
In situ alternative switching between Ti4+ and Ti3+ driven by H2O2 in TiO2 nanostructures: mechanism of pseudo-Fenton reaction†
Zhihe
Liu
 a,
Teng
Wang
b,
Xin
Yu
c,
Zhaoxin
Geng
*d,
Yuanhua
Sang
*a and
Hong
Liu
*a
a,
Teng
Wang
b,
Xin
Yu
c,
Zhaoxin
Geng
*d,
Yuanhua
Sang
*a and
Hong
Liu
*a
aState Key Laboratory of Crystal Materials, Shandong University, Jinan, Shandong 250100, China. E-mail: hongliu@sdu.edu.cn
bFaculty of Material Science and Chemistry, China University of Geosciences, Wuhan, 430070, China
cBeijing Institute of Nanoenergy and Nanosystems, Chinese Academy of Science, Beijing 100083, China
dSchool of Information Engineering, Minzu University of China, Beijing, 100081, China
First published on 31st May 2017
Abstract
Here, we report the discovery that Ti4+ in TiO2 nanostructures can be reduced in situ to Ti3+ in the presence of H2O2 to generate hydroxyl radicals for the oxidation of organic pollutants. The Ti4+ in the TiO2 nanostructure and the reduced Ti3+ driven by H2O2 can alternatively switch in situ, which endows the catalyst with a long life and excellent cyclic ability.
The ever-increasing issue of polluted water resources is currently one of the greatest concerns for science and technology.1,2 Among the various types of pollutants, organic chemicals drained into water resources are one of the most serious due to the fast growth of the manufacturing industry. Many water treatment methods and processes have been applied to eliminate organic chemicals and purify waste water, such as precipitation, bio-degradation, and film filtration.3–6 However, waste water treated by traditional water treatment methods still maintains a high chemical oxygen demand (COD), which cannot match the standard for industrial waste water disposal. Therefore, some techniques based on the oxidation or degradation of organic molecules in water have been used for the advanced treatment of waste water to further decrease COD, such as Fenton,7–10 micro-electrolysis,11 electrochemical catalytic oxidation,12 photocatalytic degradation,13–19 and photoelectrocatalytic methods.20,21 Among these oxidation methods, an advanced oxidation process called the Fenton reaction22 has strikingly contributed to purify dye waste water by applying the hydroxyl radical to destroy the C–C, C–H, C–N, C–P or C–S bonds in the organic components and turn them into inorganic micromolecular compounds. The classical Fenton oxidation process involves adding Fenton reagent (mostly a solution with +2 valence iron ions and H2O2) into polluted water and adjusting the pH of the system to acidic conditions for the oxidation of organic molecules. The usual Fenton mechanism is the oxidation of ferrous to ferric ions to decompose H2O2 into hydroxyl radicals, which is represented by eqn (1).
| Fe2+ + H2O2 → Fe3+ + OH− + ˙OH | (1) |
| Fesurf2+ + H2O2 → Fesurf3+ + OH− + ˙OH | (2) |
Titanium dioxide (TiO2), one of the most popular semiconductors, has been applied in many fields, such as electric ceramics, gas sensors, lithium-ion batteries, solar cells, supercapacitors, and photochromic materials,29–36 because of its excellent electronic band structure, abundance in the world, very good physical and chemical stability, and environmentally friendly properties.37–41 Because variable valences can exist in TiO2 nanostructures and because the valence of TiO2 can be changed by environmental conditions in the presence of H2O2, TiO2 is supposed to act as a Fenton catalyst in heterogeneous Fenton reactions.
Here, we report the discovery of the catalytic properties of TiO2 nanostructures for the degradation of organic pollutant molecules with H2O2 present in the reaction system. The catalytic ability of TiO2 with a high valence should originate from Ti3+ formed in situ by the H2O2 present in the system at a certain temperature, which differs from the heterogeneous Fenton process. Therefore, we defined this process as a pseudo-Fenton method. Compared with other Fenton catalysts, TiO2 nanomaterials possess a very high catalytic activity, perfect cyclic properties, and a high stability, and they do not consume the catalyst or form sludge during water treatment. This new concept of pseudo-Fenton catalysts and methods will have great applications in practical waste water treatment.
The material characteristics of phase composition and morphology were studied via X-ray diffraction (XRD), Raman spectroscopy, scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM), as shown in Fig. 1. As shown in Fig. 1a, all the diffraction peaks in the pattern can be unambiguously indexed to the tetragonal anatase type (JCPDS card no. 99-0008). The Raman spectrum shows that there are six characteristic Raman active modes (3Eg + 2B1g + A1g), with a Raman shift of Eg = 146.1 cm−1, which can be assigned to the anatase structure with the tetragonal space group I41/amd (Fig. 1b).42,43 The product powder is composed of aggregates of microspheres approximately 1–3 μm in diameter (Fig. 1c). The microspheres are assembled by TiO2 nanowires (Fig. 1d). The nanowires are approximately 15–20 nm in diameter and up to several hundred nanometers in length (Fig. 1e). The as-assembled TiO2 microstructure is hollow, similar to an urchin; therefore, it is called an urchin-like microstructure in this study. Moreover, the SAED pattern in Fig. 1f shows that the as-synthesized TiO2 has a polycrystalline structure, with a series of diffraction rings corresponding to the diffractions from the (101), (004), (200), (105), (204), (220), (215) and (224) planes, elucidating the formation of anatase TiO2, which is consistent with the XRD result. The lattice structure image of the assembled nanowires clearly shows excellent, successive 2D atomic lattices with a spacing of 0.35 nm, corresponding to (101) planes of anatase TiO2. The TiO2 nanowire grew along [010], which is the common growth direction of 1D TiO2 nanostructures (Fig. 1g).29
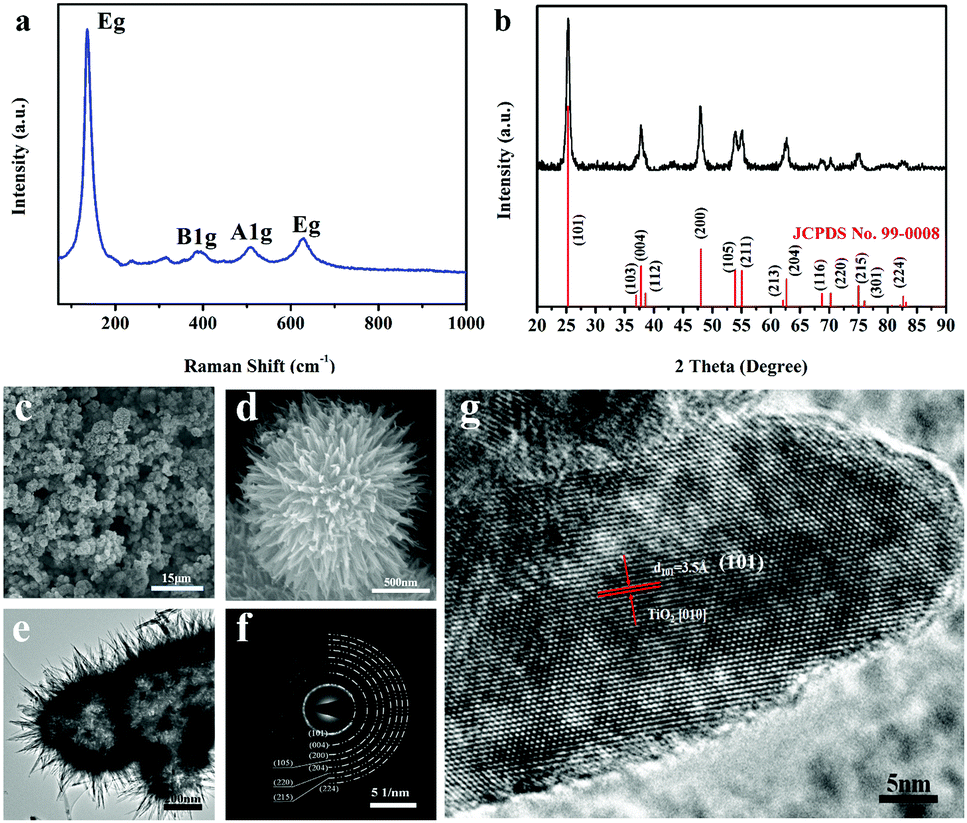 | ||
| Fig. 1 (a) XRD pattern of TiO2 microspheres; (b) Raman spectrum of TiO2 microspheres; (c and d) SEM images of TiO2 microspheres; (e) TEM images of the TiO2 microspheres and (f) the corresponding SAED pattern; (g) HRTEM image of TiO2 nanowires on the surface of the TiO2 microspheres. | ||
To evaluate the catalytic properties of TiO2 urchin-like nanostructures for H2O2 transformation, a 10 mL reaction system with methyl blue (MB) as a model pollutant was used.44 To eliminate the photocatalytic oxidation effect on TiO2 nanostructures during the pseudo-Fenton reaction process, the reaction was performed in a dark room. The MB degradation and cyclic MB degradation properties were recorded with various TiO2 catalyst amounts, H2O2 dosages, and reaction temperatures (Fig. 2). In the initial reaction system, the concentration of MB was 100 mg L−1, with 1.5 mL of H2O2. Different amounts of TiO2 urchin-like nanostructures were added in the reaction system to optimize the catalyst dosage. As shown in Fig. 2a, without TiO2 nanostructures, less than 5% of the MB is degraded in the system with 1.5 mL H2O2 and a temperature of 60 °C, even after a 50 min reaction. This finding indicates that the H2O2 is decomposed into hydroxyl radicals at a low rate at a certain temperature. Surprisingly, the degradation ratio of MB reaches 60% after a 50 min reaction by using only 1 mg of TiO2 nanostructures as the catalyst in the same reaction system. When the amount of TiO2 nanostructures increases, the MB degradation rate accelerates. It takes 30 min and 20 min to achieve the complete degradation of MB with 5 mg and 10 mg of TiO2 microspheres, respectively. This result indicates that TiO2 urchin-like nanostructures are the key factor in the oxidation reaction. Similarly, the effect of the amount of H2O2 on MB degradation was assessed by fixing the amount of TiO2 nanostructures at 5 mg and varying the amount of H2O2 in the system. Fig. 2b illustrates the degradation of MB in the reaction system when the volume of H2O2 varies from 0 to 2.0 mL. Without H2O2, TiO2 nanostructures cannot degrade MB in the dark despite a 5% physical adsorption. The degradation rate of MB can reach 92% in 50 min with only 0.5 mL of H2O2 added to the system. This result indicates that a small amount of H2O2 can endow the system with a very strong degradation rate of MB. When an increasing amount of H2O2 is added, the degradation rate of MB in the system increases. With over 1 mL of H2O2, MB is mostly removed in 50 min. Therefore, in the following experiments, we fixed the amounts of TiO2 urchin-like nanostructures at 5 mg and of H2O2 at 1.5 mL.
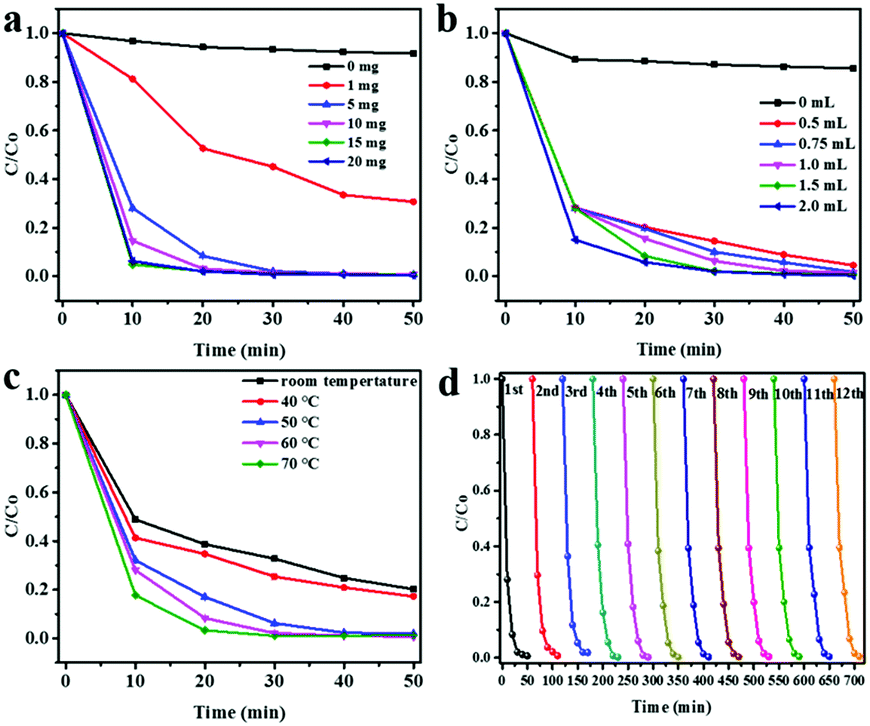 | ||
| Fig. 2 MB removal under various conditions. (a) Effect of catalyst dosage on MB degradation (10 mL of the initial MB concentration, 100 mg L−1; H2O2 1.5 mL, at 60 °C). Error bars represent standard deviation of the mean; (b) effect of H2O2 dosage on MB degradation (initial MB concentration, 100 mg L−1; TiO2, 5 mg, at 60 °C). Error bars represent standard deviation of the mean; (c) effect of temperature on MB degradation (initial MB concentration, 100 mg L−1; catalyst, 5 mg; H2O2, 1.5 mL). Error bars represent standard deviation of the mean; (d) recycling/degradation of MB in presence of TiO2 and H2O2 (10 mL of the initial MB concentration, 100 mg L−1; H2O2, 1.5 mL, at 60 °C; TiO2, 5 mg). | ||
As is well known, the solid-state catalyst-based Fenton reaction works at a certain temperature. The temperature of the system is a crucial factor in such a reaction.28 Therefore, the effect of temperature on the TiO2-based pseudo-Fenton reaction was assessed by varying the temperature (Fig. 2c). To our surprise, at room temperature the degradation rate of MB is over 70% after a 50 min reaction with 5 mg of TiO2 and 1.5 mL of H2O2, which clearly differs from that of the temperature requirement in the normal solid-state catalyst-based heterogeneous Fenton reaction system. A higher temperature results in a higher MB degradation rate. The complete degradation of MB can occur in 40 min at 50 °C. With temperatures over 60 °C, the MB in the system can be eliminated within 30 min. This TiO2-based pseudo-Fenton process is more efficient than the normal solid-state catalyst-based Fenton process.43 The cyclic performance of TiO2 urchin-like nanostructures was studied by repeating the pseudo-Fenton reaction with the same TiO2 catalyst under the same reaction conditions (Fig. 2d). After 10 cycles, the TiO2-based pseudo-Fenton reaction achieves a MB degradation rate of almost 100%. Superior to other Fenton systems, TiO2 nanostructures can be used repeatedly many times with the degradation efficiency almost unchanged. XRD patterns, SEM images, and Raman spectra of TiO2 microspheres obtained after 12 cycles show that the crystallinity and morphology hardly change even after several cycles in the TiO2-based pseudo-Fenton process, which further illustrates the stability of the catalytic properties of TiO2 (ESI,† Fig. S1). As is well known, the traditional Fenton reaction is a catalyst-consuming process, and the reaction stops if all the low valence ions change to high valence ions. Therefore, we think that TiO2 in this pseudo-Fenton reaction is not a conventional Fenton catalyst because it is a long-life, so a non-consumptive catalyst during the pseudo-Fenton process. The cyclic ability of the catalyst is a pivotal factor for its practical application. The pseudo-Fenton properties of TiO2 urchin-like nanostructures are much better than those of other solid-state catalysts such as Fe3O4, especially for their perfect cyclic activity (ESI,† Fig. S2 and Table S1). The UV-vis absorption spectra taken from the reaction system at different reaction times confirmed that MB was degraded by a mineralization process during the pseudo-Fenton process (ESI,† Fig. S3).
According to the mechanism of traditional Fenton and solid-state Fenton reactions, radical-involved oxidation of the organic pollutants should be initiated from the reaction between low valence metal ions and H2O2. In this system, TiO2 with a full 4+ valence should not possess the ability to catalytically decompose H2O2. However, the possible reaction between Ti4+ and H2O2 to form Ti3+ and HO2˙ makes the Fenton reaction become reasonable. Therefore, a pseudo-Fenton reaction mechanism in this TiO2-based system is suggested (Scheme 1). It is known that H2O2 can act as both an oxidizer and reducer.45 In this work, we take advantage of this special chemical behavior of H2O2 and the valence variation ability of the titanium element to realize Fenton reactions in this system. Inspired by the Fe3+-based Fenton process,46 when TiO2 is in contact with a H2O2 molecule, Ti4+ on the surface of TiO2 can react with H2O2 to generate HO2˙, with the reduction of Ti4+ to Ti3+. Then, the formed Ti3+ sequentially reacts with H2O2 to generate ˙OH, with the oxidation of Ti3+ to Ti4+ in TiO2 nanostructures. Both H2O˙ and ˙OH can act as oxidizers to degrade organic molecules in solution. Therefore, TiO2 coordinated with H2O2 has a very strong ability to degrade MB. During the pseudo-Fenton process driven by H2O2, Ti4+ can be reduced in situ to Ti3+, and Ti3+ can be oxidized in situ to Ti4+. The alternative switching between Ti4+ and Ti3+ ions endows TiO2 with good cyclic properties in the pseudo-Fenton process, which is the greatest difference between the TiO2 based pseudo-Fenton reaction and the conventional solid-state Fenton reaction.47 The commercial TiO2 product P25 also has a pseudo-Fenton performance but with a lower catalytic activity than that based on the urchin-like TiO2 nanostructures in this work (ESI,† Fig. S4). The enhanced catalytic activity of urchin-like TiO2 nanostructures should originate from their large surface area, which was 158.8167 m2 g−1 and 47.0438 m2 g−1 for urchin-like TiO2 nanostructures and P25, respectively (ESI,† Fig. S5).
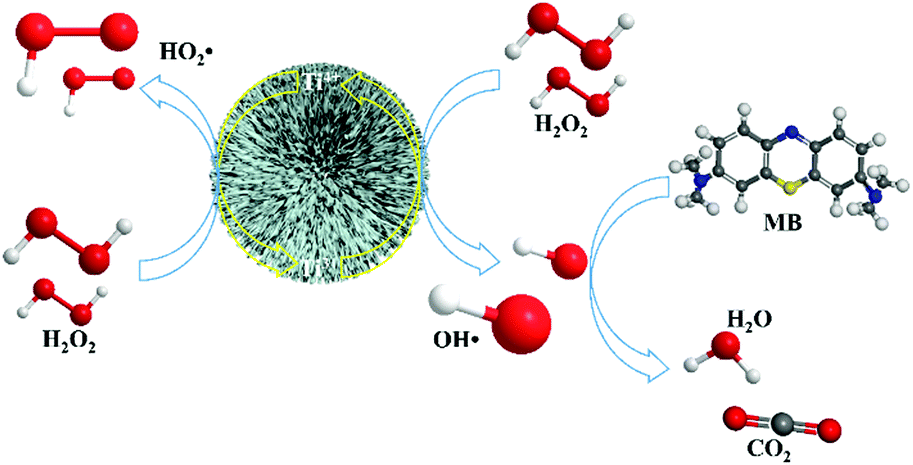 | ||
| Scheme 1 The mechanism for the pseudo-Fenton degradation of MB by TiO2 in the presence of H2O2. | ||
The existence of Ti3+ ions in the TiO2 urchin-like nanostructures in the presence of H2O2 should be fundamental to the proposed mechanism of the pseudo-Fenton process. Electron spin resonance (ESR) spectra of TiO2 urchin-like nanostructures were recorded at 90 K before and after being dipped in the H2O2 solution, focusing on the detection of Ti3+ ions. As shown in Fig. 3a, no signal of the Ti3+ ion can be detected in the as-synthesized TiO2 urchin-like nanostructures. However, the ESR spectrum of TiO2 urchin-like nanostructures treated with H2O2 gives rise to a very strong Ti3+ signal, which demonstrates the presence of Ti3+ in the sample. According to previous reports, when crystalline TiO2 is treated with a strong reductant such as N2H4, and H2 at a high temperature,42,48–51 the oxygen atoms in the lattice can be removed, and Ti3+ is naturally generated. In fact, the formation of Ti3+ in TiO2 nanostructures normally occurs under complex and harsh experimental conditions.52 However, in this work, Ti4+ ions can transfer to Ti3+ ions through a facile in situ process. This finding will be of great impact for the chemistry of titanium dioxide.
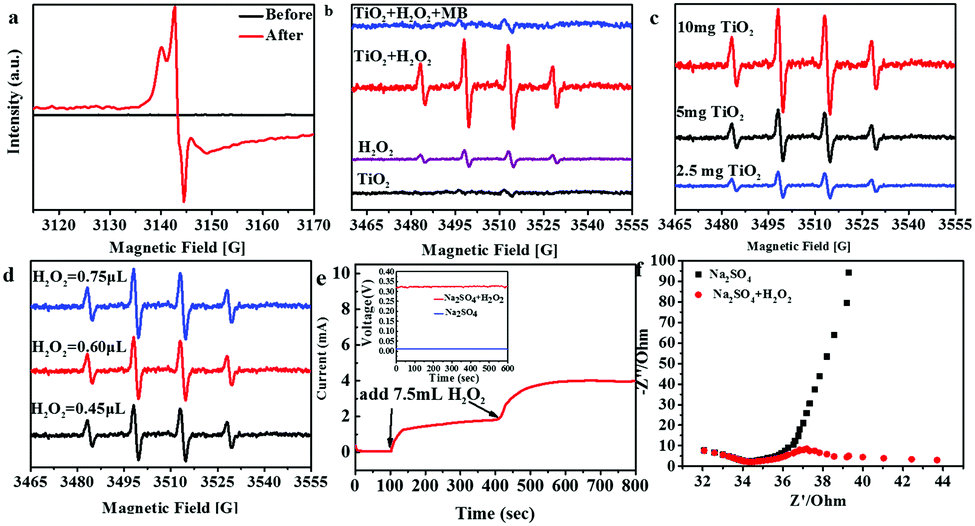 | ||
| Fig. 3 (a) The X-band ESR spectra of TiO2 before and after the reaction (T = 90 K) with H2O2; (b) 100 μL of suspension containing 0.1 mg TiO2 and 20 μL DMPO as the control; (c) all the samples were 50 μL volumes of mixtures of 7.5 μL H2O2 and 20 μL DMPO, with different quantities of TiO2; (d) all the samples were 50 μL volumes of mixtures of 0.1 mg TiO2 and 20 μL DMPO, with different concentrations of H2O2; (e) amperometric i–t curves (the initial potential is −0.35 V, and the volume of the electrolyte is 100 mL) and the corresponding open circuit potential–time curves; (f) AC impedance curves of TiO2 in different electrolytes (Na2SO4 and Na2SO4/H2O2). | ||
As proposed above, hydroxyl radicals act as putative contributors to the degradation of MB, where TiO2 was used as a catalyst to decompose H2O2.53 After the formation of Ti3+ in TiO2 urchin-like nanostructures was demonstrated, the identification of the catalytic properties of Ti3+ ions for the formation of hydroxyl radicals from H2O2 should be the final information needed for the proposed mechanism of the pseudo-Fenton process. The ESR technique was applied to monitor the formation of hydroxyl radicals in the presence of both H2O2 and urchin-like TiO2 nanostructures. In this experiment, 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was used to capture short-lived ˙OH radicals.54,55 To avoid the hydroxyl radicals generated from the photocatalytic effect of TiO2, all the experiments were performed in dark conditions. As shown in Fig. 3b, without H2O2 in the system, pure TiO2 in water hardly causes a signal in dark conditions. In addition, the pure H2O2 in water caused weak signals. With both H2O2 and TiO2 in the system, the ˙OH related signals are distinctively strong. Therefore, we conclude that TiO2 bolsters the catalytic activity by accelerating the formation of hydroxyl radicals from decomposing H2O2, which accounts with the reaction mechanism. Moreover, when MB is added to the TiO2 and H2O2 co-existing solution, the signal of DPMO/˙OH becomes very weak. This finding demonstrates that the decomposition of MB has consumed hydroxyl radicals generated from the reaction between TiO2 and H2O2. The above results are the most convincing evidence for our suggested pseudo-Fenton reaction mechanism. To further confirm the catalytic effect of TiO2 urchin-like nanostructures on the generation of hydroxyl free radicals, ESR measurements of DPMO/˙OH in the TiO2 and H2O2 co-existing solution system with different amounts of TiO2 nanostructures were recorded (Fig. 3c). The signal intensity of DPMO/˙OH obviously becomes stronger with more TiO2 added to the system. Furthermore, the effect of the dosage of H2O2 on the formation of hydroxyl radicals was studied (Fig. 3d). Obviously, more H2O2 in the reaction system can induce a higher DPMO/˙OH signal; however, this enhancement is not significant. Similarly, as discussed in Fig. 2b, a small amount of H2O2 can induce a striking enhancement in MB degradation. However, the increase in H2O2 added does not result in a noticeable enhancement. This finding implies that the conversion of H2O2 into hydroxyl radicals may possess a threshold. Moreover, H2O2 does not induce a higher DPMO/˙OH signal, despite more ˙OH overall.
To further study the valence switching of the element Ti in TiO2 urchin-like nanostructures with H2O2, electrochemical measurements of TiO2 with and without H2O2 present were carried out on a model TiO2 electrode sample through a three-electrode system by using 1 M Na2SO4 as the electrolyte. The model TiO2 electrode was synthesized by assembling a TiO2 nano-tree on the surface of a transparent conductive fluorine-doped tin oxide (FTO) glass substrate (ESI,† Fig. S7). The voltage of the three-electrode system is almost zero, but the voltage remains at ∼0.325 V when H2O2 is added to the electrolyte. Correspondingly, the current displays a stable intensity after adding H2O2 under conditions where the voltage is fixed at −0.35 V, which is beyond the opposite open circuit potential. Moreover, the electrochemical impedance spectra in Fig. 3f reflect that after H2O2 is added to the electrolyte, electron transportation is promoted. The above results confirm that TiO2 reacts with H2O2, which enhances the electrical properties and reasonably explains the mechanism. To further couple the catalytic performance of TiO2 microspheres/H2O2 with their practical application in the treatment of industry waste water, we exploited the degradation of dye waste water (initial COD, 600 mg L−1). As shown in Fig. S6 (ESI†), the COD removal remains at ∼70%, and the catalytic activity of the TiO2 microspheres is considerable after 12 cycles.
Conclusions
In summary, a new pseudo-Fenton reaction has been realized by using TiO2 urchin-like nanostructures as catalysts with the presence of H2O2 in the reaction system at a temperature below 60 °C. The proposed pseudo-Fenton process is initiated from the in situ alternative switching between Ti4+ and Ti3+ in TiO2 nanostructures driven by H2O2, and the Fenton reaction based on the reaction between Ti3+ ions reduced in situ by H2O2 in TiO2 nanostructures and H2O2 forms Ti4+ and ˙OH. This in situ switching of Ti4+ and Ti3+ in TiO2 nanostructures endows the catalyst with non-consumptive properties, excellent cyclic ability and a long life. This new discovery is dramatic for TiO2-based nanomaterials applied in environmental protection, and promotes their practical applications in the degradation of organic pollutants.Acknowledgements
The authors are thankful for funding from the National Natural Science Foundation of China (Grant No. 51372142), the 2014 Innovative Jiaxing Elite Leading Talents Program (A), and the Science and Technology Project Foundation of Jiaxing City (2015BZ12004).Notes and references
- J. Tian, Z. Zhao, A. Kumar, R. I. Boughton and H. Liu, Chem. Soc. Rev., 2014, 43, 6920–6937 RSC.
- Y. Li, H. Zhang, P. Liu, D. Wang, Y. Li and H. Zhao, Small, 2013, 9, 3336–3344 CAS.
- Y. Chen, L. Chen, H. Bai and L. Li, J. Mater. Chem. A, 2013, 1, 1992–2001 CAS.
- P. Aravind, H. Selvaraj, S. Ferro and M. Sundaram, J. Hazard. Mater., 2016, 318, 203–215 CrossRef CAS PubMed.
- S. Dervin, D. D. Dionysiou and S. C. Pillai, Nanoscale, 2016, 8, 15115–15131 RSC.
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang, Y. Wang, P. Liu, Y. Zhang, Y. Wen, L. Gu, G. Ma, Z. Su, Z. Tang, H. Zhao and D. Wang, Nat. Energy, 2016, 1, 16050 CrossRef CAS.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- R. Li, L. Zhang and P. Wang, Nanoscale, 2015, 7, 17167–17194 RSC.
- L. Yu, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2016, 28, 92–97 CrossRef CAS PubMed.
- G. Subramanian and G. Madras, Chem. Commun., 2017, 53, 1136–1139 RSC.
- Y. Li, H. Zhang, Y. Wang, P. Liu, H. Yang, X. Yao, D. Wang, Z. Tang and H. Zhao, Energy Environ. Sci., 2014, 7, 3720–3726 CAS.
- Y. Meng, W. Song, H. Huang, Z. Ren, S. Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- Y. Sang, Z. Zhao, J. Tian, P. Hao, H. Jiang, H. Liu and J. P. Claverie, Small, 2014, 10, 3775–3782 CrossRef CAS PubMed.
- H. Li, Y. Wang, G. Chen, Y. Sang, H. Jiang, J. He, X. Li and H. Liu, Nanoscale, 2016, 8, 6101–6109 RSC.
- G. Chen, S. Ji, Y. Sang, S. Chang, Y. Wang, P. Hao, J. Claverie, H. Liu and G. Yu, Nanoscale, 2015, 7, 3117–3125 RSC.
- J. Su, X. Zou, G. D. Li, Y. M. Jiang, Y. Cao, J. Zhao and J. S. Chen, Chem. Commun., 2013, 49, 8217–8219 RSC.
- W. Zhou, F. Sun, K. Pan, G. Tian, B. Jiang, Z. Ren, C. Tian and H. Fu, Adv. Funct. Mater., 2011, 21, 1922–1930 CrossRef CAS.
- W. Zhou, W. Li, J. Q. Wang, Y. Qu, Y. Yang, Y. Xie, K. Zhang, L. Wang, H. Fu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 9280–9283 CrossRef CAS PubMed.
- J. Qi, K. Zhao, G. Li, Y. Gao, H. Zhao, R. Yu and Z. Tang, Nanoscale, 2014, 6, 4072–4077 RSC.
- G. Li, L. Wu, F. Li, P. Xu, D. Zhang and H. Li, Nanoscale, 2013, 5, 2118–2125 RSC.
- J. Qi, J. Chen, G. Li, S. Li, Y. Gao and Z. Tang, Energy Environ. Sci., 2012, 5, 8937 CAS.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- Z. M. Cui, Z. Chen, C. Y. Cao, L. Jiang and W. G. Song, Chem. Commun., 2013, 49, 2332–2334 RSC.
- Z. Ma, L. Ren, S. Xing, Y. Wu and Y. Gao, J. Phys. Chem. C, 2015, 119, 23068–23074 CAS.
- J. Chen, W. Wen, L. Kong, S. Tian, F. Ding and Y. Xiong, Ind. Eng. Chem. Res., 2014, 53, 6297–6306 CrossRef CAS.
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
- Y. Wang, H. Zhao, M. Li, J. Fan and G. Zhao, Appl. Catal., B, 2014, 147, 534–545 CrossRef CAS.
- L. Peng, J. Zhang, Z. Xue, B. Han, J. Li and G. Yang, Chem. Commun., 2013, 49, 11695–11697 RSC.
- W. Zhou, H. Liu, J. Wang, D. Liu, G. Du and J. Cui, ACS Appl. Mater. Interfaces, 2010, 2, 2385–2392 CAS.
- B. Liu, H. M. Chen, C. Liu, S. C. Andrews, C. Hahn and P. Yang, J. Am. Chem. Soc., 2013, 135, 9995–9998 CrossRef CAS PubMed.
- H. Liu, W. Li, D. Shen, D. Zhao and G. Wang, J. Am. Chem. Soc., 2015, 137, 13161–13166 CrossRef CAS PubMed.
- B. Liu, L.-M. Liu, X.-F. Lang, H.-Y. Wang, X. W. Lou and E. S. Aydil, Energy Environ. Sci., 2014, 7, 2592 CAS.
- Z. Zhao, J. Tian, Y. Sang, A. Cabot and H. Liu, Adv. Mater., 2015, 27, 2557–2582 CrossRef CAS PubMed.
- W. Jiao, N. Li, L. Wang, L. Wen, F. Li, G. Liu and H. M. Cheng, Chem. Commun., 2013, 49, 3461–3463 RSC.
- J. Zhang, H. Ren, J. Wang, J. Qi, R. Yu, D. Wang and Y. Liu, J. Mater. Chem. A, 2016, 4, 17673–17677 CAS.
- N. Yang, Y. Zhang, J. E. Halpert, J. Zhai, D. Wang and L. Jiang, Small, 2012, 8, 1762–1770 CrossRef CAS PubMed.
- J. Chen, Y.-F. Li, P. Sit and A. Selloni, J. Am. Chem. Soc., 2013, 135, 18774–18777 CrossRef CAS PubMed.
- S. K. Balasingam, M. G. Kang and Y. Jun, Chem. Commun., 2013, 49, 11457–11475 RSC.
- H. Ren, R. Yu, J. Wang, Q. Jin, M. Yang, D. Mao, D. Kisailus, H. Zhao and D. Wang, Nano Lett., 2014, 14, 6679–6684 CrossRef CAS PubMed.
- H. Ren, J. Sun, R. Yu, M. Yang, L. Gu, P. Liu, H. Zhao, D. Kisailus and D. Wang, Chem. Sci., 2016, 7, 793–798 RSC.
- H. Tang, C. M. Hessel, J. Wang, N. Yang, R. Yu, H. Zhao and D. Wang, Chem. Soc. Rev., 2014, 43, 4281–4299 RSC.
- X. Xin, T. Xu, J. Yin, L. Wang and C. Wang, Appl. Catal., B, 2015, 176-177, 354–362 CrossRef CAS.
- S. Xing, Z. Zhou, Z. Ma and Y. Wu, Appl. Catal., B, 2011, 107, 386–392 CrossRef CAS.
- X. Yang, W. Chen, J. Huang, Y. Zhou, Y. Zhu and C. Li, Sci. Rep., 2015, 5, 10632 CrossRef PubMed.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., 2013, 52, 1280–1284 CrossRef CAS PubMed.
- I. S. X. Pinto, P. H. V. V. Pacheco, J. V. Coelho, E. Lorençon, J. D. Ardisson, J. D. Fabris, P. P. de Souza, K. W. H. Krambrock, L. C. A. Oliveira and M. C. Pereira, Appl. Catal., B, 2012, 119-120, 175–182 CrossRef CAS.
- N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang and J. C. Diniz da Costa, Sci. Rep., 2014, 4, 4594 CrossRef PubMed.
- C. Mao, F. Zuo, Y. Hou, X. Bu and P. Feng, Angew. Chem., 2014, 53, 10485–10489 CrossRef CAS PubMed.
- J. Huo, Y. Hu, H. Jiang and C. Li, Nanoscale, 2014, 6, 9078–9084 RSC.
- S. Hoang, S. P. Berglund, N. T. Hahn, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2012, 134, 3659–3662 CrossRef CAS PubMed.
- Z. Shi, J. Wang, W. Wang, Y. Zhang, B. Li, Z. Lu and Y. Li, Nano Res., 2015, 9, 353–362 CrossRef.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., 2016, 55, 2230–2234 CrossRef CAS PubMed.
- L. D. Sánchez, S. F. M. Taxt-Lamolle, E. O. Hole, A. Krivokapić, E. Sagstuen and H. J. Haugen, Appl. Catal., B, 2013, 142–143, 662–667 CrossRef.
- T. Wen, H. Zhang, Y. Chong, W. G. Wamer, J.-J. Yin and X. Wu, Nano Res., 2016, 9, 1663–1673 CrossRef CAS.
- Y. Qiu, Z. Wang, A. C. Owens, I. Kulaots, Y. Chen, A. B. Kane and R. H. Hurt, Nanoscale, 2014, 6, 11744–11755 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00163k |
| This journal is © the Partner Organisations 2017 |