
A microporous yttrium metal–organic framework of an unusual nia topology for high adsorption selectivity of C2H2 and CO2 over CH4 at room temperature†
Kang
Liu
a,
Xu
Li
b,
Dingxuan
Ma
a,
Yi
Han
a,
Baiyan
Li
c,
Zhan
Shi
 c,
Zhenjiang
Li
d and
Lei
Wang
*a
c,
Zhenjiang
Li
d and
Lei
Wang
*a
aKey Laboratory of Eco-chemical Engineering, Ministry of Education, Laboratory of Inorganic Synthesis and Applied Chemistry, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: inorchemwl@126.com; Fax: +86 532-840-22681
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 117576, Singapore
cState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China
dKey Laboratory of Polymer Material Advanced Manufacturings Technology of Shandong Provincial, College of Electromechanical Engineering, College of Sino-German Science and Technology, Qingdao University of Science and Technology, Qingdao 266061, P. R. China
First published on 5th June 2017
Abstract
A new microporous yttrium metal–organic framework Y-H3TDPAT [H6TDPAT = 2,4,6-tris(3,5-dicarboxylphenyl-amino)-1,3,5-triazine] with Lewis basic sites on the pore surface was solvothermally synthesized and structurally determined as an unusual (6,6)-connected nia topology. The small pores and the Lewis basic sites within the desolvated Y-H3TDPAT have improved the affinity for C2H2 and CO2 which has been revealed in their large isosteric heats of adsorption (38.2 and 30.9 kJ mol−1). In addition, highly selective separation of C2H2/CH4 and CO2/CH4 at room temperature makes Y-H3TDPAT possess a potential application in natural gas purification.
Introduction
Nowadays, the energy crisis has caused worldwide concern and some global issues need to be resolved with urgency. What is worrying the world greatly now is the fossil fuel shortage in the near future, including coal, oil and natural gas. Improving the energy efficiency of fossil fuel would provide a roadmap to utilize resources rationally. Among them, natural gas purification has drawn more attention from materials scientists and chemists in recent years.1–3 Methane (CH4), as the main ingredient of natural gas and biogas, is a clean fuel for residential use and a useful C1 feedstock for the production of various chemicals in petrochemical industries, such as chloromethane and formaldehyde.4,5 However, impure CH4 as a starting material may complicate the separation processes of products for petrochemical and chemical applications. The major impurities in naturally occurring CH4 are C2 hydrocarbons, typically C2H2. Furthermore, CO2 can also be found in natural gas in large quantities. Some impurities may inhibit the conversion of CH4. For example, CO2 poisons the Li/MgO catalyst that converts CH4 into ethane and ethylene.6 Materials that can preferentially capture C2H2 and CO2 over CH4 are very desirable. The commonly used cryogenic distillation technology for the separation of C2H2 and CO2 from CH4 is very energy consuming. Therefore, it is urgent to develop new effective adsorbents for reduction of the cost to produce high-purity natural gas.Metal–organic frameworks (MOFs) are a unique class of crystalline porous materials that have attracted growing interest on account of their extensive application scope in gas storage,7–9 separation,10–12 catalysis13–15 and optical devices.16–18 Owing to their high porosities, adjustable chemistry and tunable pores/shapes, a number of crucial and required gas separations, such as CO2/N2 separation,19,20 O2/N2 separation,21,22 light hydrocarbon separation,23,24etc., have been achieved by utilizing novel MOFs as absorbents. Among the diverse gas separations, separation of C2H2 and CO2 over CH4 is practical and needs to be further developed.
The establishment of the permanent porosity is absolutely vital to explore the gas separation performance of Ln-MOF materials. Lanthanide ions have higher coordination numbers than transition-metal ions, which is beneficial to stabilize the frameworks.25–27 To date, compared with the huge amounts of transition-metal MOFs, there are obviously less porous Ln-MOFs that have been exploited for the application of gas storage and separation.28,29 In addition, the incorporation of specific sites, such as Lewis basic sites (LBSs) onto the walls of porous MOFs can dramatically improve their gas storage and separation capacities.30–32 Although the present research of porous MOFs for the separation of C2H2/CH4 and CO2/CH4 has seen some progress, examples of Ln-MOFs with rich LBSs for high adsorption selectivity of these gas molecules at room temperature are relatively scarce.
Recently, we have prepared a dendritic six-carboxylate ligand, 2,4,6-tris(3,5-dicarboxylphenylamino)-1,3,5-triazine (H6TDPAT).33 Self-assembly of the H3TDPAT3− ligand with Y3+ ions under solvothermal conditions afforded a 3D nia-type Ln-MOF called Y-H3TDPAT. The resultant microporous Y-H3TDPAT features rich LBSs, which endow it with interesting performances in the separation of C2H2/CH4 and CO2/CH4 at room temperature.
Experimental section
Materials and measurements
All solvents and reagents used in this study are commercially available and were used as received without further purification. Fourier-transform (FT-IR) spectra were acquired on a Bruker IFS-66 V/S FT-IR spectrometer. Thermogravimetric measurement was carried on a pre-weighed sample in a N2 stream using a Netzsch STA449C apparatus with a heating rate of 10 °C min−1. Elemental analyses for C, H, and N were performed on a vario MICRO CHN Analyzer. Powder X-ray diffraction (PXRD) data were obtained using a Bruker D8 ADVANCE diffractometer (Cu-Kα, 1.5418 Å).Gas sorption measurements
A Micromeritics ASAP 2020 instrument was used to measure gas adsorption characteristics. The guest solvent molecules in the pores of the freshly prepared sample of Y-H3TDPAT were exchanged with acetone molecules. Then the sample was activated at 393 K for 10 h with high vacuum (the outgas rate was <5 μm Hg min−1). The adsorption experiment was maintained at 273 K with an ice-water bath and 77 K with liquid nitrogen, respectively. As the circulation constant temperature water bath was set up, the temperature for the adsorption isotherms was constant at 298 K.The virial equation is used to analyze the isotherm data:34
| ln(n/p) = A0 + A1n + A2n2 +⋯ | (1) |
Prediction of the gas adsorption selectivity by IAST
Ideal adsorption solution theory (IAST) was employed for prediction of the binary mixture adsorption from the experimental single-component isotherms.35,36 In order to carry out the integrations required by IAST, the pure-gas isotherms should be fitted by a suitable model. Practices show that several methods are available to process the curve fitting. We found that the dual-site Langmuir–Freundlich equation was successful in fitting for this set of data. As can be seen in Fig. S19–S21 and Table S3 (ESI†), the results from the model match the isotherms very well (R2 > 0.99999).Here, N is the adsorbed amount per unit weight of adsorbing substance (mmol g−1), Nmax1 and Nmax2 are the saturation adsorption capacities on sites 1 and 2 (mmol g−1), P is the pressure of the bulk gas in a state of equilibrium with the adsorbed phase (kPa), b1 and b2 represent the coefficients of adsorption affinity on sites 1 and 2 (1 kPa−1), and n1 and n2 are the deviations from an ideal uniform surface. The model parameters were then utilized for prediction of a multi-component adsorption process with IAST.
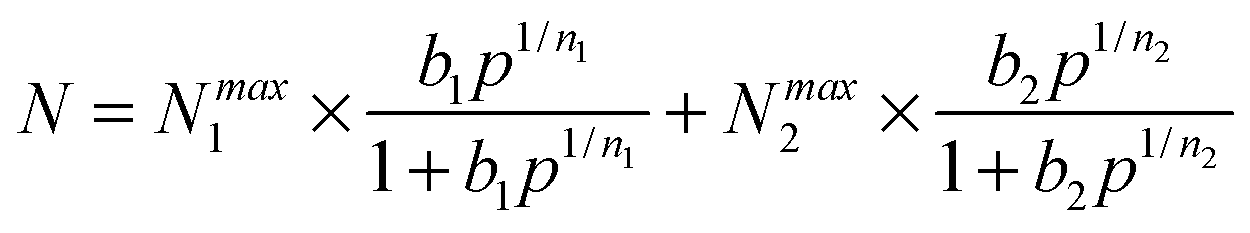 | (2) |
GCMC simulation
The density distribution of C2H2H molecules was calculated through Grand Canonical Monte Carlo (GCMC) simulation at 1 bar and 298 K. The periodic boundary conditions and Universal force field (UFF) were used.37 The VDW interactions between C2H2 molecules and the MOF were calculated using Lennard-Jones potentials. Electrostatic interaction was evaluated through the Ewald summation method. Charge equilibration uses the specified method to redistribute the overall charge on the C2H2 molecules and atoms of the framework.38 All the GCMC simulations were carried out using the Sorption module in Material Studio software.39Synthesis of Y-H3TDPAT
Ultrasonic dissolution with Y(NO3)3·6H2O (19 mg, 0.05 mmol) and H6TDPAT (15 mg, 0.025 mmol) was performed in 3 mL of DMF and 2 mL of H2O. Then 0.10 mL of nitric acid was added, and the mixture was placed in a teflon-lined stainless steel vessel and heated at 120 °C for 48 h, then cooled to room temperature over 24 h. Block colorless crystals were obtained to give Y-H3TDPAT. Anal. calcd for C27H15N6O12Y (%): C, 46.00; H, 2.13; N, 11.93. Found: C, 45.64; H, 2.20; N, 12.09. IR (cm−1): 3645(w), 3282(m), 3074(m), 2961(m), 2772(m), 2467(m), 1573(s), 1499(s), 1372(s), 1106(m), 1025(m), 846(m), 783(s), 725(m), 609(m), 499(w), 427(m) (Fig. S3, ESI†).X-ray single-crystal diffraction
Data collection for Y-H3TDPAT was carried out on a Rigaku RAXIS-RAPID-based diffractometer with graphite monochromatized Mo-Kα radiation (λ = 0.71073 Å) at 293 K. The SADBAS program was applied to the data for the corrections of incident and diffracted ray absorption effects.40 The SHELXTL program was used to solve the crystal structure by a direct method and refine the data by the full matrix least-squares against F2 values.41 The non-hydrogen atoms were located from Fourier maps successfully and refined through anisotropic thermal parameters. On account of some disordered solvent molecules in Y-H3TDPAT, their diffraction contributions were removed by utilizing the PLATON/SQUEEZE.42 Crystal data of Y-H3TDPAT are listed in Table S2 (ESI†).Results and discussion
Crystals of Y-H3TDPAT suitable for X-ray analysis were obtained by carefully adjusting the pH, solvent and temperature using a solvothermal method. Single crystal X-ray crystallographic analysis reveals that Y-H3TDPAT crystallizes in a non-centrosymmetric trigonal P31c space group. The asymmetric unit contains one Y3+ ion and one-sixth of the ligand H3TDPAT3− (Fig. S1, ESI†). In every H3TDPAT3− ligand, the dihedral angles of the central triazine ring with three terminal benzene rings are all 54.66°. The larger torsion dihedral angles in the ligand are probably due to the semi-rigid connection effect. As shown in Fig. 1a, each Y3+ ion is nine-coordinated adopting a distorted monocapped square antiprismatic molecular geometry and among them there are nine oxygen atoms from the six carboxylate groups of H3TDPAT3− ligands. The H3TDPAT3− ligand acts as a μ6-bridge to link six Y3+ ions to construct a 3D structure via three bidentate chelating (μ1-η1:η1) carboxyl groups and three monodentate carboxyl groups (Fig. 1b). Their coordination bond lengths are 2.2595(19), 2.3559(19) and 2.754(2) Å, respectively.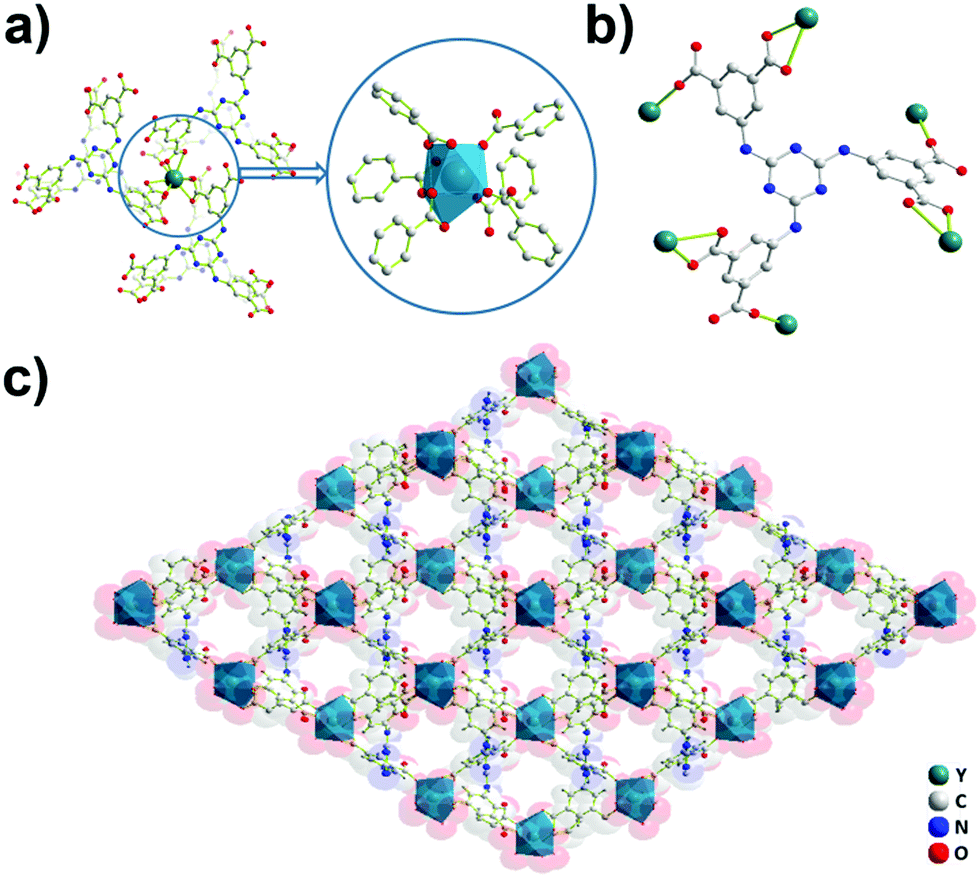 | ||
| Fig. 1 Single crystal structure of Y-H3TDPAT indicating (a) the coordination environment of Y3+ ions in Y-H3TDPAT, (b) the coordination mode of H3TDPAT3−, and (c) the 3D framework of Y-H3TDPAT along the b axis. | ||
There are open channels along the b direction in Y-H3TDPAT, with a diameter of about 7 Å (excluding van der Waals radii) (Fig. 1c and Fig. S2, ESI†). The void space accounts for approximately 51.4% of the whole crystal volume (1154.7 Å3 out of the 2246.4 Å3 per unit cell volume) by PLATON analysis.42 To achieve a better insight into the structure of Y-H3TDPAT, topological analysis was carried out.43 Each simplified framework in Y-H3TDPAT can be topologically represented as a binodal (6,6)-connected nia topology by reducing Y3+ ions and H3TDPAT3− ligands as two different 6-connected nodes (Fig. 2a–c). Yaghi et al. reported that a nia-type porous MOF can be generated by connecting an octahedral Zn4O cluster as an inorganic building block (IBB) with a trigonal prismatic six-node ligand.44 Conversely, Zhu et al. reported that a porous nia-MOF can also be formed by connecting a trigonal prismatic In3O cluster IBB with an octahedral six-node ligand.45 However, nia-type MOFs with a lanthanide metal center and a six-node ligand are uncommon. One should explicitly recognize that the hexatopic linker can be simplified as four 3-coordinated (3-c) branch points as there are a few distinct types of octahedral node in basic nia nets (Fig. 2d).46 In this case, the derived net is jjt shown in Fig. 2e.
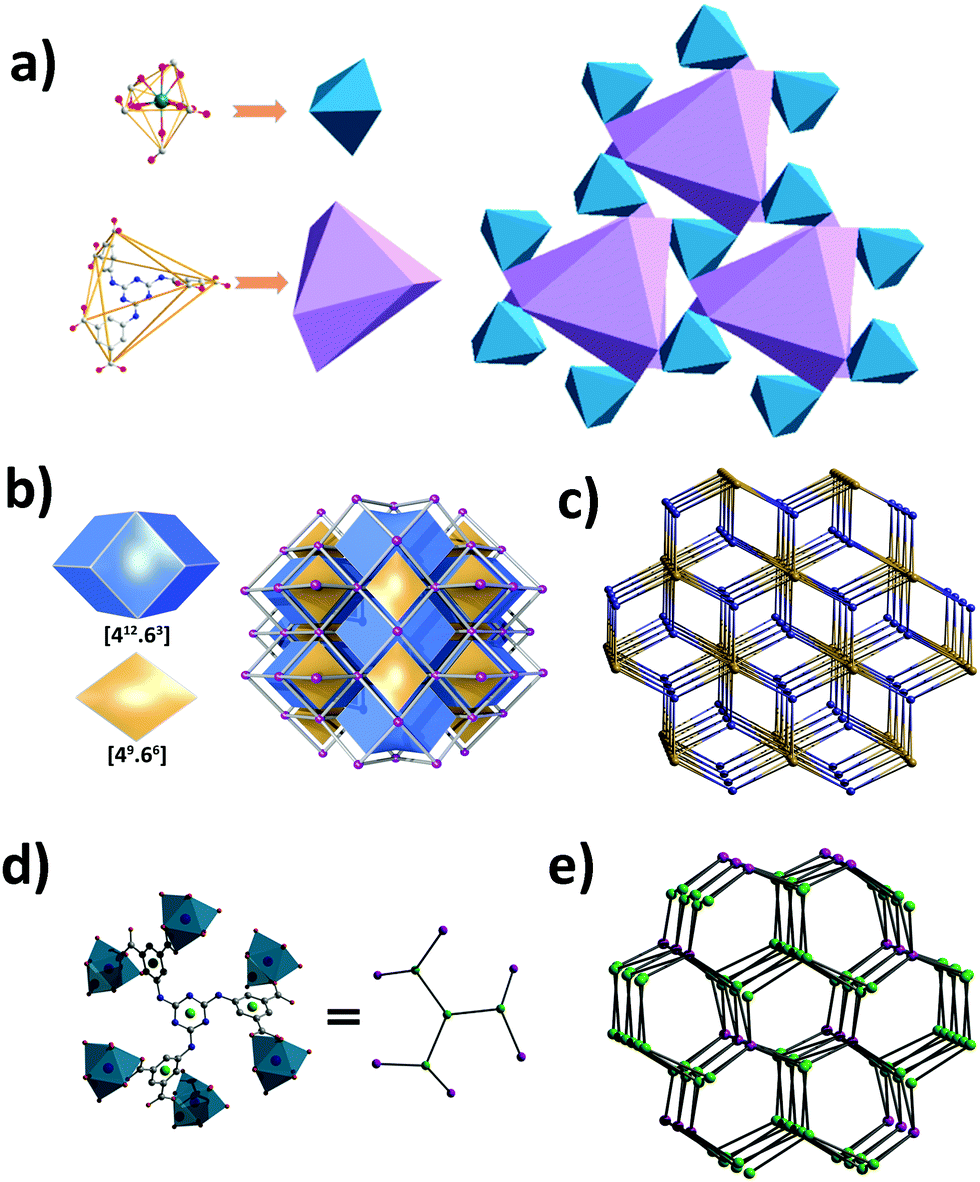 | ||
| Fig. 2 (a) Polyhedral representation of nia topology; (b) two types of tiles with a face symbol and natural tiling; (c) the (6,6)-connected nia topology of Y-H3TDPAT; (d) the hexatopic linker with branch points of the underlying net shown as green and magenta balls (respectively 3-c and 4-c vertices). C, gray; O, red; Y, magenta; (e) the jjt topology of Y-H3TDPAT. | ||
The combined feature of the small pore and the LBSs on the pore surfaces highlights the potential strong binding and high storage of gas molecules. The N2 sorption isotherm at 77 K showed that Y-H3TDPAT displayed type-I sorption behaviour with a BET surface area of 962 m2 g−1 (Langmuir surface area, 1105 m2 g−1) (Fig. 3 and Fig. S4, ESI†). Comparison of PXRD patterns of the as-synthesized sample, guest-free sample and sample after adsorption was made carefully (Fig. S5, ESI†). Moreover, the stabilities of Y-H3TDPAT in several common organic solvents (CH2Cl2, CHCl3, acetone, MeOH and EtOH) were also tested (Fig. S22, ESI†). Clearly, the main original skeleton of Y-H3TDPAT remained intact after these tests. The total pore volume calculated from the maximum amount of N2 adsorbed is 0.42 cm3 g−1, which is in good agreement with the theoretical pore volume (0.48 cm3 g−1).
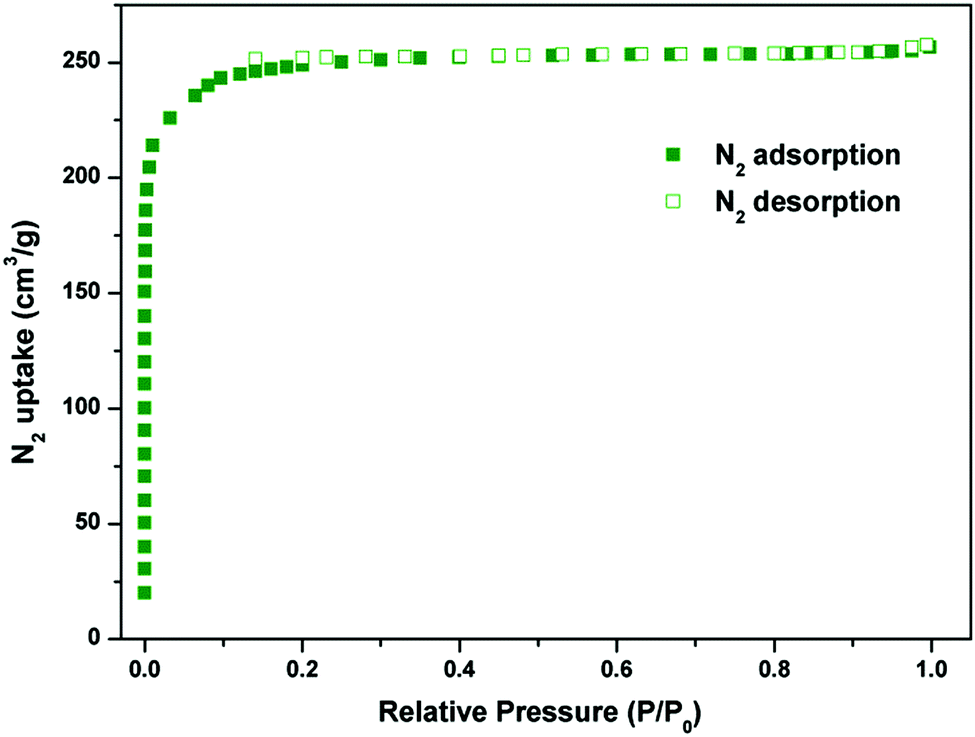 | ||
| Fig. 3 N2 sorption isotherm at 77 K. | ||
The above structural features of Y-H3TDPAT encouraged us to evaluate its potential application in gas capture. As shown in Fig. 4a and b, Y-H3TDPAT can take the amount of C2H2 (153.7 cm3 g−1), CO2 (129.2 cm3 g−1) and CH4 (25.3 cm3 g−1) at 1 bar and 273 K; and C2H2 (100.0 cm3 g−1), CO2 (70.1 cm3 g−1) and CH4 (17.5 cm3 g−1) at 1 bar and 298 K. The uptake capacity for C2H2 in Y-H3TDPAT at 298 K is lower than some famous MOFs with high density unsaturated metal sites, such as HKUST-1 (201 cm3 g−1),47 CoMOF-74 (197 cm3 g−1)48 and UTSA-20 (150 cm3 g−1).49 However, this value is higher than the ones with LBSs such as UTSA-5 (59.8 cm3 g−1)50 and [Zn4(dmf)(ur)2(ndc)4] (32.5 cm3 g−1).51 It is also significantly higher than some MOFs with steady large pores and high surface areas such as MOF-5 (26 cm3 g−1) and ZIF-8 (25 cm3 g−1),47 which represent the best C2H2 adsorption property among the MOFs with small pores and LBSs. This illustrates that the combined feature of LBSs (triazine and imino groups) and small pore size lead to the efficient capture of C2H2 molecules in Y-H3TDPAT. A comparison of C2H2 adsorption by some other porous MOFs is given in Table S1 (ESI†).
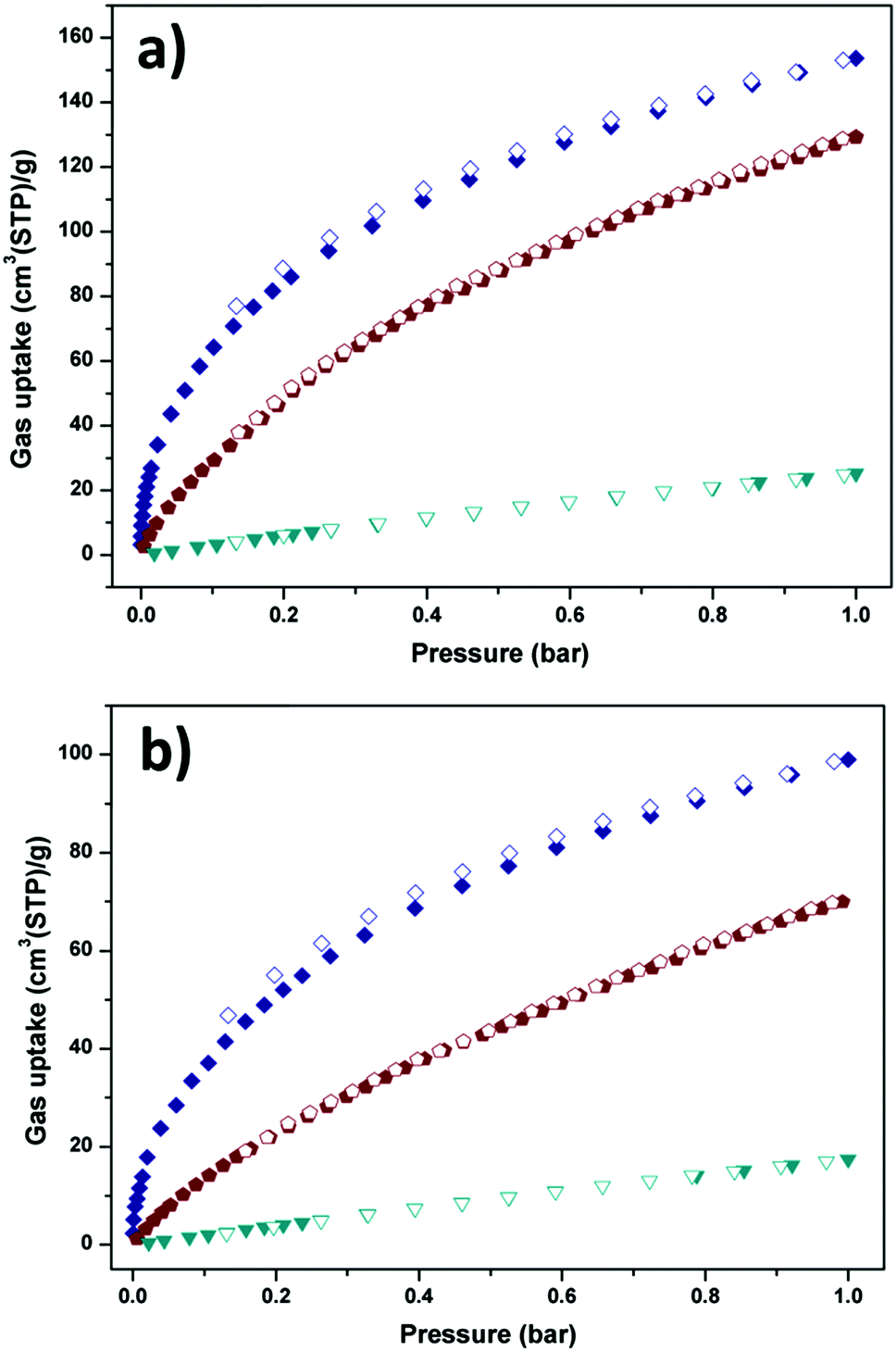 | ||
| Fig. 4 C2H2 (navy), CO2 (wine) and CH4 (dark cyan) sorption isotherms of Y-H3TDPAT at (a) 273 K and (b) 298 K. Solid symbols: adsorption, open symbols: desorption. | ||
The coverage-dependent isosteric heats of adsorption (Qst) for C2H2, CO2 and CH4 were calculated using the virial method, which is a dependable and well-established methodology fitting from the pure component isotherms at 298 K and 273 K, as given in Table 1 and Fig. S7–S12 (ESI†). The isosteric heats of adsorption at zero coverage are 38.2, 30.9 and 13.5 kJ mol−1 for C2H2, CO2 and CH4, respectively. It is noteworthy that Qst for C2H2 is higher than that for USTA-20 (30.8 kJ mol−1)49 and HKUST-1 (30.4 kJ mol−1)47 with high density unsaturated metal sites and ZJU-5 (35.8 kJ mol−1)52 with both unsaturated metal sites and LBSs. The Qst for CO2 is also comparable with those for the MOFs with LBSs.53 Furthermore, the Qst for C2H2 and CO2 is nearly 25 kJ mol−1 and 17 kJ mol−1 larger than that for CH4 (13.5 kJ mol−1), respectively. The marked feature of Y-H3TDPAT is the fairly higher Henry's law selectivity of 77.2 for C2H2/CH4 at 298 K. To the best of our knowledge, the C2H2/CH4 adsorption selectivity of 77.2 is the highest one ever reported among the MOFs containing only LBSs (Table 2 and Fig. S13–S18, ESI†). The highly selective adsorption for C2H2/CH4 and CO2/CH4 highlights the potential of Y-H3TDPAT in practical nature gas purification. GCMC simulation reveals that the adsorbed C2H2 molecules are associated with particular adsorption sites. Upon adsorption, C2H2 can be accommodated between the two 1,3,5-triazine rings (Fig. 5a). These two face to face π(C2H2)⋯π(-triazine) stacking interactions cause relatively strong binding between C2H2 and Y-H3TDPAT. The density distribution of the center-of-mass of C2H2 molecules obtained from GCMC simulation illustrates that the C2H2 molecules prefer to locate at the electron-dense aromatic rings within the framework (Fig. 5b). Moreover, the 1D channel also allows other C2H2 molecules to locate inside, and these adsorbed C2H2 molecules are distributed in a wave type arrangement with intermolecular C–H⋯π interactions (Fig. S23, ESI†). The π(C2H2)⋯π(-triazine) and intermolecular C–H⋯π interactions do not exist between CH4 and the framework. The difference in the guest/framework interaction strength is likely the reason why the performance of separation for C2H2/CH4 is outstanding in Y-H3TDPAT at room temperature.
| Adsorbate | T [K] | K H [mol g−1 Pa−1] | A 0 [ln(mol g−1 Pa−1)] | A 1 [g mol−1] | R 2 | S ij | [kJ mol−1] |
|---|---|---|---|---|---|---|---|
| a The Henry's law selectivity for the gas component i over CH4 at the speculated temperature is calculated based on the equation: Sij = KH(i)/KH(CH4). | |||||||
| C2H2 | 273 | 2.157 × 10−6 | −13.047 ± 0.036 | −791.594 ± 21.697 | 0.99700 | 147.5 | 38.2 |
| 298 | 6.838 × 10−7 | −14.196 ± 0.044 | −885.088 ± 33.351 | 0.99434 | 77.2 | ||
| CO2 | 273 | 2.211 × 10−7 | −15.325 ± 0.019 | −436.273 ± 18.240 | 0.99304 | 15.1 | 30.9 |
| 298 | 8.561 × 10−8 | −16.273 ± 0.008 | −633.389 ± 21.019 | 0.99561 | 9.7 | ||
| CH4 | 273 | 1.462 × 10−8 | −18.041 ± 0.002 | −307.759 ± 11.384 | 0.99455 | — | 13.5 |
| 298 | 8.850 × 10−9 | −18.543 ± 0.000 | −158.466 ± 1.228 | 0.99976 | — | ||
| Compounds | Uptake of C2H2a | Uptake of CH4 | Q st (CH4)b | Q st (C2H2) | S ij | Ref. |
|---|---|---|---|---|---|---|
| a The unit for the gas uptake is cm3 (STP) per gram. b Q st (kJ mol−1) is the enthalpy at zero coverage. c S ij is the Henry's law selectivity. | ||||||
| Cu-TDPAT | 178 | 28.3 | 20.7 | 42.5 | 127.1 | 33 |
| Cu-TDPAH | 155 | 22.1 | 13.8 | 23.5 | 80.9 | 54 |
| Y-H3TDPAT | 100.0 | 17.5 | 13.5 | 38.2 | 77.2 | This work |
| UTSA-50a | 90.6 | 18.8 | 18.6 | 39.4 | 68 | 55 |
| UTSA-15a | 34 | 2.5 | 13.6 | 39.5 | 55.6 | 56 |
| UTSA-36a | 57 | 13 | 24.4 | 29.0 | 13.8 | 57 |
| Zn5(BTA)6(TDA)2 | 44 | 10 | 26.1 | 37.3 | 15.5 | 58 |
| [Zn4(OH)2(1,2,4-BTC)2] | 53 | 10 | 14.8 | 28.2 | 14.7 | 59 |
| Cu(BDC-OH) | 43 | 13 | 18.5 | 25.7 | 9.26 | 60 |
| UTSA-38a | 64 | — | 18.9 | 24.7 | 5.6 | 61 |
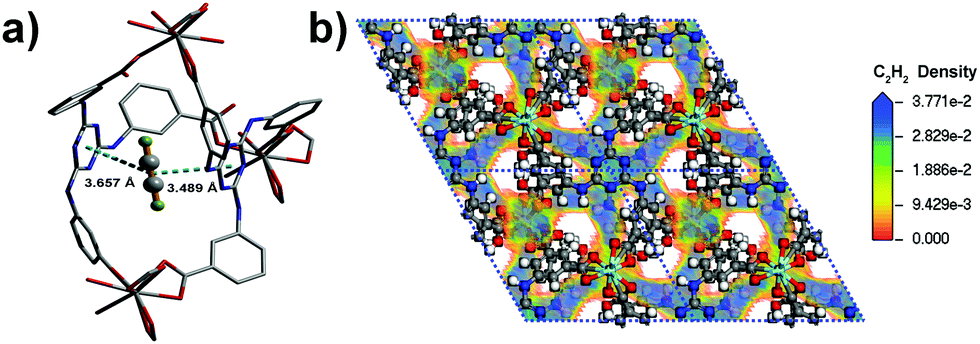 | ||
| Fig. 5 (a) The C2H2 molecule sits right between the two 1,3,5-triazine rings. Multiple-point interactions of the C2H2 with the framework: d[center(C2H2)⋯center(-triazine)(left)] = 3.657 Å, d[center(C2H2)⋯center(-triazine)(right)] = 3.489 Å; (b) density distribution of the center-of-mass of C2H2 molecules in the unit cell of Y-H3TDPAT at 298 K and 1 bar simulated by GCMC. | ||
Since the binary mixture adsorption isotherms cannot be quickly and conveniently measured, it is essential to use a proper adsorption model, such as ideal adsorbed solution theory (IAST), for prediction of the bi-component adsorption from the experimental single-component isotherms. Fig. 6 shows the IAST calculations of the adsorption selectivity for equimolar C2H2/CH4 and CO2/CH4 mixtures at 298 K in Y-H3TDPAT. The C2H2/CH4 selectivity (∼188) and the CO2/CH4 selectivity (∼11) have their maximum values at a relatively low pressure and decrease sharply as the pressure rises. The selectivity reaches a plateau at about 40 kPa and stays fairly stable under higher pressures. The values at 100 kPa are estimated to be ∼32 for C2H2/CH4 and ∼7 for CO2/CH4, respectively. Fractionation of these gaseous mixtures is expected to be practically feasible.
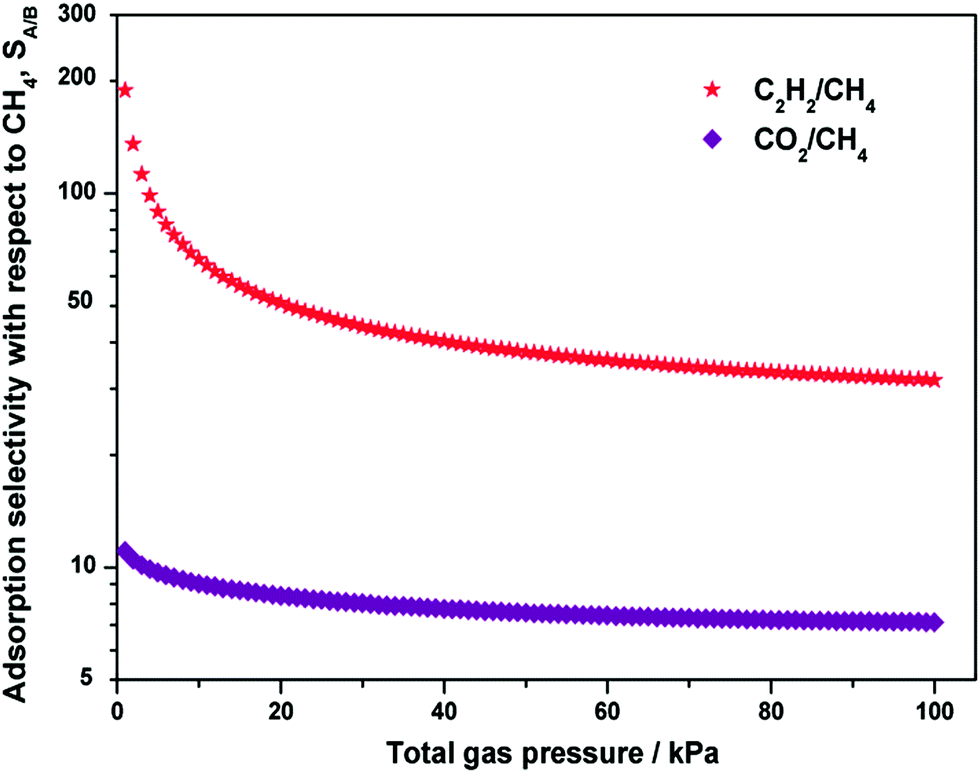 | ||
| Fig. 6 Calculations of the adsorption selectivity, SA/B, for equimolar C2H2/CH4 and CO2/CH4 mixtures at 298 K in Y-H3TDPAT using IAST. | ||
Conclusions
In summary, we have successfully synthesized a porous Ln-MOF Y-H3TDPAT with a high density of LBSs on the pore surface. The collaborative enforcement of the small pores and LBSs has enabled Y-H3TDPAT to take up a moderately high amount of C2H2 gas molecules of 100 cm3 g−1 at 298 K and 1 bar, and furthermore to exhibit highly selective C2H2/CH4 and CO2/CH4 gas sorption with the Henry's law selectivity of 77.2 and 9.7, respectively. The selective adsorption properties of Y-H3TDPAT make it a promising candidate for CH4 purification and recycling. Further applications of this MOF, the combination of a dendritic hexacarboxylate linker and other lanthanide metal ions, and other related works are ongoing.Acknowledgements
We acknowledge National Supercomputing Center in Shenzhen for providing the computational resources and materials studio (version 6.1, module DMol3). This work was supported by the National Natural Science Foundation of China (No. 21601103, 51372125, 21571112 and 51572136), the Taishan Scholars Program of Shandong Province, the Natural Science Foundation of Shandong Province, China (No. ZR2016BP04 and ZR2016BQ28) and the Scientific and Technical Development Project of Qingdao (No. 17-1-1-78-jch).Notes and references
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- M. Tagliabue, D. Farrusseng, S. Valencia, S. Aguado, U. Ravon, C. Rizzo, A. Corma and C. Mirodatos, Chem. Eng. J., 2009, 155, 553–566 CrossRef CAS.
- Y. He, W. Zhou, R. Krishna and B. Chen, Chem. Commun., 2012, 48, 11813–11831 RSC.
- U. Olsbye, O. V. Saure, N. B. Muddada, S. Bordiga, C. Lamberti, M. H. Nilsen, K. P. Lillerud and S. Svelle, Catal. Today, 2011, 171, 211–220 CrossRef CAS.
- E. Mcfarland, Science, 2012, 338, 340–342 CrossRef CAS PubMed.
- S. Al-Zahrani, Q. Song and L. L. Lobban, Ind. Eng. Chem. Res., 1994, 33, 251–258 CrossRef CAS.
- O. K. Farha, A. O. Yazaydin, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- D. Alezi, Y. Belmabkhout, M. Suyetin, P. M. Bhatt, L. J. Weselinski, V. Solovyeva, K. Adil, I. Spanopoulos, P. N. Trikalitis, A. Emwas and M. Eddaoudi, J. Am. Chem. Soc., 2015, 137, 13308–13318 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- Y. Bae, C. Y. Lee, K. C. Kim, O. K. Farha, P. Nickias, J. T. Hupp, S. T. Nguyen and R. Q. Snurr, Angew. Chem., Int. Ed., 2012, 51, 1857–1860 CrossRef CAS PubMed.
- H. Liu, Y. He, J. Jiao, D. Bai, D. Chen, R. Krishna and B. Chen, Chem. – Eur. J., 2016, 22, 14988–14997 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- C. M. McGuirk, M. J. Katz, C. L. Stern, A. A. Sarjeant, J. T. Hupp, O. K. Farha and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 919–925 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- Y. Cui, T. Song, J. Yu, Y. Yang, Z. Wang and G. Qian, Adv. Funct. Mater., 2015, 25, 4796–4802 CrossRef CAS.
- H. Zhang, P. Lin, X. Shan, F. Du, Q. Li and S. Du, Chem. Commun., 2013, 49, 2231–2233 RSC.
- Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed.
- R. Vaidhyanathan, S. S. Iremonger, G. K. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- P. S. Nugent, V. L. Rhodus, T. Pham, K. Forrest, L. Wojtas, B. Space and M. J. Zaworotko, J. Am. Chem. Soc., 2013, 135, 10950–10953 CrossRef CAS PubMed.
- S. Shimomura, M. Higuchi, R. Matsuda, K. Yoneda, Y. Hijikata, Y. Kubota, Y. Mita, J. Kim, M. Takata and S. Kitagawa, Nat. Chem., 2010, 2, 633–637 CrossRef CAS PubMed.
- A. Demessence and J. R. Long, Chem. – Eur. J., 2010, 16, 5902–5908 CrossRef CAS PubMed.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed.
- J. Pires, M. L. Pinto and V. K. Saini, ACS Appl. Mater. Interfaces, 2014, 6, 12093–12099 CAS.
- B. Li, H. Wen, Y. Cui, G. Qian and B. Chen, Prog. Polym. Sci., 2015, 48, 40–84 CrossRef CAS.
- K. Liu, X. Zhang, X. Meng, W. Shi, P. Cheng and A. K. Powell, Chem. Soc. Rev., 2016, 45, 2423–2439 RSC.
- V. Guillerm, Ł. J. Weseliński, Y. Belmabkhout, A. J. Cairns, V. D'Elia, Ł. Wojtas, K. Adil and M. Eddaoudi, Nat. Chem., 2014, 6, 673–680 CAS.
- S. Roy, A. Chakraborty and T. K. Maji, Coord. Chem. Rev., 2014, 273, 139–164 CrossRef.
- Y. Chen and S. Ma, Rev. Inorg. Chem., 2012, 32, 81–100 CrossRef CAS.
- L. Li, S. Tang, C. Wang, X. Lv, M. Jiang, H. Wu and X. Zhao, Chem. Commun., 2014, 50, 2304–2307 RSC.
- C. Song, J. Hu, Y. Ling, Y. Feng, R. Krishna, D. Chen and Y. He, J. Mater. Chem. A, 2015, 3, 19417–19426 CAS.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Angew. Chem., Int. Ed., 2012, 51, 1826–1829 CrossRef CAS PubMed.
- B. Li, Z. Zhang, Y. Li, K. Yao, Y. Zhu, Z. Deng, F. Yang, X. Zhou, G. Li, H. Wu, N. Nijem, Y. J. Chabal, Z. Lai, Y. Han, Z. Shi, S. Feng and J. Li, Angew. Chem., Int. Ed., 2012, 51, 1412–1415 CrossRef CAS PubMed.
- X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159–2171 CrossRef CAS PubMed.
- Y. S. Bae, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, J. T. Hupp and R. Q. Snurr, Chem. Commun., 2008, 4135–4137 RSC.
- M. Heuchel, R. Q. Snurr and E. Buss, Langmuir, 1997, 13, 6795–6804 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10039 CrossRef CAS.
- A. K. Rappe and W. A. Goddard, J. Phys. Chem., 1991, 95, 3358–3363 CrossRef CAS.
- Materials Studio V4.0, Accelrys Software Inc., San Diego, 2005 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction for Area Detector Data, University of Gottingen, Gottingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL Version 5.1 Software Reference Manual, Bruker AXS. Inc., Madison, WI, 1997 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- V. A. Blatov and A. P. Shevchenko, TOPOS-Version 4.0 Professional, Samara State University, Samara, Russia, 2006 Search PubMed.
- H. K. Chae, M. Eddaoudi, J. Kim, S. I. Hauck, J. F. Hartwig, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 11482–11483 CrossRef CAS PubMed.
- J. Jia, F. Sun, T. Borjigin, H. Ren, T. Zhang, Z. Bian, L. Gao and G. Zhu, Chem. Commun., 2012, 48, 6010–6012 RSC.
- M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343–1370 CrossRef CAS PubMed.
- S. Xiang, W. Zhou, J. M. Gallegos, Y. Liu and B. Chen, J. Am. Chem. Soc., 2009, 131, 12415–12419 CrossRef CAS PubMed.
- S. Xiang, W. Zhou, Z. Zhang, M. A. Green, Y. Liu and B. Chen, Angew. Chem., Int. Ed., 2010, 49, 4615–4618 CrossRef CAS PubMed.
- Y. He, R. Krishna and B. Chen, Energy Environ. Sci., 2012, 5, 9107–9120 CAS.
- G. Chen, Z. Zhang, S. Xiang and B. Chen, CrystEngComm, 2013, 15, 5232–5235 RSC.
- S. A. Sapchenko, D. N. Dybtsev, D. G. Samsonenko, R. V. Belosludov, V. R. Belosludov, Y. Kawazoe, M. Schröder and V. P. Fedin, Chem. Commun., 2015, 51, 13918–13921 RSC.
- X. Rao, J. Cai, J. Yu, Y. He, C. Wu, W. Zhou, T. Yildirim, B. Chen and G. Qian, Chem. Commun., 2013, 49, 6719–6721 RSC.
- Z. Zhang, Y. Zhao, Q. Gong, Z. Li and J. Li, Chem. Commun., 2013, 49, 653–661 RSC.
- K. Liu, B. Li, Y. Li, X. Li, F. Yang, G. Zeng, Y. Peng, Z. Zhang, G. Li, Z. Shi, S. Feng and D. Song, Chem. Commun., 2014, 50, 5031–5033 RSC.
- H. Xu, Y. He, Z. Zhang, S. Xiang, J. Cai, Y. Cui, Y. Yang, G. Qian and B. Chen, J. Mater. Chem. A, 2013, 1, 77–81 CAS.
- Z. Chen, S. Xiang, H. D. Arman, J. U. Mondal, P. Li, D. Zhao and B. Chen, Inorg. Chem., 2011, 50, 3442–3446 CrossRef CAS PubMed.
- M. C. Das, H. Xu, S. Xiang, Z. Zhang, H. D. Arman, G. Qian and B. Chen, Chem. – Eur. J., 2011, 17, 7817–7822 CrossRef CAS PubMed.
- Z. Zhang, S. Xiang, Y. Chen, S. Ma, Y. Lee, T. Phely-Bobin and B. Chen, Inorg. Chem., 2010, 49, 8444–8448 CrossRef CAS PubMed.
- Z. Zhang, S. Xiang, X. Rao, Q. Zheng, F. R. Fronczek, G. Qian and B. Chen, Chem. Commun., 2010, 46, 7205–7207 RSC.
- Z. Chen, S. Xiang, H. D. Arman, P. Li, S. Tidrow, D. Zhao and B. Chen, Eur. J. Inorg. Chem., 2010, 3745–3749 CrossRef CAS.
- M. C. Das, H. Xu, Z. Wang, G. Srinivas, W. Zhou, Y. F. Yue, V. N. Nesterov, G. Qian and B. Chen, Chem. Commun., 2011, 47, 11715–11717 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1475480. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00164a |
| This journal is © the Partner Organisations 2017 |