
Cobalt(III)-catalyzed cross-coupling of enamides with allyl acetates/maleimides†
Wenlong
Yu
a,
Wei
Zhang
a,
Yue
Liu
a,
Zhanxiang
Liu
a and
Yuhong
Zhang
*ab
aDepartment of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: yhzhang@zju.edu.cn
bState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, China
First published on 10th October 2016
Abstract
Cp*Co(III)-catalyzed direct allylation of enamides has been accomplished with the exclusive formation of allylated Z-enamides with high efficiency. In addition, the employment of maleimides as the reaction partner under the same catalytic conditions provides a series of succinimide-substituted Z-enamides.
The allyl moiety is among the most synthetically useful motifs in organic synthesis, and is easily functionalized by numerous methods. Consequently, methodological studies relating to it have attracted the attention of synthetic chemists for many years, and efficient approaches for incorporating the allyl group into a molecule are of great value.1 In recent years, in terms of atom economy and efficiency, the direct allylation of inert C–H bonds under transition metal catalysis has drawn much attention, and various allylating reagents, such as allyl acetates,2 carbonates,3 phosphates,4 allylic halides,5 allylic ethers,6 and allylic alcohols7 have been successfully exploited in direct coupling reactions with C(sp2)–H bonds under transition metal catalysis. Enamides are versatile building blocks and key precursors for catalytic asymmetric C–C bond forming processes.8 As important synthons, enamides have been studied as key coupling partners for vinyl C–H bond activation, and their arylation,9 alkenylation,10 alkylation,11 alkynylation,12 acylation,13 and acetoxylation14 have been demonstrated. Despite these indisputable advances, the highly selective allylation of enamides through direct vinyl C–H functionalization has not been realized.
The use of inexpensive earth-abundant first-row transition metals to perform established transition metal-catalyzed C–H functionalizations has recently gained significant attention because of their low cost, nontoxicity, and novel catalytic properties.15 Among them, high-valent Cp*Co(III) catalysts have been proven to be robust, powerful, and cheap metal catalysts for C–H activation by the groups of Kanai and Matsunaga,7b,c,16 Ackermann,2c,17 Glorius,3b,18 Ellman,19 Chang,20 Li,21 Cheng,22 and others.23 Our group has been devoted to enlarging the application of cobalt catalysis in the field of C–H activation.24 Considering the practical importance of enamides, we were attracted to exploring the reaction conditions for cobalt-catalyzed C–H allylation of enamides with readily available allyl acetate. In this context it is noteworthy that the reaction proceeds efficiently with high stereoselectivity to deliver the allylated enamides as absolute Z-isomers. Additionally, succinimide, a central pharmacophore of many pharmaceuticals,25 could be easily installed into enamides under cobalt catalysis when maleimides were employed as the coupling partner (Table 1).
|
|
|||
|---|---|---|---|
| Entry | Additive | Solvent | Yieldb (%) |
a Reactions were carried out using 1a (0.1 mmol), 2a (0.15 mmol), Cp*Co(CO)I2 (10 mol%), additive (20 mol%), solvent (1.0 mL), 90 °C, air, 10 h.
b Isolated yield.
c ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2a (0.3 mmol).
d 2a (0.45 mmol).
e [Cp*RhCl2]2/AgOAc (5 mol%/10 mol%) was used as the catalyst.
f Cp*Co(CO)I2 was not used. 2a (0.3 mmol).
d 2a (0.45 mmol).
e [Cp*RhCl2]2/AgOAc (5 mol%/10 mol%) was used as the catalyst.
f Cp*Co(CO)I2 was not used.
|
|||
| 1 | AgOAc | CH3CN | NR |
| 2 | AgOAc | DCE | NR |
| 3 | AgOAc | PhMe | NR |
| 4 | AgOAc | Dioxane | NR |
| 5 | AgOAc | MeOH | NR |
| 6 | AgOAc | CF3CH2OH | 40 |
| 7c | AgOAc | CF3CH2OH | 63 |
| 8d | AgOAc | CF3CH2OH | 73 |
| 9d | KOAc | CF3CH2OH | 45 |
| 10d | Zn(OAc)2 | CF3CH2OH | 33 |
| 11d | Cu(OAc)2 | CF3CH2OH | 56 |
| 12d,e | AgOAc | CF3CH2OH | 60 |
| 13f | AgOAc | CF3CH2OH | NR |
| 14 | — | CF3CH2OH | Trace |
We initiated our optimization experiments with enamide 1a and allyl acetate (2a) as model substrates for the allylation reaction. It was found that solvents had a significant influence on the reaction. Common solvents that are widely used in C–H activation reactions, including DCE, PhMe, CH3CN, dioxane and MeOH, were all ineffective (entries 1–5). To our delight, when the reaction was conducted in CF3CH2OH, the desired product was obtained in 40% yield (entry 6). Increasing the amount of allyl acetate (2a) was necessary to improve the yield, which may be attributed to the volatilization loss of the allyl acetate (2a) (entries 6–8). AgOAc gave the optimal result among the acetate salts screened (entries 8–11), providing 3aa in 73% yield. Other acetate salts such as KOAc, Zn(OAc)2, and Cu(OAc)2 were not as effective as AgOAc. Replacing Cp*Co(CO)I2 with [Cp*RhCl2]2 afforded the product 3aa in a lower yield (entry 12), indicating that Cp*Co(III) had a better catalytic activity than Cp*Rh(III) in this transformation. No reaction was observed in the absence of the cobalt catalyst or AgOAc (entries 13 and 14).
With the set of optimized reaction conditions in hand, the scope of substrates in this reaction was explored as illustrated in Scheme 1. It was observed that the reaction efficiency was dependent on electronic effects. Enamides bearing electron-donating substituents, such as methyl (3ba), methoxyl (3ca), phenyl (3da), and meta-methyl (3ia), displayed good reactivity, providing the desired products in 65–86% yields. Comparably, enamides with electron-withdrawing groups, such as fluorine (3ea), chlorine (3fa), and bromine (3ga), showed better reactivity in this reaction to deliver the corresponding products in 85–93% yields. Steric effects also had a significant impact on the reaction. Enamides with substituents at the ortho position of the phenyl ring had poor reactivity, giving the products (3ha, 3ja) in 52–60% yields. Naphthyl enamides participated in the reaction smoothly to furnish the allylation products (3ka). Notably, heteroarene-substituted enamides (3la, 3ma) were also effective substrates, generating the allylation products in moderate yields. Using allyl acetate (2a) as the reaction partner, methyl 2-acetamidoacrylate (1n) could not be successfully transformed into the corresponding allylation product under the established reaction conditions. Fortunately, the allylation product (3na) was obtained in 80% yield when the allyl acetate (2a) was replaced by allyl methyl carbonate (2a′).
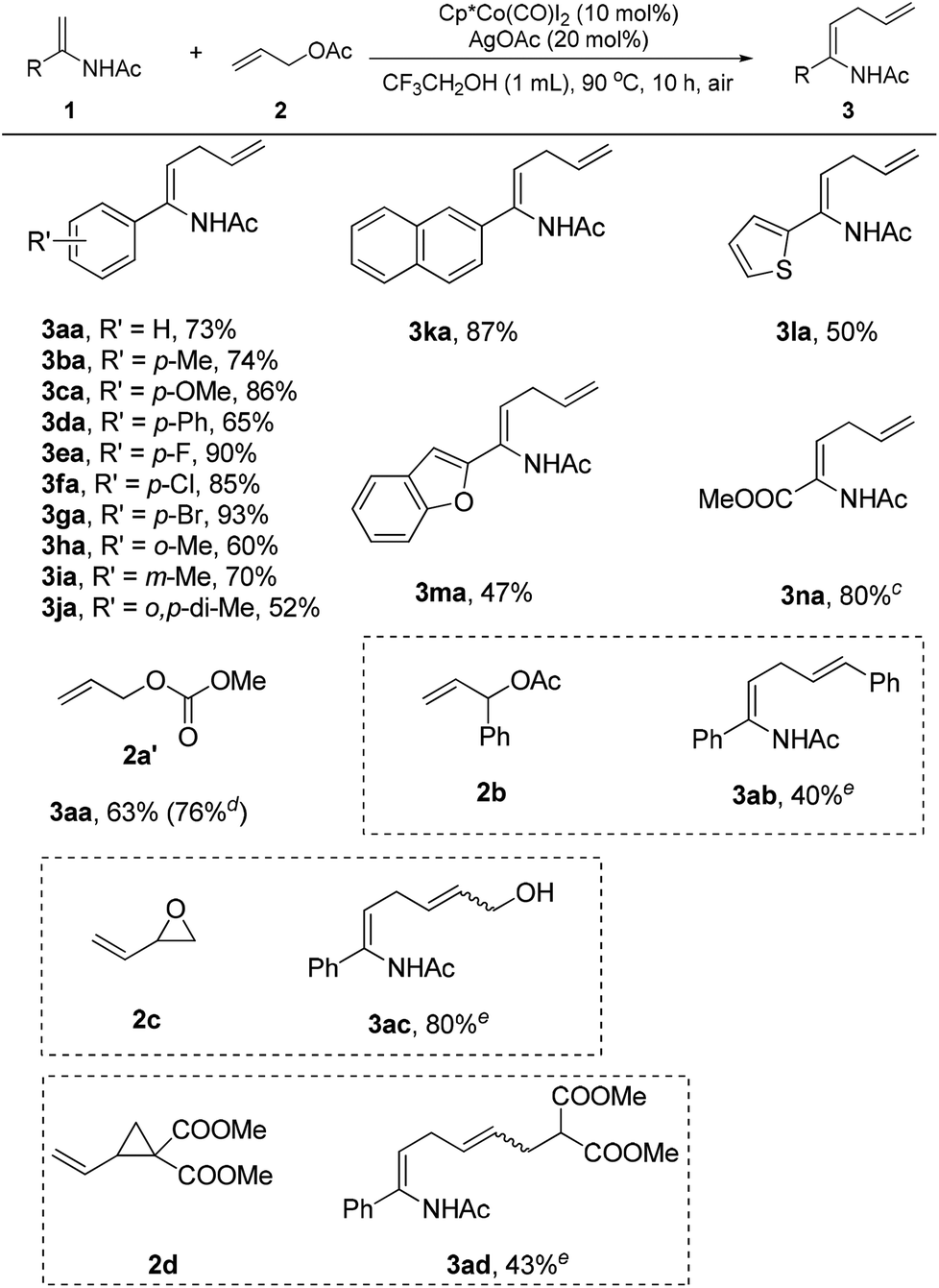 | ||
| Scheme 1 Scope of substrates in the allylation reaction.a,b aReaction conditions: enamide 1 (0.1 mmol), allyl acetate 2 (0.45 mmol), Cp*Co(CO)I2 (10 mol%), AgOAc (20 mol%), CF3CH2OH (1 mL), 90 °C, air, 10 h. bIsolated yields. cAllyl methyl carbonate 2a′ (0.2 mmol) was used as the allylation reagent. dEnamide 1a (1 mmol), allyl methyl carbonate 2a′ (1.5 mmol), Cp*Co(CO)I2 (5 mol%), AgOAc (10 mol%), CF3CH2OH (3 mL), 90 °C, air, 24 h. eThe amount of allylation reagent was 2 equiv. | ||
Different allylation reagents were also investigated under the reaction conditions. The coupling of enamide 1a with allyl methyl carbonate (2a′) proceeded smoothly to give the product (3aa) in 63% yield. Interestingly, the desired product was obtained in higher yield when the reaction was performed on a 1 mmol scale with a lower catalyst loading. The introduction of a phenyl group into the α-position of the allyl acetate (2b) was unfavorable for the reaction, and the corresponding product (3ab) was isolated in low yield. The allylation reagents were not restricted to allyl acetates. The employment of 2-vinyloxirane (2c) as the coupling partner delivered a novel allylic alcohol-substituted enamide (3ac) in good yield. In particular, vinylcyclopropane (2d) was also an effective allylation reagent,17c providing a ζ-amino acid precursor (3ad) in moderate yield. Cinnamyl acetate and allyl alcohol were unreactive in this transformation.
Encouraged by these results, we turned our attention to studying the coupling of enamides with maleimides, which are effective coupling partners to introduce the useful succinimide motif into biomolecules in organic synthesis.26 To our delight, the enamide (1a) reacted with N-methyl maleimide (4a) smoothly under the established reaction conditions to afford the desired product (5aa) in 78% yield. The structure of 5aa was further confirmed by X-ray crystal analysis. Surprisingly, when Cp*Rh(III) was employed as the catalyst in this transformation, the desired product was obtained in trace amounts, implying the unique reactivity of the Cp*Co(III) catalyst. Enamides bearing different groups on the phenyl such as methyl, methoxyl, phenyl, fluoro, chloro, bromo, and meta-methyl were all well tolerated and provided the corresponding products (5ba–5ga, 5ia) in 42–91% yields. It was found that steric effects in the enamide seemingly exerted a negligible influence on the efficiency of the reaction. The reaction of an ortho-enamide (5ha) gave the product in 70% yield. A disubstituted enamide also showed high reactivity, providing the corresponding product (5ja) in 85% yield. Enamides bearing naphthalene and heteroarene groups (5ka–5ma) also provided good results. N-Vinylacetamide (1n) was sluggish in this reaction, giving the product (5na) in low yield. Interestingly, when methyl 2-acetamidoacrylate (1o) was employed as the substrate, a mixture (5oa/5oa′ = 1.4/1), including the olefin migration product (5oa′), was obtained in moderate yield. Cyclic enamides could not react with maleimides in this reaction (Scheme 2).
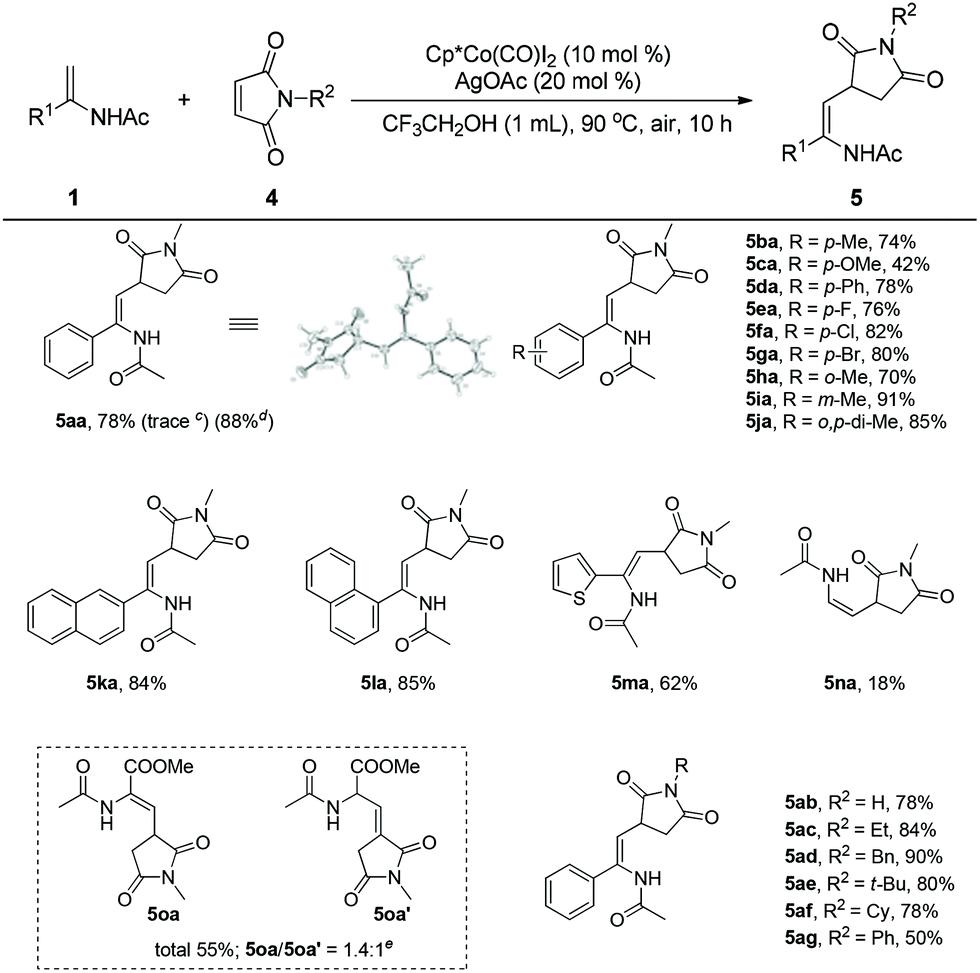 | ||
| Scheme 2 Cobalt-catalyzed direct coupling of various enamides and maleimide.a,b aReaction conditions: enamide 1 (0.2 mmol), maleimide 4 (0.3 mmol), Cp*Co(CO)I2 (10 mol%), AgOAc (20 mol%), CF3CH2OH (1 mL), 90 °C, air, 10 h. bIsolated yields. c[Cp*RhCl2]2 (5 mol%)/AgOAc (10 mol%) was used as the catalyst. dReactions were performed on a 0.5 mmol scale with Cp*Co(III) (5 mol%) and AgOAc (10 mol%). eDetermined by 1H NMR. | ||
To further evaluate the scope of this process, a range of maleimides were investigated under the optimal reaction conditions. The N-substituents of the maleimide had an obvious influence on the coupling reaction. The unprotected maleimide was tolerated to deliver the product (5ab) in 78% yield. Maleimides bearing different N-alkyl groups such as ethyl, benzyl, cyclohexyl, and t-butyl were all effective coupling partners, affording the final products in good to excellent yields (5ac–5af). However, when N-phenyl maleimide was employed as the substrate, the corresponding product (5ag) was isolated in moderate yield. Some common olefins such as butyl acrylate, styrene, benzoquinone and norbornene were ineffective substrates in this reaction.
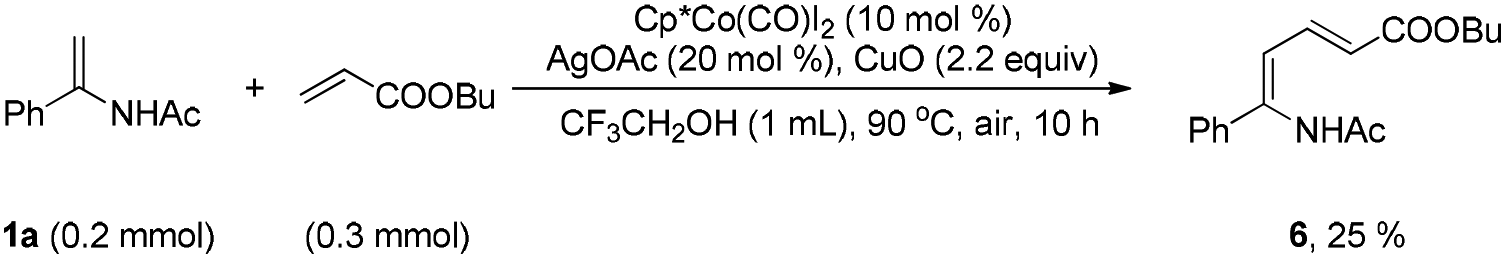 | (1) |
 | (2) |
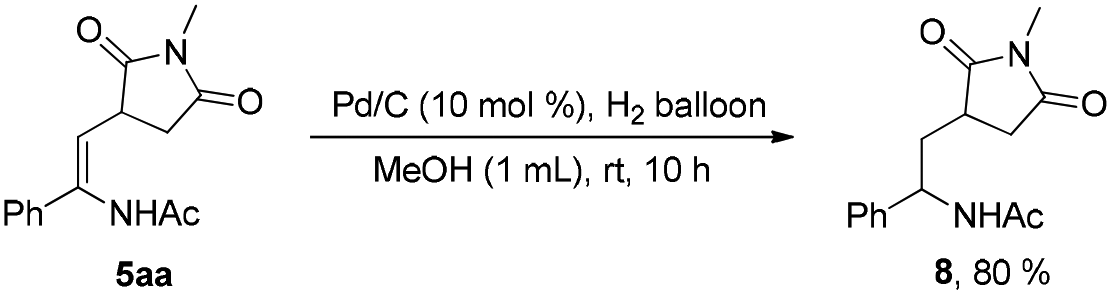 | (3) |
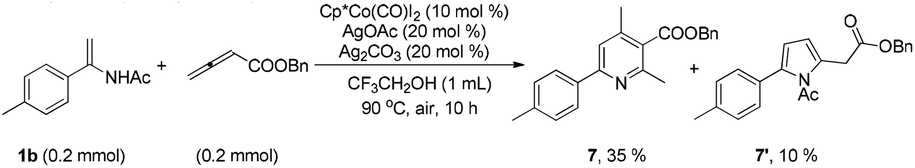
Although electron-deficient butyl acrylate could not provide the alkylation product under the optimal conditions, it was found that the addition of copper oxide promoted the generation of the alkenylation product (6) (eqn (1)). Furthermore, the allene as a coupling partner was tested in this reaction and no desired product was detected under the established reaction conditions. To our delight, after 20 mol% Ag2CO3 was added to the reaction, a mixture of pyridine (7) and pyrrole (7′) was obtained in moderate yield (eqn (2)). The structure of 7 was further characterized by X-ray crystal analysis (see ESI†). In addition, the enamide 5aa was further transformed into a γ-amino acid derivative (8) in good yield with a Pd/C catalyst under a hydrogen atmosphere (eqn (3)).
Conclusions
In conclusion, we have developed an efficient cobalt(III)-catalyzed cross-coupling of enamides with allyl acetates/maleimides. This reaction proceeds efficiently under mild conditions and displays good functional group compatibility. Besides allyl acetates, 2-vinyloxirane and even vinylcyclopropane can also serve as allyl sources. Maleimides were also effective coupling partners to introduce the useful succinimide motif into enamides under the established conditions. Notably, Cp*Co(III) exhibited a better catalytic activity than Cp*Rh(III) in this transformation.Acknowledgements
Funding from the Natural Science Foundation of China (No. 21272205, 21472165) and the Program for Zhejiang Leading Team of S&T Innovation (2011R50007) is acknowledged.Notes and references
- (a) R. M. Magid, Tetrahedron, 1980, 36, 1901 CrossRef CAS; (b) F. C. Pigge, Synthesis, 2010, 1745 CrossRef CAS.
- (a) C. Feng, D. Feng and T.-P. Loh, Org. Lett., 2013, 15, 3670 CrossRef CAS PubMed; (b) C. Feng, D. Feng and T.-P. Loh, Chem. Commun., 2015, 51, 342 RSC; (c) M. Moselage, N. Sauermann, J. Koeller, W. Liu, D. Gelman and L. Ackermann, Synlett, 2015, 1596 CAS.
- (a) S.-S. Zhang, J.-Q. Wu, Y.-X. Lao, X.-G. Liu, Y. Liu, W.-X. Lv, D.-H. Tan, Y.-F. Zeng and H. Wang, Org. Lett., 2014, 16, 6412 CrossRef CAS PubMed; (b) D.-G. Yu, T. Gensch, F. Azambuja, S. Vasquez-Cespedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722 CrossRef CAS PubMed; (c) H. Wang, N. Schroder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386 CrossRef CAS PubMed.
- X. Cong, Y. Li, Y. Wei and X. Zeng, Org. Lett., 2014, 16, 3926 CrossRef CAS PubMed.
- Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308 CrossRef CAS PubMed.
- S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 17755 CrossRef CAS PubMed.
- (a) X. Cong and X. Zeng, Org. Lett., 2014, 16, 3716 CrossRef CAS PubMed; (b) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944 CrossRef CAS PubMed; (c) Y. Bunno, N. Murakami, Y. Suzuki, M. Kanai, T. Yoshino and S. Matsunaga, Org. Lett., 2016, 18, 2216 CrossRef CAS PubMed.
- (a) D. R. Carbery, Org. Biomol. Chem., 2008, 6, 3455 RSC; (b) R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292 CrossRef CAS PubMed; (c) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599 CrossRef CAS PubMed; (d) G. R. Dake, Synlett, 2012, 814 CrossRef CAS; (e) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS PubMed.
- (a) S. Pankajakshan, Y.-H. Xu, J. K. Cheng, M. T. Low and T.-P. Loh, Angew. Chem., Int. Ed., 2012, 51, 5701 CrossRef CAS PubMed; (b) F. Bartoccini, D. M. Cannas, F. Fini and G. Piersanti, Org. Lett., 2016, 18, 2762 CrossRef CAS PubMed.
- (a) Y.-H. Xu, Y. K. Chok and T.-P. Loh, Chem. Sci., 2011, 2, 1822 RSC; (b) T. Besset, N. Kuhl, F. W. Patureau and F. Glorius, Chem. – Eur. J., 2011, 17, 7167 CrossRef CAS PubMed.
- (a) C. Feng and T.-P. Loh, Chem. Sci., 2012, 3, 3458 RSC; (b) R. Ding, Z.-D. Huang, Z.-L. Liu, T.-X. Wang, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2016, 52, 5617 RSC.
- C. Feng, D. Feng and T.-P. Loh, Chem. Commun., 2014, 50, 9865 RSC.
- (a) K. D. Hesp, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 11430 CrossRef CAS PubMed; (b) R. Ding, Q.-C. Zhang, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2014, 50, 11661 RSC.
- W. Yu, J. Chen, K. Gao, Z. Liu and Y. Zhang, Org. Lett., 2014, 16, 4870 CrossRef CAS PubMed.
- (a) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743 CrossRef CAS; (b) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498 CrossRef CAS; (c) E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061 CrossRef CAS PubMed.
- (a) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207 CrossRef CAS PubMed; (b) H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424 CrossRef CAS PubMed; (c) B. Sun, T. Yoshino, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2015, 54, 12968 CrossRef CAS PubMed.
- (a) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 8551 CrossRef CAS PubMed; (b) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 3635 CrossRef CAS PubMed; (c) D. Zell, Q. Bu, M. Feldt and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7408 CrossRef CAS PubMed; (d) H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 10386 CrossRef CAS PubMed; (e) N. Sauermann, M. J. Gonzalez and L. Ackermann, Org. Lett., 2015, 17, 5316 CrossRef CAS PubMed.
- (a) D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508 CrossRef CAS PubMed; (b) T. Gensch, F. J. R. Klauck and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 11287 CrossRef CAS PubMed.
- (a) J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490 CrossRef CAS PubMed; (b) J. A. Boerth, J. R. Hummel and J. A. Ellman, Angew. Chem., Int. Ed., 2016, 55, 12650 CrossRef CAS PubMed.
- J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103 CrossRef CAS PubMed.
- (a) L. Li, H. Wang, S. Yu, X. Yang and X. Li, Org. Lett., 2016, 18, 3662 CrossRef CAS PubMed; (b) L. Kong, S. Yu, G. Tang, H. Wang, X. Zhou and X. Li, Org. Lett., 2016, 18, 3802 CrossRef CAS PubMed; (c) F. Wang, H. Wang, Q. Wang, S. Yu and X. Li, Org. Lett., 2016, 18, 1306 CrossRef CAS PubMed; (d) L. Kong, S. Yu and X. Li, Org. Lett., 2016, 18, 588 CrossRef CAS PubMed; (e) L. Kong, X. Yang, X. Zhou, S. Yu and X. Li, Org. Chem. Front., 2016, 3, 813 RSC.
- S. Prakash, K. Muralirajan and C.-H. Cheng, Angew. Chem., Int. Ed., 2016, 55, 1844 CrossRef CAS PubMed.
- (a) Y. Liang, Y.-F. Liang, C. Tang, Y. Yuan and N. Jiao, Chem. – Eur. J., 2015, 21, 16395 CrossRef CAS PubMed; (b) X.-G. Liu, S.-S. Zhang, C.-Y. Jiang, J.-Q. Wu, Q. Li and H. Wang, Org. Lett., 2015, 17, 5404 CrossRef CAS PubMed; (c) Z.-Z. Zhang, B. Liu, C.-Y. Wang and B.-F. Shi, Org. Lett., 2015, 17, 4094 CrossRef CAS PubMed; (d) D. Kalsi and B. Sundararaju, Org. Lett., 2015, 17, 6118 CrossRef CAS PubMed; (e) R. Mei, H. Wang, S. Warratz, S. A. Macgregor and L. Ackermann, Chem. – Eur. J., 2016, 22, 6759 CrossRef CAS PubMed; (f) N. Thrimurtulu, A. Dey, D. Maiti and C. M. R. Volla, Angew. Chem., Int. Ed., 2016, 55, 12361 CrossRef CAS PubMed; (g) Z.-Z. Zhang, B. Liu, C.-Y. Wang and B.-F. Shi, Org. Lett., 2015, 17, 4094 CrossRef CAS PubMed; (h) W. Song and L. Ackermann, Angew. Chem., Int. Ed., 2012, 124, 8376 CrossRef.
- (a) J. Zhang, H. Chen, C. Lin, Z. Liu, C. Wang and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 12990 CrossRef CAS PubMed; (b) W. Yu, W. Zhang, Z. Liu and Y. Zhang, Chem. Commun., 2016, 52, 6837 RSC; (c) Q. Yan, T. Xiao, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2016, 358, 2707 CrossRef CAS; (d) Q. Yan, Z. Chen, Z. Liu and Y. Zhang, Org. Chem. Front., 2016, 3, 678 RSC; (e) W. Yu, W. Zhang, Y. Liu, Y. Zhou, Z. Liu and Y. Zhang, RSC Adv., 2016, 6, 24768 RSC.
- (a) C. A. Miller and L. M. Long, J. Am. Chem. Soc., 1951, 73, 4895 CrossRef CAS; (b) F. A. Luzzio, D. Y. Duveau, E. R. Lepper and W. D. Figg, J. Org. Chem., 2005, 70, 10117 CrossRef CAS PubMed; (c) T. Ishiyama, K. Tokuda, T. Ishibashi, A. Ito, S. Toma and Y. Ohno, Eur. J. Pharmacol., 2007, 572, 160 CrossRef CAS PubMed.
- (a) F. Wang, G. Song, Z. Du and X. Li, J. Org. Chem., 2011, 76, 2926 CrossRef CAS PubMed; (b) C. Zhu and J. R. Falck, Chem. Commun., 2012, 48, 1674 RSC; (c) V. Lanke, K. R. Bettadapur and K. R. Prabhu, Org. Lett., 2015, 17, 4662 CrossRef PubMed; (d) K. R. Bettadapur, V. Lanke and K. R. Prabhu, Org. Lett., 2015, 17, 4658 CrossRef PubMed; (e) W. Miura, K. Hirano and M. Miura, Org. Lett., 2015, 17, 4034 CrossRef CAS PubMed; (f) S. Sharma, S. H. Han, Y. Oh, N. K. Mishra, S. H. Lee, J. S. Oh and I. S. Kim, Org. Lett., 2016, 18, 2568 CrossRef CAS PubMed; (g) P. Keshri, K. R. Bettadapur, V. Lanke and K. R. Prabhu, J. Org. Chem., 2016, 81, 6056 CrossRef CAS PubMed; (h) Q. Wang, Y. Li, Z. Qi, F. Xie, Y. Lan and X. Li, ACS Catal., 2016, 6, 1971 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of compounds 3aa–3na, 3ab–3ad, 5aa–5oa, 5ab–5ag, 6, 7, 7′ and 8; X-ray crystal data for compounds 5aa and 7. CCDC 1486236 and 1492223. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00479b |
| This journal is © the Partner Organisations 2017 |