S. Muthusubramanian and N. Bhuvanesh, Org. Lett., 2014, 16, 744 CrossRef CAS PubMed; (e) K. Rajaguru, A. Mariappan, S. Muthusubramanian and N. Bhuvanesh, ChemistrySelect, 2016, 1, 1636 CrossRef CAS.
- T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831 CAS.
- (a) A. González, F. Güell, J. Marquet and M. M. Mañas, Tetrahedron Lett., 1985, 26, 3735 CrossRef; (b) M. Kimura, A. Ezoe, S. Tanaka and Y. Tamaru, Angew. Chem., Int. Ed., 2001, 40, 3600 CrossRef CAS; (c) M. Taillefer, N. Xia and A. Ouali, Angew. Chem., Int. Ed., 2007, 46, 934 CrossRef CAS PubMed; (d) S. L.-F. Chan, K.-H. Low, C. Yang, S. H.-F. Cheung and C.-M. Che, Chem. – Eur. J., 2011, 17, 4709 CrossRef CAS PubMed; (e) Q. Chen, L. Ilies, N. Yoshikai and E. Nakamura, Org. Lett., 2011, 13, 3232 CrossRef CAS PubMed; (f) T. P. A. Goossens, F. A. C. Vrielynck, N. G. C. Hosten and K. J. A. V. Aken, Transesterification process using mixed salt acetylacetonates catalysis, US2013/0090492A1, 2013 Search PubMed; (g) C. Pan, J. Han, H. Zhang and C. Zhu, J. Org. Chem., 2014, 79, 5374 CrossRef CAS PubMed; (h) H. E. Ho, K. Oniwa, Y. Yamamoto and T. Jin, Org. Lett., 2016, 18, 2487 CrossRef CAS PubMed.
- (a) H. Hemetsberger and D. Knittel, Monatsh. Chem., 1972, 103, 194 CrossRef CAS; (b) B. J. Stokes, H. Dong, B. E. Leslie, A. L. Pumphrey and T. G. Driver, J. Am. Chem. Soc., 2007, 129, 7500 CrossRef CAS PubMed; (c) S. Chiba, Y.-F. Wang, G. Lapointe and K. Narasaka, Org. Lett., 2008, 10, 313 CrossRef CAS PubMed; (d) F. Lehmann, M. Holm and S. Laufer, Tetrahedron Lett., 2009, 50, 1708 CrossRef CAS; (e) B. J. Stokes, B. Jovanović, H. Dong, K. J. Richert, R. D. Riell and T. G. Driver, J. Org. Chem., 2009, 74, 3225 CrossRef CAS PubMed; (f) D. Hong, Z. Chen, X. Lin and Y. Wang, Org. Lett., 2010, 12, 4068 Search PubMed; (g) J. Bonnamour and C. Bolm, Org. Lett., 2011, 13, 2012 CrossRef CAS PubMed; (h) E. P. J. Ng, Y.-F. Wang, B.-W. Q. Hui, G. Lapointe and S. Chiba, Tetrahedron, 2011, 67, 7728 CrossRef CAS; (i) H. G. Bonacorso, F. M. Libero, G. M. D. Forno, E. P. Pittaluga, L. M. F. Porte, M. A. P. Martins and N. Zanatta, Tetrahedron Lett., 2015, 56, 5190 CrossRef CAS; (j) I. T. Alt and B. Plietker, Angew. Chem., Int. Ed., 2016, 55, 1519 CrossRef CAS PubMed; (k) C. Pooran, C. Jason, C. Brian, T. Gilles, T. Sucheta and T. John, Small molecule inhibitors of pfkfb3 and glycolytic flux and their methods of use as anti-cancer therapeutics, WO2011103557, 2011 Search PubMed.
- V. Nair and T. G. George, Tetrahedron Lett., 2000, 41, 3199 CrossRef CAS.
- C. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68 RSC.
- G. L. Sorella, G. Strukul and A. Scarso, Green Chem., 2015, 17, 644 RSC.
- (a) S. Tu, C. Li, G. Li, L. Cao, Q. Shao, D. Zhou, B. Jiang, J. Zhou and M. Xia, J. Comb. Chem., 2007, 9, 1144 CrossRef CAS PubMed; (b) S. Tu, S. Wu, S. Yan, W. Hao, X. Zhang, X. Cao, Z. Han, B. Jiang, F. Shi, M. Xia and J. Zhou, J. Comb. Chem., 2009, 11, 239 CrossRef CAS PubMed; (c) N. Ma, B. Jiang, G. Zhang, S.-J. Tu, W. Weverb and G. Li, Green Chem., 2010, 12, 1357 RSC; (d) C. Cheng, B. Jiang, S.-J. Tu and G. Li, Green Chem., 2011, 13, 2107 RSC.
- R. L. Prasad, A. Kushwaha and B. P. S. Gautam, J. Coord. Chem., 2009, 62, 2983 CrossRef CAS.
- CCDC number for 2b: 1482069; 2j: 1482068.
- I. W. Davies, V. A. Guner and K. N. Houk, Org. Lett., 2004, 6, 743 CrossRef CAS PubMed.
- S. Jana, M. D. Clements, B. K. Sharp and N. Zheng, Org. Lett., 2010, 12, 3736 CrossRef CAS PubMed.
- CCDC number for 3c: 1482067.
Footnote |
| † Electronic supplementary information (ESI) available: The copies of 1H and 13C NMR spectra for all the synthesized compounds. CCDC 1482067–1482069. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00541a |
| This journal is © the Partner Organisations 2017 |