
Controlling solid-state optical properties of stimuli responsive dimethylamino-substituted dibenzoylmethane materials†
Tristan
Butler
a,
Fang
Wang
a,
Michal
Sabat
b and
Cassandra L.
Fraser
 *a
*a
aDepartment of Chemistry, University of Virginia, McCormick Road, Charlottesville, Virginia 22904, USA. E-mail: fraser@virginia.edu
bDepartment of Materials Science and Engineering, University of Virginia, 395 McCormick Road, Charlottesville, Virginia 22904, USA
First published on 11th May 2017
Abstract
The mechanochromic luminescent (ML) properties of boron coordinated β-diketones (BF2bdks) have been widely studied, however the stimuli responsive properties of uncoordinated β-diketones (bdks) are less known. While bdk dyes show promising properties, including high contrast ML, rapid room temperature self-erasure, solvatochromism, and aggregation induced emission (AIE), previously reported dyes exhibit emission over a narrow range of wavelengths (∼420–500 nm). To tune luminescence over a broader color range, dimethylamino (DMA) substituted β-diketones were synthesized with a series of electron donating and withdrawing substituents. Solvatochromism, aggregation induced emission (AIE), ML, and thermochromism were investigated. Most dyes showed positive solvatochromic shifts and were responsive to mechanical and thermal stimuli. The solid-state emission wavelengths correlated with the electron withdrawing strength of the para substituent and ranged from blue to orange (488–578 nm). Structural and thermal characterization was performed by powder X-ray diffraction (XRD) and differential scanning calorimetry (DSC) respectively. These data indicate that stimuli responsive properties are the result of a crystalline to amorphous phase transition. Additionally, the aggregation induced emission (AIE) properties of iodo and cyano substituted dyes were measured in THF/H2O solutions. Both showed intense emission resulting from aggregation. The sensitivity of these dyes toward matrix polarity was further investigated by fabricating polystyrene films with increasing concentrations of dye and camphoric anhydride, a non-emissive polar dopant. A red-shift in emission was observed in these films which demonstrated that matrix effects can be used to tune the emission of DMA-substituted diketones.
Introduction
Stimuli responsive luminescent materials have garnered attention given their potential for optical memory storage, security inks, sensing materials, bioprobes and other applications.1–7 Many luminescent materials show emission changes when exposed to thermal,8–10 mechanical,11,12 chemical13–15 and other stimuli. However, their emissive properties (i.e. wavelength, quantum yield, lifetime) must often be tailored for specific applications.16,17 Many different design strategies such as substitution with donor and acceptor groups and heavy atom substitution have been employed to rationally modulate the properties of materials.18–21Emissive solids are required for many applications however dyes that are highly emissive in solution often suffer from aggregation caused quenching (ACQ) and thus, are non-emissive in the solid state.22 A strategy for designing materials with efficient solid state emission has been developed by Tang et al. While emission can be quenched for fluorophores that rotate or vibrate in solution,23–26 upon dye aggregation, these molecular motions are restricted resulting in solid state emission.27,28 Dyes utilizing aggregation induced emission (AIE) have been incorporated into materials ranging from bioprobes29 to organic light emitting diodes and chemical sensors.30,31
While AIE dyes display efficient solid state emission, many applications also require a wide range of emission wavelengths.32,33 Substitution of luminescent dyes with donor and acceptor groups is an effective strategy for tuning emission colors of stimuli responsive fluorophores.34–36 Depending on the position and strength of the electron donating or withdrawing substituent, the relative molecular orbital energies of a luminescent dye can be greatly affected.37 Often this strategy leads to intramolecular charge transfer (ICT) that red shifts emission.38 The electronic properties of a given substituent can be parameterized using their Hammett constants.39 Comparison of the para-substituted Hammett constants of each substituent can help to explain trends in the optical properties of donor and acceptor substituted dyes.40
Another byproduct of donor and acceptor substitution and associated large molecular dipoles is solvent sensitive emission.41 Known as solvatochromism, the excited states of these dyes can be stabilized by solvent molecules through dipole–dipole interactions.42,43 Therefore, longer wavelength emission is observed when solvatochromic dyes are dissolved in more polar solvents. An analogous effect can be observed for dyes emissive in the solid state.44 The emission of polar dyes in a polymer matrix can be modulated through the addition of polar dopant molecules such as camphoric anhydride (CA) or tris(8-hydroxyquinoline)aluminum (Alq3).45,46 This strategy, known as solid-state solvation, has been used to improve the efficiency of luminescent solar concentrators.47 Additionally, Bulovic et al., recently manipulated the emission of doped organic thin films through the application of pressure, which forced dyes and dopant molecules into closer proximity and red-shifted emission.46
Boron difluoride β-diketonate (BF2bdk) compounds are an intensely studied family of dyes due to their diverse stimuli responsive optical properties and facile synthesis from commercially available starting materials.48,49 Their mechanochromic luminescent (ML) properties have been tuned through a variety of different methods,50,51 including alkyl chain substitution.52 Dyes substituted with heavy atoms exhibit quenching upon mechanical stimulus53–55 and heteroatom substituted BF2bdk derivatives show thermochromic responses in addition to ML.56 Mechanistic studies of BF2bdk ML by Zhang et al., suggest that the application of a shear force to dye thin films forms low energy H-aggregates with red-shifted emission.57
Recently, stimuli responsive behavior has been reported in dinaphthoyl-substituted bdk ligands absent boron coordination.58,59 Compared to their boronated counterparts, bdks are even easier to synthesize and exhibit both AIE and faster rates of recovery after smearing. Additionally, bdks in the keto–enol form have hydrogen bonds in the ground state; thus, excited state intramolecular proton transfer (ESIPT) may be accessible for asymmetric bdks.60–62 Dyes with ESIPT show particularly large Stokes shifts, of interest for bioprobes38 and display technologies.63,64
While bdks possess many characteristics promising for application, their stimuli responsive properties have been little investigated. Previously reported ML active bdk dyes showed a narrow (∼420–500 nm) emission range, and their thermochromic properties were not reported.59,65 To produce a broader wavelength range of stimuli responses, in this study, a series of dimethylamino (DMA) substituted diketones were prepared with different electron donating and withdrawing groups. The effect of donor and acceptor substitution on optical properties was tested in solution, and their mechanical and thermochromic responses were measured in the solid state. In addition, the AIE properties of DMA-I and DMA-CN were investigated in THF/H2O solutions. Polystyrene (PS) thin films of these dyes were used to examine dye loading and polar dopant effects. Structural characterization was performed using powder X-ray diffraction (XRD) and the crystal structure of DMA-I is reported. Thermal properties were investigated using differential scanning calorimetery (DSC).
Experimental details
Materials and methods
THF was dried over molecular sieves activated at 300 °C as previously described. Reactions were monitored using silica TLC plates. Compounds purchased from Sigma-Aldrich and TCI were reagent grade and used without further purification. The dimethylamino-substituted β-diketones were synthesized via Claisen condensation using a previously described method.48 Data for DMA-H,61,66 DMA-OMe,67 and DMA-CN,68 are in accord with previous reports. Characterization data for new derivatives are provided in the ESI.†Methods
1H NMR (600 MHz) spectra were recorded on a Varian VRMS/600 spectrometer in deuterated DMSO and CDCl3. Spectra were referenced to the signals for residual protio-DMSO at 2.50 ppm and protio-CDCl3 at 7.27 ppm. Coupling constants were reported in Hz. Mass spectra were recorded using a Micromass Q-TOF Ultima spectrometer, using electrospray ionization (ESI) MS/MS techniques. Absorption spectra were collected on a Hewlett-Packard 8452A diode-array UV-vis spectrophotometer. A Horiba Fluorolog-3 Model FL3-22 spectrofluorometer (double-grating excitation and double-grating emission monochromator) was used to measure steady-state emission spectra. Time-correlated single-photon counting (TCSPC) fluorescence lifetime measurements were performed with a NanoLED-370 (λex = 369 nm) excitation source and a DataStation Hub as the SPC controller. Lifetime analysis was done with DataStation v2.4 software from Horiba Jobin Yvon. Fluorescence quantum yields, φF, in CH2Cl2 were calculated versus a standard of dilute quinine sulfate solution in 0.1 M H2SO4 using a previously described method51 and the following values: φF quinine sulfate in 0.1 M H2SO4 = 0.54,69nD70 0.1 M H2SO4 = 1.33, nD70 CH2Cl2 = 1.424. Optically dilute CH2Cl2 solutions of all samples were prepared in 1 cm path length quartz cuvettes with absorbances <0.1 (a.u.). A F-3029 Quanta-Φ Integrating Sphere from Horiba Scientific was used to measure solid state quantum yields and data were analyzed using FluorEssence software. A Laurel Technologies WS-65OS spin-coater was used to fabricate polystyrene (PS) films for dye loading and polarity studies. Differential scanning calorimetry (DSC) was performed on the pristine powders using a TA Instruments 2920 Modulated DSC. Data were analyzed using Universal Analysis software V 2.3 from TA Instruments. Thermograms were recorded using the standard mode and a constant heating rate of 5 °C min−1. A cooling rate of 10 °C min−1 was used during the initial cycle compared to a cooling rate of 1 °C min−1 for subsequent cycles. Powder X-ray diffraction (XRD) patterns were obtained using a Panalytical X'Pert Pro MPD diffractometer operating at 40 kV and 40 mA using Mo Kα radiation.Dye loading and dopant effects on emission properties were investigated using spin cast polystyrene (PS) films. For dye loading experiments (dye/PS films), films were comprised of PS and increasing concentrations of a single dye (DMA-I, DMA-CN). Films for dopant studies (CA/dye/PS) contained constant amounts of PS and dye with increasing quantities of camphoric anhydride (CA). Two sets of CA/dye/PS films were made for each dye, both dilute (3 × 10−7 mol) and concentrated (4.5 × 10−6 mol). Generally, PS films were fabricated by adding 10 drops of dye/PS or CA/dye/PS dichloromethane solutions to 18 × 18 mm glass microscope coverslips rotating at 3000 rpm. Solutions for spin casting dye/PS films were prepared in sample vials by mixing dichloromethane stock solutions of PS (0.01 M), DMA-I (0.03 M), or DMA-CN (0.03 M) in the proper ratios to produce dye/PS films ranging from 0.019 to 28.0 mol%. The resulting solutions were evaporated in air before dilution with 1 mL of dichloromethane for spin casting. Films for testing the effect of matrix polarity on properties (i.e. CA/dye/PS films) were made using an analogous procedure except that different volumes of a CA stock solution (0.33 M) was added to each vial in order to fabricate CA/dye/PS films ranging from 0 to 63.5 mol% CA. Films were dried under vacuum for 20 min prior to measurement.
To investigate the thermal and mechanical responses, nitrile examination gloves were used to smear a small amount of dye (∼2 mg) onto 5 × 5 cm squares of weighing paper (WP). Films were annealed at 110 °C for 10 min prior to measuring their emission. Mechanochromic luminescence was measured after gentle smearing of annealed films with a cotton swab. Thermochromic properties were observed after melting with a heat gun and cooling in air (i.e. melt quenching).
Results and discussion
Synthesis
In order to modulate the solution and solid state optical properties of stimuli responsive β-diketonate dyes, a series of 4-dimethylamino (DMA) substituted derivatives was prepared with various electron donating and withdrawing groups located in the 4′-para position. Substituents were chosen based on their para-substituted Hammett constants, σp, in order to assess the effect of donor/acceptor substitution on the optical properties of bdk dyes (OMe = −0.27, H = 0, F = 0.06, I = 0.18, Br = 0.23, CF3 = 0.42, COOMe = 0.45, CN = 0.66, Scheme S1, ESI†). Compounds were synthesized via Claisen condensation with dimethylamino substituted ketones and the corresponding para-substituted ester in the presence of NaH. All derivatives in this set show enol proton chemical shifts in their NMR spectra (∼17.0 ppm) which indicates that dyes occupy keto–enol configurations in the ground state.Solution and computational characterization
In order to gauge how these different donor/acceptor moieties affect the optical properties of DMA substituted diketones in solution, the absorption and emission spectra were measured in dichloromethane (1 × 10−5 M) (Table 1 and Fig. 1). Two distinct peaks were observed in the UV-vis spectra of each dye. With the exception of DMA-H, where the most intense peak was highest in energy, the peak absorbance (λabs) of other dyes corresponded to the most red-shifted band. Though bimodal absorption spectra has been attributed to absorption from different tautomeric species that exist in previous studies,61,66 calculations indicate that that bimodal absorption profiles could arise via transitions to the excited state from lower energy orbitals (see computational details in ESI†). Comparison of the peak absorbance (λabs) of each dye showed a general trend toward longer wavelengths (i.e. lower energy) as the electron withdrawing ability of the para substituent was increased. Substituent effects were also observed on the molar absorptivity, as compounds showed extinction coefficients ranging from 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 (DMA-Br) to 44500 M−1 cm−1 (DMA-OMe), however no general trend could be established.
000 M−1 cm−1 (DMA-Br) to 44500 M−1 cm−1 (DMA-OMe), however no general trend could be established.
| λ abs (nm) | ε (M−1 cm−1) | λ em (nm) | Φ (%) | τ (ns) | τ rad (ns) | |
|---|---|---|---|---|---|---|
| a Excited at 385 nm, room temperature, air. b Absorbance maximum. c Emission maximum. d Fluorescence quantum yield. e Pre-exponential weighted fluorescence lifetime. f Radiative lifetime = τ/Φ. | ||||||
| DMA-H | 314 | 36000 |
482 | 30 | 1.32 | 4.4 |
| DMA-OMe | 403 | 44500 |
469 | 51 | 1.58 | 3.1 |
| DMA-F | 403 | 28000 |
482 | 37 | 1.42 | 4.0 |
| DMA-Br | 408 | 18000 |
495 | 16 | 1.00 | 6.3 |
| DMA-I | 409 | 35000 |
494 | 18 | 0.99 | 5.5 |
| DMA-CF3 | 411 | 34500 |
511 | 3 | 0.56 | 18.7 |
| DMA-COOMe | 414 | 38500 |
532 | <1 | 0.30 | N/A |
| DMA-CN | 420 | 39500 |
541 | 1 | 0.59 | 59 |
 | ||
| Fig. 1 Absorption (a) and emission (b) spectra of diketones in 1 × 10−5 M dichloromethane solution. | ||
A clear substituent effect was observed in the emission properties for dyes in dichloromethane solution. The highest energy peak emission (λem) was observed for the methoxy substituted derivative, DMA-OMe (λem = 469 nm), which glowed blue under UV excitation. Upon substitution with stronger electron withdrawing substituents (i.e. larger Hammett constants), a gradual red-shift in λem was observed. The lowest energy emission was detected for DMA-CN (λem = 541 nm), which exhibited faint yellow-green emission when exposed to UV light. Additionally, a high-energy shoulder was present in the emission spectrum of DMA-CN, which wasn’t observed in the spectra of other dyes. As bimodal emission has often been attributed to ESIPT in dyes with ground state intramolecular hydrogen bonds, this blue-shifted transition could be evidence of a proton transfer process occurring for the cyano substituted diketone.62 Comparison of the fluorescence lifetime (τ) and quantum yield (Φ) of each dye showed a similar trend. The largest quantum yield and longest lifetime was measured for DMA-OMe (τ = 1.58 ns, Φ = 51%), however both the fluorescence lifetime and quantum yield decreased considerably when substituted with progressively stronger electron withdrawing groups, such that emission is barely observable to the eye for DMA-CF3 (τ = 0.56 ns, Φ = 3%), DMA-COOMe (τ = 0.30 ns, Φ = 1%), and DMA-CN (τ = 0.59 ns, Φ < 1%). Since previously reported diketones are non-emissive in dichloromethane solution,59 these results demonstrate that donor and acceptor substitution is an effective strategy for designing bdk dyes that are emissive in solution.
According to the emission of DMA dyes in dichloromethane, there is a strong correlation between the emission properties of each dye and the electron withdrawing ability of the para substituent. With the exception of DMA-OMe, all dyes exhibit radiative lifetimes (τrad > 4.0 ns) corresponding to ICT emission.72 The radiative lifetime observed in DMA-OMe solutions was borderline (3.1 ns). Furthermore, previous investigations show that a twisted intramolecular charge transfer transition (TICT) may result from rotation of the DMA substituent.66 In order to further investigate these trends, density functional theory (DFT) calculations with a Tomasi polarized continuum for dichloromethane solvent were performed. The B3LYP/6-31G(d) basis set was used to produce HOMO and LUMO molecular orbitals (MOs) and simulated UV-vis spectra for all but the hydrogen substituted derivative (Fig. 2 and Fig. S1, ESI†), which has been investigated previously.66 The experimental and computational UV-vis spectra are in close agreement which indicates that computations reasonably approximate optical properties in solution.
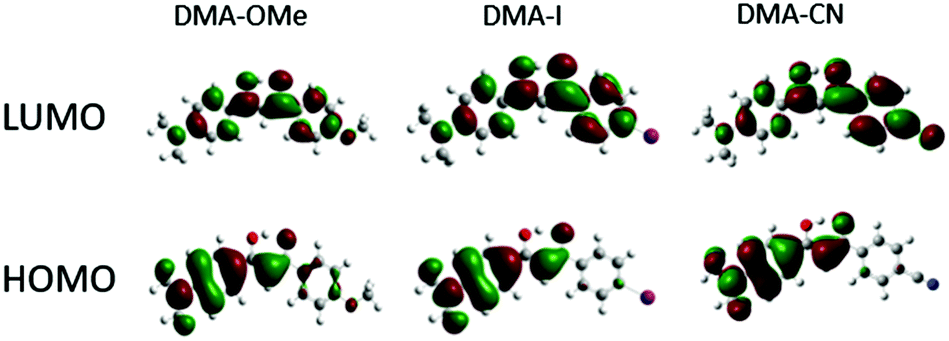 | ||
| Fig. 2 HOMO and LUMO molecular orbitals for methoxy (DMA-OMe), iodo (DMA-I) and cyano (DMA-CN) substituted diketones. | ||
The ground state optimized geometries for the dyes are mostly planar, however slight distortions in planarity were observed. Additionally, calculations indicate that the lowest energy conformation of each dye is a keto–enol with the hydroxyl group positioned closest to the DMA substituted phenyl ring. Examination of the HOMO for each dye indicates that electron density is concentrated on the side of the molecule containing the highly electron donating DMA substituent, however a small amount of electron density can be found on the methoxy substituted phenyl ring for DMA-OMe. In the respective LUMOs, the electron density is delocalized throughout the entire molecule for each dye. Close inspection of the LUMO for DMA-CN shows a gradient in electron density throughout the molecule, where the majority is located on the cyano-substituted phenyl ring. A similar distribution in electron density was observed in the LUMO for DMA-COOMe which indicates that stronger electron withdrawing groups tend to attract more electron density in the LUMO. The localized electron density observed in the HOMOs of each dye and the delocalized electron density in the LUMO, also point to ICT transitions, as expected for donor acceptor systems and based on radiative lifetime data.
Optical properties in different solvents
Relatively large Stokes shifts, long radiative lifetimes, and asymmetric distribution of electron density in molecular orbitals are observed in DMA dyes and are often associated with changes in the excited state molecular geometry as a result of TICT.51,73,74 However large Stokes shifts have also been observed in systems that experience excited state intramolecular proton transfer (ESIPT). Since ESIPT has previously been observed in asymmetric diketones, it is possible that these DMA substituted derivatives also exhibit ESIPT.64 Additionally, solvatochromism is often present in dyes that undergo ICT transitions. Solvatochromism and ESIPT emission can be observed by measuring the optical properties of a given dye in a variety of different solvents. A red-shift in emission with increasing solvent polarity is often associated with solvatochromic processes, whereas the nature of the solvent (e.g. polar, protic) determines the emission of ESIPT dyes.Generally, polar or protic solvents disrupt the proton transfer process, which can result in a variety of spectroscopic features depending on dye structure and conformation. It is often difficult to decouple ESIPT from ICT processes since they can occur in conjunction with one another in certain dye systems. The solvent dependent emission of DMA-H was previously studied in great detail using computational and spectroscopic techniques including steady state and ultrafast transient absorption and fluorescence techniques. These studies show that solvent effects not only determine the ground state conformation of DMA-H but also govern the excited state dynamics (i.e. ESIPT and/or ICT/TICT) affecting emission.61,66 While the solvent dependent properties of DMA-H dyes are highly complex (i.e. both ESIPT and/or ICT/TICT occur depending on solvent polarity and ground state conformation) they offer insight into the optical properties of this set of dyes in different solvents.
Investigation of solvent effects on the emission of DMA diketones was performed by measuring the absorption and emission spectra of each dye dissolved in a series of organic solvents at 1 × 10−5 M concentration (Fig. 3 and Fig. S2–S6, ESI†). With the exception of DMA-CN, all dyes showed a red-shift in emission as the solvent polarity was increased, which is consistent with solvatochromism. Comparison of solvatochromic dyes indicates that the degree of red-shift is dependent on the electron donating or withdrawing ability of the para substituent. To the eye, the emission of the electron donating methoxy substituted derivative, DMA-OMe, changed from blue when dissolved in toluene (λem = 437 nm) to green in acetone (λem = 478 nm). As the solvent polarity was further increased, emission was not visible and could only be detected spectroscopically. For DMA-COOMe, which is substituted with an electron withdrawing acetate group, the emission appeared green in toluene solution (λem = 483 nm), and shifted to yellow (λem = 513 nm) in chloroform, before becoming non-emissive in more polar solvents. The quenching of emission in polar solvents is typically observed and can be attributed to increased non-radiative decay as solvent stabilization results in a smaller energy gap between ground and excited states, however it could also be due to the formation of a non-radiative TICT state.74,75
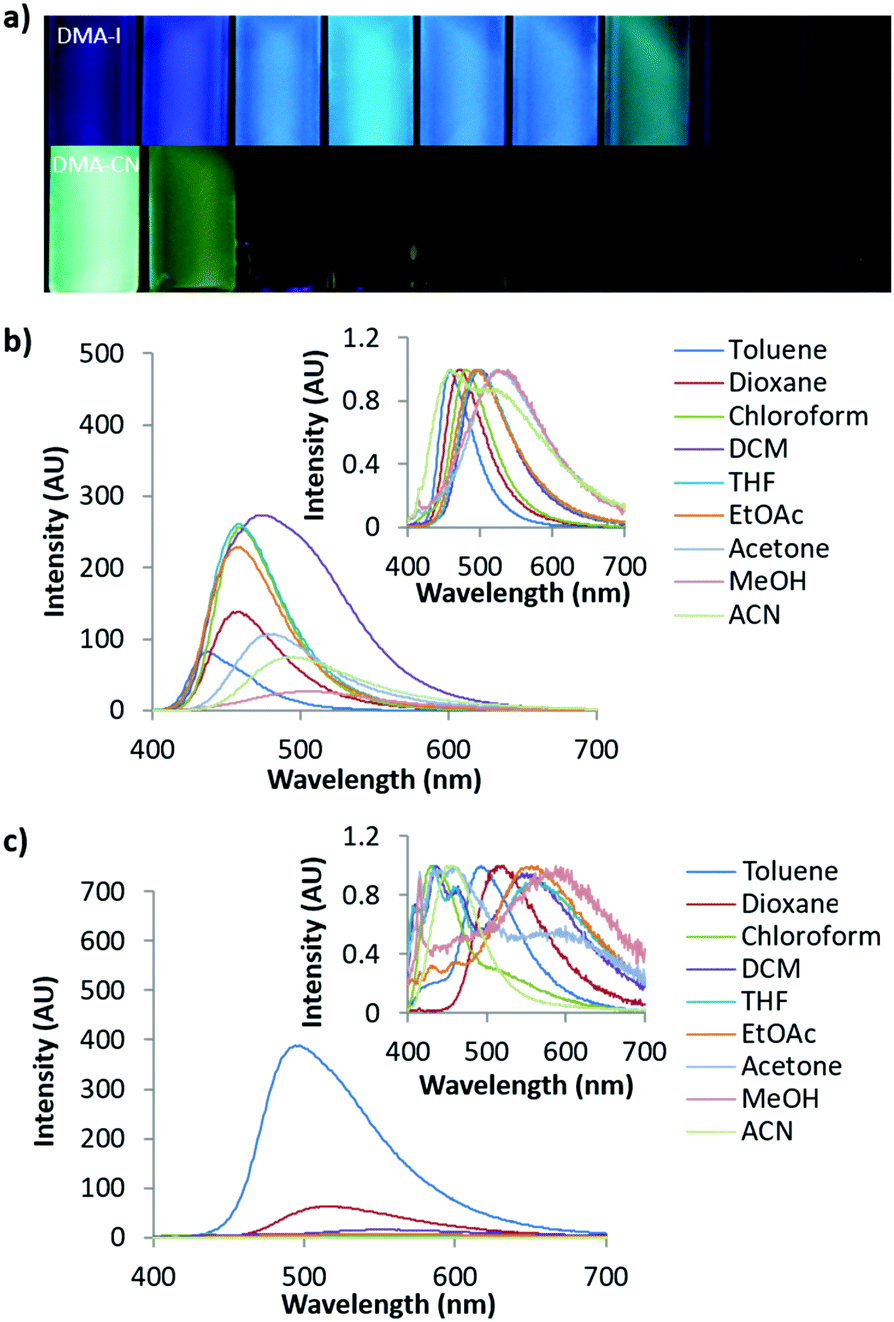 | ||
| Fig. 3 Image under UV irradiation (from left to right: toluene, 1,4-dioxane, chloroform, dichloromethane, ethyl acetate, acetone, methanol, acetonitrile, λex = 365 nm) (a) and emission spectra of DMA-I (b) and DMA-CN (c) dissolved in different solvents (1 × 10−5 M, λex = 369 nm). Insets of (b) and (c) depict emission spectra normalized to their peak emission. | ||
The slope observed in the Lippert–Mataga plots (Stokes shift, Δν, versus solvent polarity parameter, Δf) describes the sensitivity of a dye toward solvent polarity.42,43 Aside from DMA-CF3 (1750), the slope is also correlated with the electronics of the para substituent, however, deviations from this trend could be the result of ESIPT (Fig. S7, ESI†). The slopes of the most strongly donating and withdrawing dyes, DMA-OMe (1150) and DMA-COOMe (2800), bracket those observed for DMA-F (1500), DMA-I (2100), and DMA-Br (2200). This trend indicates that dye sensitivity towards solvent polarity can be effectively tuned through simple donor/acceptor substitutions, consistent with related studies.41 The anomalous behavior of DMA-CF3 may result from the dual emission profiles measured in chloroform, THF, and acetone, which could be due to ESIPT and/or TICT processes.66,76 Dual emission is often observed for dyes with TICT as radiation can occur from locally excited (LE) and TICT states. Additionally, emission from both tautomeric excited state species of ESIPT dyes also produces multiple emission peaks. As previously mentioned, these processes are often intertwined so it is difficult to assign the origin of dual emission, however the relatively shallow slope observed for DMA-CF3 is likely the result of dual emission.
The cyano substituted derivative, DMA-CN, shows red-shifted emission when dissolved in dioxane (λem = 519 nm) compared to toluene (λem = 492 nm) but clear solvatochromic trends were not observed in corresponding Lippert–Mataga plots (Fig. S7, ESI†). Like DMA-CF3, dual emission profiles were present when dissolved in certain solvents (toluene, chloroform, dichloromethane, THF, and acetonitrile). In acetonitrile solution, the maximum emission of DMA-CN (λem = 456 nm) was blue-shifted relative to the peak emission in the non-polar solvents, toluene (λem = 492 nm), and dioxane (λem = 519 nm). Previously, these spectroscopic features have been ascribed to the formation of a TICT state,68 however investigation of the solvent mediated excited state dynamics of DMA-H indicate that ESIPT could also occur for DMA-CN. Despite deviating from Lippert–Mataga theory, the emission shifts observed in toluene and dioxane solutions indicate that DMA-CN is also sensitive to polarity.
Aggregation studies
Aggregation induced emission (AIE) was previously observed in dinaphthyl and tetraphenyl ethylene substituted bdk systems.59,65 While AIE/AIEE has been detected for certain derivatives, these systems exhibit relatively small emission enhancement factors and a narrow range of emission wavelengths. To investigate potential aggregation dependent emission properties and assess donor and acceptor effects on bdk systems, the excitation and emission spectra were measured for moderately (DMA-I) and strongly (DMA-CN) withdrawing dyes (3 × 10−4 M) dissolved in THF/H2O solutions of varying THF and H2O ratios (Fig. 4 and Fig. S8, S9, ESI†). These dyes were chosen based on their relative difference in electron withdrawing substituent as well as their high solubility in THF. While H2O acts as a non-solvent that forces these organic dyes closer together eventually forming aggregate species and enabling visualization of AIE/AIEE, it also increases the polarity of the system. Based on the positive solvatochromic shifts observed for DMA-I and DMA-CN, longer emission wavelengths should be expected as the water fraction is increased.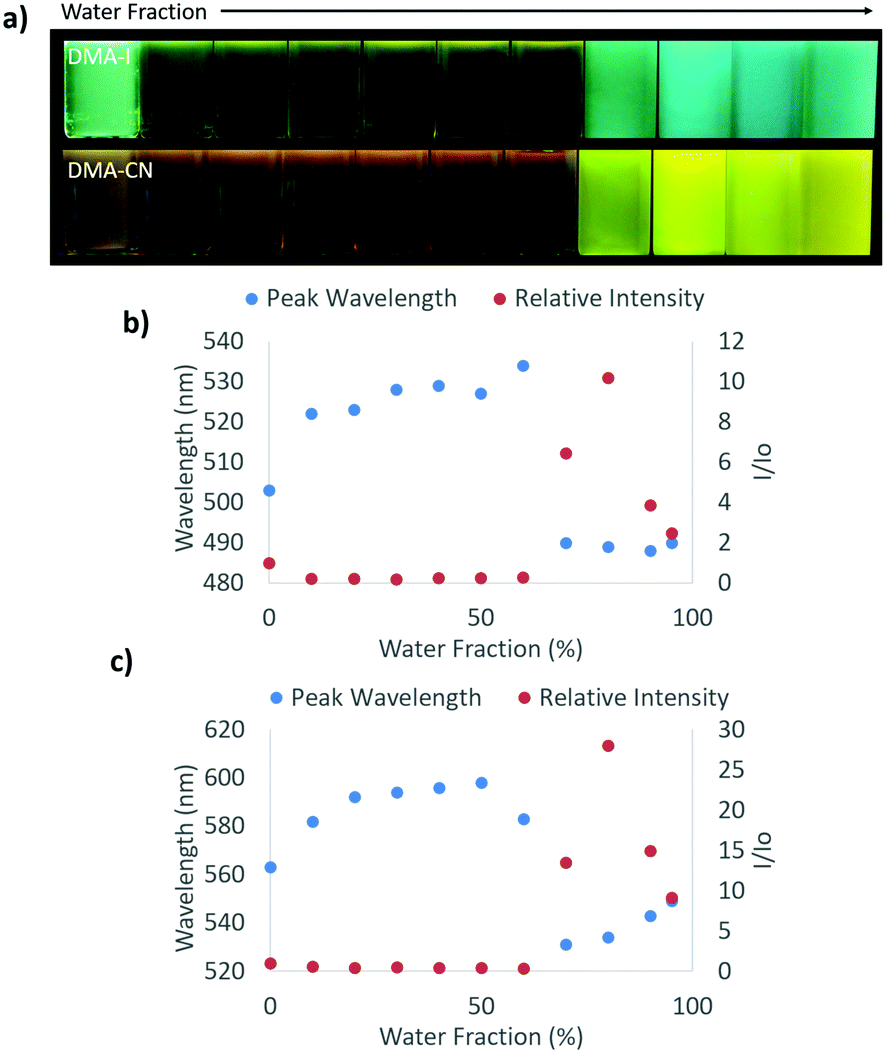 | ||
| Fig. 4 Image under UV irradiation (λex = 365 nm) (a). Plots of peak emission wavelength and intensity relative to emission in pure THF for DMA-I (b) and DMA-CN (c) dissolved in different THF/H2O solutions with increasing H2O concentrations (from left to right: 0%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%) (3 × 10−4 M, λex = 369 nm). | ||
In pure THF, DMA-I displayed blue-green emission under UV irradiation (λem = 503 nm) that was more intense than the dim orange emission observed for DMA-CN (λem = 563 nm). These differences can be explained based on the increased electron withdrawing ability of the cyano group relative to the iodo substituent. The addition of H2O resulted in diminished emission intensity and red-shifts in emission wavelengths for both dyes. For water fractions between 10–60%, the emission intensity remained relatively constant however a continual shift towards longer wavelengths was detected before reaching a maximum at 50% H2O for DMA-CN (λem = 598 nm) and 60% for DMA-I (λem = 534 nm). At higher water concentrations (i.e. >60%), both dyes exhibited sharp intensity increases along with blue-shifted wavelengths relative to emission in pure THF, which is indicative of AIEE. Furthermore, these solutions are highly turbid which indicates that aggregate species are formed in solutions with sufficiently large H2O concentrations.
For solutions between 70–95% H2O, DMA-I showed very little deviation in wavelength (λem = 488–490 nm) and a maximal 10× increase in emission intensity compared to pure THF. Over the same concentration range, DMA-CN showed a further red-shift in emission (λem = 531–549 nm) and a maximal intensity enhancement of 28×. The difference in emission enhancement between iodo and cyano substituted diketones can be explained by the greater initial intensity of DMA-I in pure THF, however it is also possible that aggregate species of DMA-CN exhibit larger quantum yields. This is expected given iodo-substituted dyes show faster rates of intersystem crossing to non-radiative triplet states via the heavy atom effect, which leads to quenching under ambient conditions and therefore lower quantum yields.54 Both dyes also exhibit trends toward longer wavelengths upon the addition of H2O which is likely due to stabilization of the polar excited states in more polar THF/H2O mixtures.
One of the largest differences in AIEE behavior between the iodo and cyano dyes is that the emission wavelength of DMA-CN gradually red-shifted even after aggregation whereas the wavelength of DMA-I was constant. Examination of the emission spectra for solutions that show evidence of aggregate formation (70–95% H2O) reveals that both DMA-I and DMA-CN initially exhibit highly structured emission. However, broad emission profiles that were devoid of structure were detected for the cyano substituted dye at 90 and 95% H2O (Fig. S8, ESI†). Comparatively, structured emission was noted for DMA-I even in solutions with the largest H2O concentrations. Previous investigation of the phase and morphology of AIE dyes suggests that this can be attributed to the formation of different emissive species (i.e. multiple crystalline or amorphous phases) depending on the THF/H2O ratio.25
To better understand AIE properties of DMA-CN and DMA-I, the excitation spectra, monitored at peak emission wavelength, were measured for all THF/H2O solutions (Fig. S9, ESI†). Low energy peaks attributed to ICT transitions were observed for both DMA-I and DMA-CN when dissolved in pure THF. These peaks were ascribed to ICT emission based on computational evidence and literature precedent (i.e. TICT in DMA-H).66 As the H2O fraction was increased, the ICT transitions were still clearly visible but with decreased intensity. Upon dye aggregation, a sharp decrease in intensity of ICT transitions was observed in the excitation spectra. For both derivatives, a hypsochromic shift in emission was observed in solutions with large H2O fractions, which is correlated with dye aggregation. It is possible that this wavelength shift is due to the minimization of the ICT transition for both dyes. Similar behavior was observed for α-substituted BF2 coordinated bdks dyes, which was attributed to the prevention of TICT upon aggregation.51 Despite the absence of boron coordination, the trends in emission wavelength of DMA-I and DMA-CN could be explained by an analogous mechanism.
Polystyrene films
Solvatochromism and AIE measurements indicate that DMA dyes are sensitive to the polarity of their surrounding environments. Furthermore, the degree of sensitivity appears to be correlated with the electron donating/withdrawing ability of the para substituent for a given derivative. Manipulation of the surrounding environment and donor/acceptor substitution are general strategies to tune dye emission in solution, however the roles that they play in determining the solid-state emission of ML active diketones are less clear. For example, previously reported luminescent bdk materials have a rather narrow color palette compared to emissive boron complex materials.59 In addition, the solid-state emission intensity of bdk materials is relatively weak compared to similar boron systems. Therefore, design strategies for tuning solid state emission color and intensity are important to improve the performance of stimuli responsive bdk materials.The effect of matrix polarity on the solid-state emission of DMA dyes was explored by fabricating polystyrene (PS) thin films of iodo (moderately withdrawing) and cyano (strongly withdrawing) substituents. Polystyrene was used for these experiments, because it provides a non-polar medium in which both diketones are soluble. Because these dyes have large ground-state dipoles (>10 D), the polarity of the surrounding medium increases at high dye loadings. However, increased concentration can also lead to aggregation and in many cases, red-shifted emission. Both aggregation and polarity effects were explored by measuring the emission of DMA-I and DMA-CN doped PS films with increasing dye concentration. In an attempt to decouple the magnitude of these competing processes, the excitation and emission properties were also measured for PS films containing dye and camphoric anhydride (CA), a non-emissive polar molecule. For these studies, the dye concentration was held constant while the concentration of CA was increased. This experiment was conducted using dilute (3 × 10−7 mol) and concentrated (4.5 × 10−6 mol) dye loadings. Films were fabricated via spin coating from dichloromethane solutions containing dye/PS or dye/CA/PS in varying ratios.
The emission of PS films was highly sensitive to dye loading (Fig. 5 and Fig. S10, ESI†). At the lowest dye concentration (0.019 mol% dye), films were faintly emissive to the eye, however via fluorescence spectroscopy, blue emission was detected for DMA-I/PS films (λem = 448 nm) compared to the blue-green emission (λem = 464 nm) observed in films containing DMA-CN. Both DMA-I and DMA-CN films exhibited a red-shift in emission even after relatively small changes in dye concentration (0.019–5.0 mol%). At higher dye loadings (>7.0 mol%), the emission wavelength was less sensitive to concentration as only modest changes in emission were detected. While maximally red-shifted emission (DMA-I: λem = 511 nm, DMA-CN: λem = 543 nm) was observed for both sets of films at the highest dye concentration measured (28 mol% dye), the trend in emission wavelengths suggests that further increases in concentration would have little effect on emission energy.
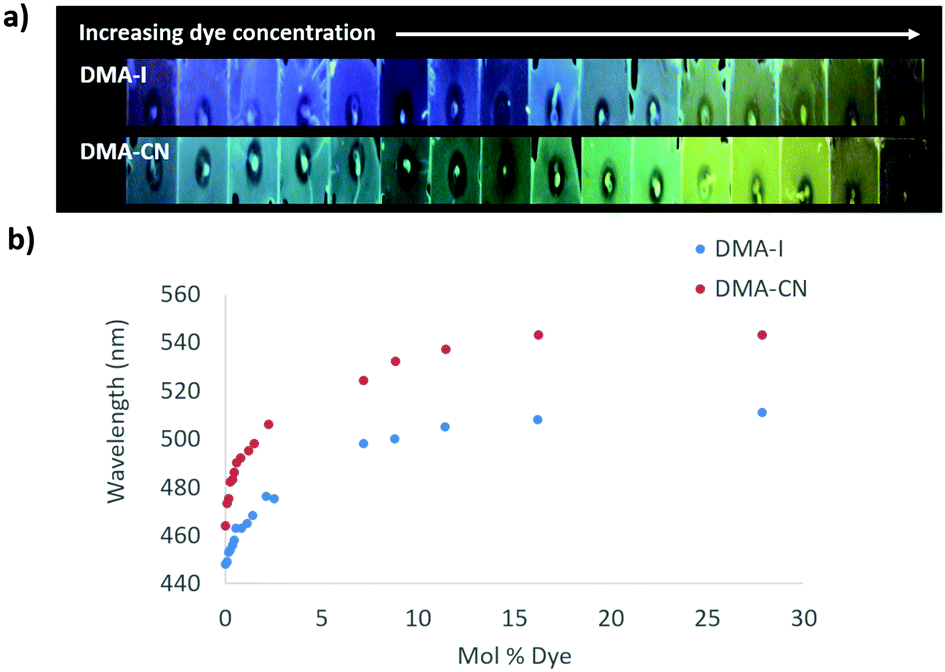 | ||
| Fig. 5 Image under UV irradiation of dye/polystyrene (PS) films with increasing dye concentration (λex = 369) (a). Plots of peak emission (λmax) vs. mol% of DMA-I and DMA-CN in PS (λex = 369 nm) (b). | ||
One potential explanation for the emission trends of dye/PS films is that the polarity of the matrix is higher due to the presence of dye molecules. Emission can be red-shifted due to stabilization of localized excited states by surrounding dye molecules which results in a phenomenon known as the solid-state solvation effect (SSSE). Evidence for SSSE dye/PS films can be seen in their red wavelength shifts (Δλem) with increasing dye concentration. The wavelength shift was greater for DMA-CN (Δλem = 79 nm, μ = 12.6 D) compared to DMA-I (Δλem = 63, μ = 10.4 D), which may be attributable to variances in matrix polarity given a larger dipole moment (μ) for the cyano substituted dye. (Estimates of μ were obtained via DFT calculations.) However, it is also possible that the red-shift is due, in part, to aggregation of DMA-I and DMA-CN dyes at higher concentrations.
In order to probe potential aggregation effects, the excitation spectra monitored at peak emission wavelength were measured for each film (Fig. S11, ESI†). The excitation spectra for all DMA-CN films were similar, which indicated that emission was produced by the same emissive species regardless of dye concentration. Examination of the excitation spectra for DMA-I films shows different behavior. Films with low dye concentration show broad peaks in the excitation spectra, however a blue shift in peak wavelength as well as the emergence of structure was observed at higher dye concentrations. This indicates that different emissive species formed as the concentration of DMA-I was increased, and could be indicative of aggregation. The difference in excitation spectra may be attributed to lower solubility of the iodo-substituted dye in the PS matrix.
To decouple the wavelength trends induced by SSSE from those produced by aggregation, dye/PS films were doped with increasing amounts of camphoric anhydride (μ ∼ 6 D)45 (Fig. 6 and Fig. S12, ESI†). When no CA dopant was added, films of DMA-I (λem = 454 nm) and DMA-CN (λem = 482 nm) glowed blue and blue-green, respectively. Similar to the dye loading experiments, a red-shift in emission was observed at low CA loadings (<10 mol% CA) followed by a more gradual change as the concentration of CA was increased. When the mole fraction of CA was increased to 46%, green emission was observed for DMA-I films (λem = 487 nm), that was significantly higher in energy compared to films of DMA-CN (λem = 523 nm), which glowed yellow. As the concentration of CA was further increased, a hypsochromic shift in emission was observed, which can be attributed to phase separation of CA molecules within PS films. Phase separation was confirmed through examination of the XRD patterns for dye/PS/films, which showed crystalline peaks at high CA loadings (63 mol% CA), but were amorphous for maximally red-shifted dye/CA/PS films (46 mol% CA) (Fig. S13, ESI†). Comparison of the wavelength shifts in CA doped films showed that DMA-CN (Δλem = 41 nm) was more sensitive to matrix polarity effects than DMA-I (Δλem = 33). This result is consistent with dye loading studies which also showed larger wavelength shifts for DMA-CN.
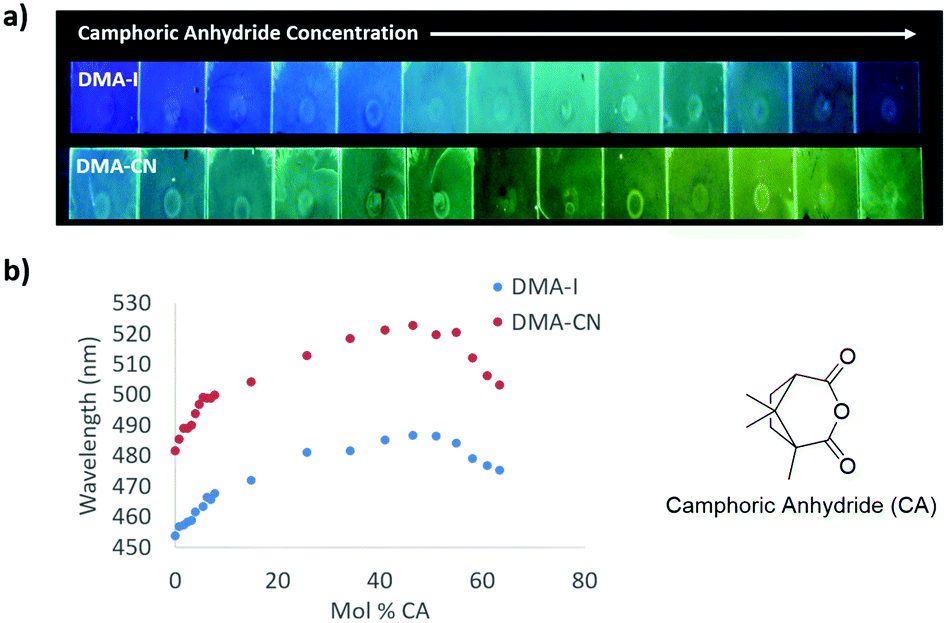 | ||
| Fig. 6 Image under UV irradiation of dye/camphoric anhydride/polystyrene (dye/CA/PS) films with increasing camphoric anhydride (CA) concentration and dilute (3 × 10−7 mol) dye concentration (λex = 365 nm) (a). Plots of peak emission (λmax) vs. mol% of CA in DMA-I and DMA-CN dye/CA/PS films (λex = 369 nm) (b). | ||
To investigate dopant effects on films with initially red-shifted emission, films with higher dye loadings and increasing CA concentration were produced (Fig. 7). The emission without the CA dopant was much more red shifted for both DMA-I (λem = 502 nm) and DMA-CN (λem = 532 nm) compared to the results discussed above, presumably due to increased dye–dye interactions. Upon the addition of CA, a red shift in emission was observed for both dyes. The maximally red-shifted emissions for DMA-I (λem = 510 nm) and DMA-CN (λem = 553 nm) were observed at CA mole fractions of 12% and 6%, respectively. Further increase of CA concentration resulted in a blue shift in peak wavelength due to phase separation. Compared to dye loading experiments, the maximally red shifted emission observed for DMA-I/CA/PS films with high initial dye concentrations was nearly identical. However, the emission of corresponding DMA-CN/CA/PS films was lower in energy than maximally red-shifted DMA-CN/PS films. This indicates that increasing matrix polarity with external dopants can further reduce the energy of emission in films with high dye–dye interactions.
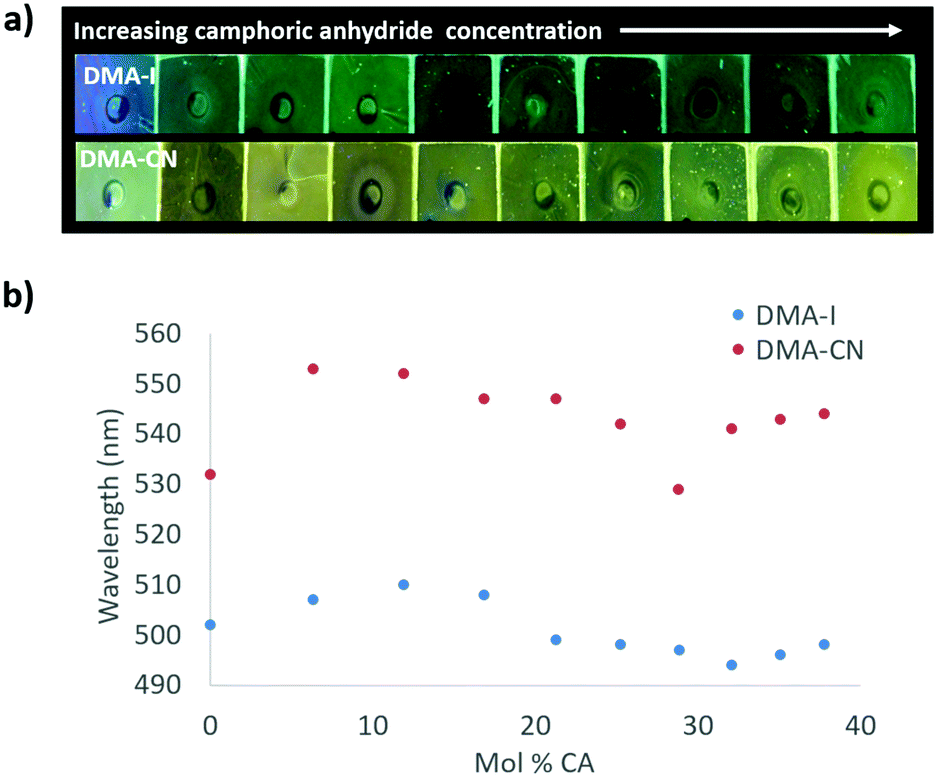 | ||
| Fig. 7 Image under UV irradiation of dye/camphoric anhydride/polystyrene (dye/CA/PS) films with increasing CA concentration and concentrated (4.5 × 10−6 mol) amounts of dye (λex = 369 nm) (a). Plots of peak emission (λmax) vs. mol% of CA in DMA-I and DMA-CN dye/CA/PS films (λex = 369 nm) (b). | ||
Excitation spectra of dye/CA/PS films were measured in order to monitor potential dopant effects on emissive species in these films (Fig. S14, ESI†). Spectra showed almost no change as the amount of CA was increased, which indicated that additional ground-state species were not formed in dye/CA/PS films with high CA concentration. Therefore, the shift in emission as the matrix polarity of DMA-I and DMA-CN films was increased may be attributable to stabilization of their respective excited states by neighboring polar molecules. Though emission can be modulated through the addition of a non-polar dopant, the wavelength shift was much smaller compared to the corresponding films with increased dye loading. Additionally, greater concentrations of CA were required to produce red-shifted emission in comparison to dye/PS films, which required smaller amounts of dye to elicit a red-shifted response. This can be explained by the relative polarity of the dopant, as DMA-I and DMA-CN both have larger ground state dipoles than CA.
Dye loading and CA doping experiments suggest that SSSE is a contributing factor toward the red-shift emission observed for these films. However, evidence of aggregation was also observed in DMA-I/PS films; thus, it is impossible to decouple aggregation from SSSE in these films. Yet, no evidence of aggregation was observed for PS films with increasing DMA-CN concentration, which indicates that the spectral shifts for those films is likely due to higher matrix polarity as opposed to aggregation effects. While it is clear that these dyes behave differently in PS films, the emissions of these dyes are sensitive to polarity effects through the addition of external dopants as well as the dyes themselves. Therefore, the relative polarity of dye molecules could also impact the emission of their corresponding stimuli-responsive solids.
Solid-state optical properties
In order to probe donor and acceptor effects on the response of DMA substituted diketones to thermal and mechanical stimuli, thin films were made by smearing a small amount of dye (∼2 mg) across 5 cm × 5 cm squares of weigh paper. Films were thermally annealed at 110 °C (TA), then were subsequently smeared with a cotton swab to generate the smeared state (SM). The effect of melt quenching was also investigated by heating films above the melting point and cooling in air to produce the melted (MT) state. After melt quenching, the TA state could be regenerated via subsequent heating at 110 °C. Emission spectra were measured for films in the TA, SM and MT states (Table 2, Fig. 8 and Fig. S15, ESI†).| Compound | Annealed | Smeared | Melted | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| λ TA (nm) | τ TA (ns) | FWHMTAd (nm) | Φ TA | λ SM (nm) | FWHMSMd (nm) | λ MT (nm) | τ MT (ns) | FWHMMTd (nm) | Φ MT | |
| a Samples were annealed at 110 °C and λes = 385 nm. b Peak emission wavelength in thermally annealed (TA) smeared (SM) and melted (MT) states. c Pre-exponential weighted lifetime in TA and MT states. d Full width at half maximum in TA, SM and MT states. e Quantum yield in TA and MT states. | ||||||||||
| DMA-H | 519 | 1.47 | 114 | 18.8 | 518 | 109 | 538 | — | 112 | 14.8 |
| DMA-OMe | 488 | 0.91 | 82 | 53.4 | 496 | 90 | 514 | 1.74 | 100 | 51.2 |
| DMA-F | 488 | 0.69 | 57 | 33.5 | 497 | 93 | 522 | 1.57 | 101 | 26.4 |
| DMA-Br | 492 | 0.44 | 68 | 25.2 | 517 | 109 | 522 | 1.48 | 121 | 30.1 |
| DMA-I | 492 | 0.13 | 63 | 14.1 | 494 | 90 | 540 | 0.85 | 106 | 18.6 |
| DMA-CF3 | 522 | 1.77 | 75 | 59.3 | 525 | 96 | 528 | 1.33 | 86 | 29.9 |
| DMA-COOMe | 542 | 1.23 | 103 | 29.9 | 550 | 111 | 576 | 1.96 | 117 | 21.7 |
| DMA-CN | 532 | 1.47 | 91 | 20.4 | 549 | 112 | 578 | 2.54 | 118 | 10.3 |
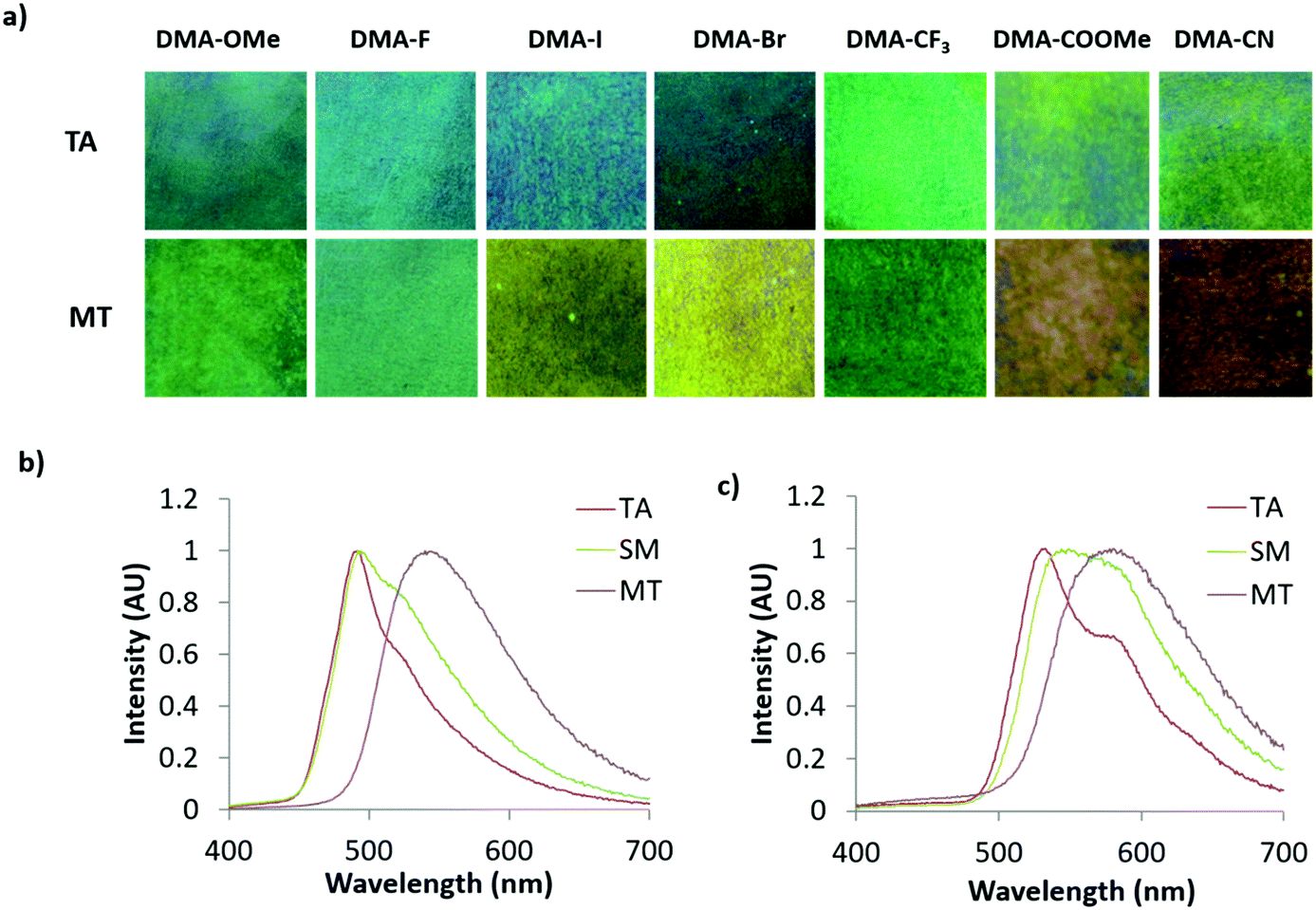 | ||
| Fig. 8 Images under UV irradiation of diketones on weigh paper (WP) films in the thermally annealed (TA) and melted (MT) states (a). Emission spectra of DMA-I (b) and DMA-CN (c) WP films in TA, smeared (SM), and MT states. | ||
Since similar behavior was observed for all DMA dyes, exemplary emission spectra for DMA-I and DMA-CN are shown in Fig. 8. While all dyes show visible solid-state emission in the TA state ranging from blue (DMA-H, DMA-OMe, DMA-F, DMA-Br, DMA-I) to green (DMA-CF3, DMA-COOMe, DMA-CN), the emission of DMA-H is only faintly visible to the eye and is therefore not pictured. The emission of all films in the TA state was red-shifted compared to their emission in dichloromethane solution, however a similar trend toward longer emission wavelengths was observed as the electron withdrawing ability of the para-substituent was increased (Table 2). For example, the highest energy emission was observed in both methoxy and fluoro substituted derivatives (λem = 488 nm), and DMA-COOMe, a strongly withdrawing dye, exhibited the most red-shifted emission (λTA = 542 nm). In addition, both solution and TA measurements indicated that the lowest quantum yield and shortest lifetime was observed for iodo substituted derivatives. This is likely due to increased intersystem crossing (ISC) to a non-emissive triplet state via the heavy atom effect.
Though comparable trends were observed for emission in dichloromethane solution and the annealed state, some aberrations were detected in TA emission trends. For example, only a small deviation in TA emission (ΔλTA = 4 nm) was observed for DMA-OMe, DMA-F, DMA-Br, and DMA-I despite large changes in electron withdrawing strength. Also, the emission of DMA-COOMe was more redshifted compared to DMA-CN (λTA = 532 nm) despite being substituted with a weaker withdrawing substituent. The DMA-H dye was a further exception to this trend because the emission of annealed DMA-H films (λTA = 514 nm) was significantly higher in energy when compared to substituents with greater electron withdrawing ability (i.e. F, Br, and I). While evidence of a trend between the electron donation and withdrawing ability and the emission of TA thin films can be established, there is much more deviation compared to dichloromethane solution. This difference in behavior likely results from the more complex optical properties of emissive solids which are often determined by molecular packing effects and often form multiple emissive species.58,77
With the exception of dim DMA-H, the emission of all DMA substituted bdks were modulated via smearing. Despite visual conformation of mechanically produced color changes, narrow ML shifts (ΔλML < 10 nm) were measured for several DMA derivatives (DMA-OMe, DMA-F, DMA-I, DMA-CF3, DMA-COOMe). The emission change for many of these analogues can be attributed to spectral broadening as increases in full width at half maximum (FWHM) were detected for each compound. Larger mechanical responses were observed for cyano (ΔλML = 17 nm) and bromo (ΔλML = 30 nm) derivatives, however they were still significantly smaller when compared to other previously studied bromo and methoxy substituted dinaphthoyl diketones (ΔλML > 50 nm).59 The absence of a mechanical response for DMA-H could indicate that thermal treatment of films at 110 °C was insufficient to fully anneal weigh paper films. However, no change in emission could be produced upon subsequent heating, despite annealing at a variety of different temperatures (80 °C, 90 °C, 100 °C, 120 °C, 130 °C). This may be indicative of similar solid state morphologies before and after annealing.
Although ML was detectable for all dyes, it was less vibrant when compared to other bdk systems with dinaphthoyl methane scaffolds.59 However, emission changes could be detected for all DMA analogues after melting and cooling in air (i.e. melt quenching). The degree of wavelength shift after melt quenching (ΔλMT) varied depending on the electron withdrawing substituent. Large shifts were observed for DMA-I (ΔλMT = 48 nm) and DMA-CN (ΔλMT = 46 nm) whereas melt quenching of DMA-CF3 only resulted in a 6 nm shift. As observed for samples in dichloromethane solution and the TA state, a general trend toward longer wavelengths is observed with greater electron withdrawing ability of the para substituted groups. The emissions of dyes with strongly withdrawing substituents, such as DMA-COOMe (λMT = 576 nm) and DMA-CN (λMT = 578 nm), were much lower in energy compared to the other para-substituted dyes. The peak emission in the MT state of the remaining derivatives fluctuated between a minimum for DMA-OMe (λMT = 515 nm) and maximum for DMA-I (λMT = 540 nm). While melt quenching resulted in an increase in fluorescence lifetimes (τMT) compared to τTA, quantum yields (ΦMT) changed very little. An increase in lifetime has previously been correlated with the formation of a longer lived amorphous state upon smearing for other ML active bdk materials.55,58 Because similar behavior was observed in MT films, it is possible that the emission change is also due to the formation of an amorphous state.
Comparison of the solid-state optical properties of DMA-I and DMA-CN with the trends observed in their corresponding PS thin films indicates that the emission of DMA-substituted diketones could be due to changes in the matrix polarity in addition to dye aggregation. To summarize, both the emission of DMA-I/PS and DMA-CN/PS films showed sensitivity toward matrix polarity which was increased through increasing dye loading as well as the addition of the polar, non-emissive dopant, camphoric anhydride. As solid films, the emissions of iodo and cyano substituted derivatives in the TA state were blue-shifted compared to their maximally red shifted dye/PS counterparts (DMA-I: λem = 511 nm; DMA-CN: λem = 543) despite closer proximity to neighboring dyes. Since dyes are typically crystalline after annealing, these results may indicate that the packing arrangement of molecular dipoles leads to diminished matrix polarity compared to the randomly oriented dyes in amorphous dye/PS films. Yet melt quenched thin films of pure dye exhibited more red-shifted emission compared to dye/PS films. Because MT films are typically amorphous, dyes are isotropic which could lead to larger matrix polarities. Therefore, the longer wavelengths observed for these films could be due, in part, to stabilization of localized excited states by randomly oriented neighboring dye molecules.
While the emission wavelength of bdk dyes is likely a combination of SSSE and aggregation effects, these data nonetheless indicate that designing solid-state emissive molecules with large molecular dipoles is an effect strategy for modulating the color of stimuli responsive bdk materials.
Thermal properties
Previously reported bdk and BF2bdk materials have shown thermochromic properties. Specifically, some methoxy substituted ligands formed transparent supercooled liquid states upon rapid cooling.78 Alternatively, certain furan substituted BF2bdks formed opaque amorphous solids after melt quenching.56 Differential scanning calorimetry (DSC) was used to distinguish these distinct behaviors and here, to probe the thermal properties of DMA diketones (Fig. S16, ESI†). All compounds were subjected to two heating/cooling cycles. While the heating rate was held constant between scans (5 °C min−1), the rate of cooling was varied from Cycle 1 (10 °C min−1) to Cycle 2 (1 °C min−1). A fast cooling rate was used to simulate the melt quenching process, whereas slower cooling scans were utilized to promote crystallization.The thermal properties of all dyes are described in Table 3. All samples showed almost no change in melting temperature (Tm) from Cycle 1 to Cycle 2 and very little deviation in crystallization temperature (Tc) despite the change in cooling rate. The presence of Tc transitions in both cycles indicates that supercooled liquid phases are not formed even at faster cooling rates. The melting temperatures of dyes can give insight into the strength of the intermolecular interactions that determine packing. The lowest melting temperatures were observed when dyes were substituted with electron withdrawing fluoro (Tm = 131.5 °C) and electron donating methoxy (Tm = 132.1 °C) substituents. Comparatively, the other halogenated derivatives DMA-Br (Tm = 188.3 °C) and DMA-I (Tm = 204.9 °C) showed relatively high melting points. Strongly withdrawing derivatives DMA-CN (Tm = 209.5 °C) and DMA-COOMe (Tm = 198.0 °C) exhibited high melting points, however the melting point of DMA-CF3 (Tm = 147.4 °C) was considerably lower. With the exception of DMA-CF3, there is a trend toward higher melting points as the electron withdrawing ability of the substituent increases. Thus, the introduction of donor/acceptor effects can be used to tune Tm of DMA dyes. The anomalous behavior of DMA-CF3 can be rationalized by the increased steric interactions of the CF3 moiety which could disrupt crystal packing and lead to weaker intermolecular associations.
| Compound | T m | T c |
|---|---|---|
| a All data was taken from the 2nd cycle. b Melting point given in °C as the peak of the major endothermic transition. c Crystallization point given in °C as the peak of the major exothermic transition. | ||
| DMA-H | 140.4 | 80.1 |
| DMA-OMe | 132.1 | 99.4 |
| DMA-F | 131.5 | 89.2 |
| DMA-Br | 188.3 | 134.4 |
| DMA-I | 204.9 | 138.9 |
| DMA-CF3 | 147.4 | 137.0 |
| DMA-COOMe | 198.0 | 170.0 |
| DMA-CN | 209.5 | 164.2 |
Structural characterization
To gain insight into the different phases that determine the stimuli responsive properties of DMA substituted dyes, powder X-ray diffraction (XRD) patterns were measured for the bulk dyes, and films on glass in TA and MT states. Due to sample removal upon smearing, patterns were not obtained for glass films in the SM state. Films for XRD were fabricated by drop casting from a dye/DCM solution (1 × 10−5 M) with evaporation in air, followed by drying under vacuum, before annealing and melting each film to access the TA and MT states, respectively. Powder XRD patterns for DMA-I and DMA-CN are shown in Fig. 9, and the data for the remaining diketones can be found in the ESI† (Fig. S17). Single crystal XRD analysis of DMA-I was also performed to investigate potential packing effects. Crystals were grown via slow evaporation of a concentrated DMA-I/DCM solution. Though we attempted to grow crystals of all previously unexplored DMA dyes, only DMA-I formed crystals suitable for diffraction.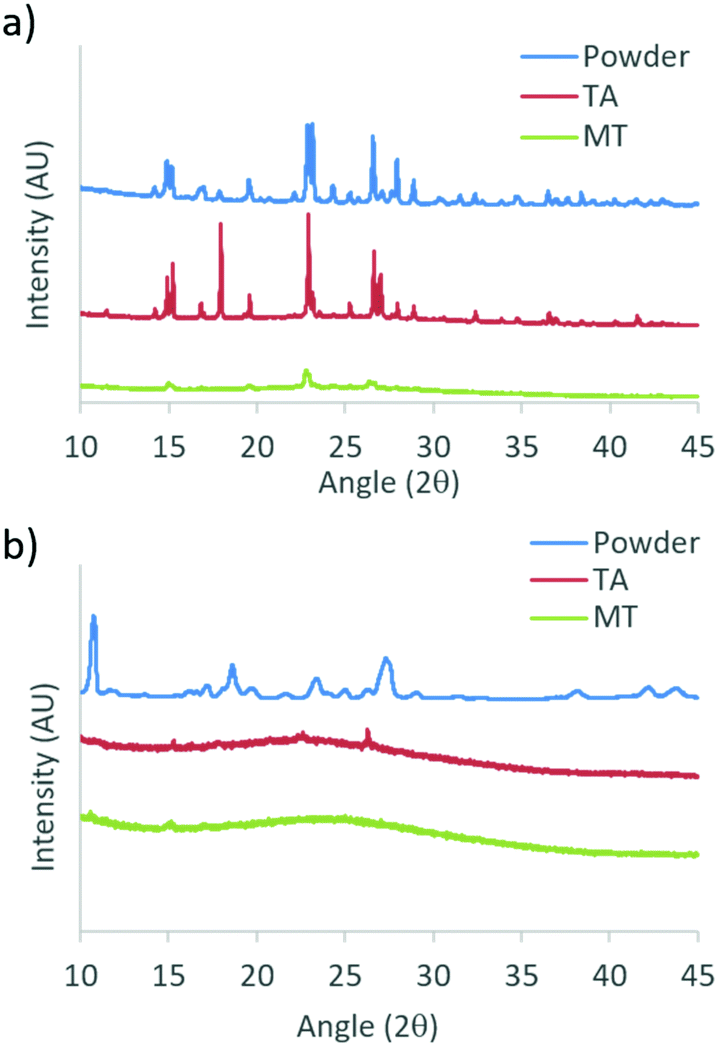 | ||
| Fig. 9 Powder X-ray diffraction patterns of (a) DMA-I and (b) DMA-CN bulk powders and films on glass in thermally annealed (TA) and melted (MT) states. | ||
The sharp diffraction peaks found in the powder XRD patterns indicate that all compounds were crystalline as bulk powders. Also, all samples showed diffraction peaks as films on glass in the TA states which corresponded to peaks in the bulk powder. Comparison of TA patterns for all dyes indicated that the relative peak intensity and number of peaks was substituent dependent, however the observation of peak patterns of TA glass films indicated the emission was produced by crystalline species. Data taken for melted films were largely devoid of peaks suggesting that they were amorphous, however small peaks were observed in bromo and iodo heavy atom substituted MT films. For example, small diffraction peaks are evident for DMA-I samples, indicating this dye has a greater propensity to crystallize, similar to previously reported iodo substituted BF2bdk derivatives.
Single crystal analysis of DMA-I revealed that dyes adopt a nearly planar conformation with the DMA substituent and phenyl rings in roughly the same plane (Fig. 10a). A slight deviation from planarity is observed in the twisting of the phenyl rings about the diketone core. Examination of the unit cell shows that DMA-I packs in a herringbone motif with C–H⋯I, and C–H⋯arene interactions (Fig. 10b). Iodine–iodine interactions, which were previously observed in crystal structures of iodo-substituted BF2bdks, were not detected in the crystal packing of DMA-I.54 Additionally, there is no evidence of dimer formation, as neighboring dyes are stacked at slight angles relative to each other. While DMA-I does show a visible ML shift upon smearing, it is less dramatic compared to previously studied dinapthoyl substituted bdks.54 The emission spectrum of DMA-I shows that the color change is mostly due to a broadening of the emission profile rather than a shift in peak emission.
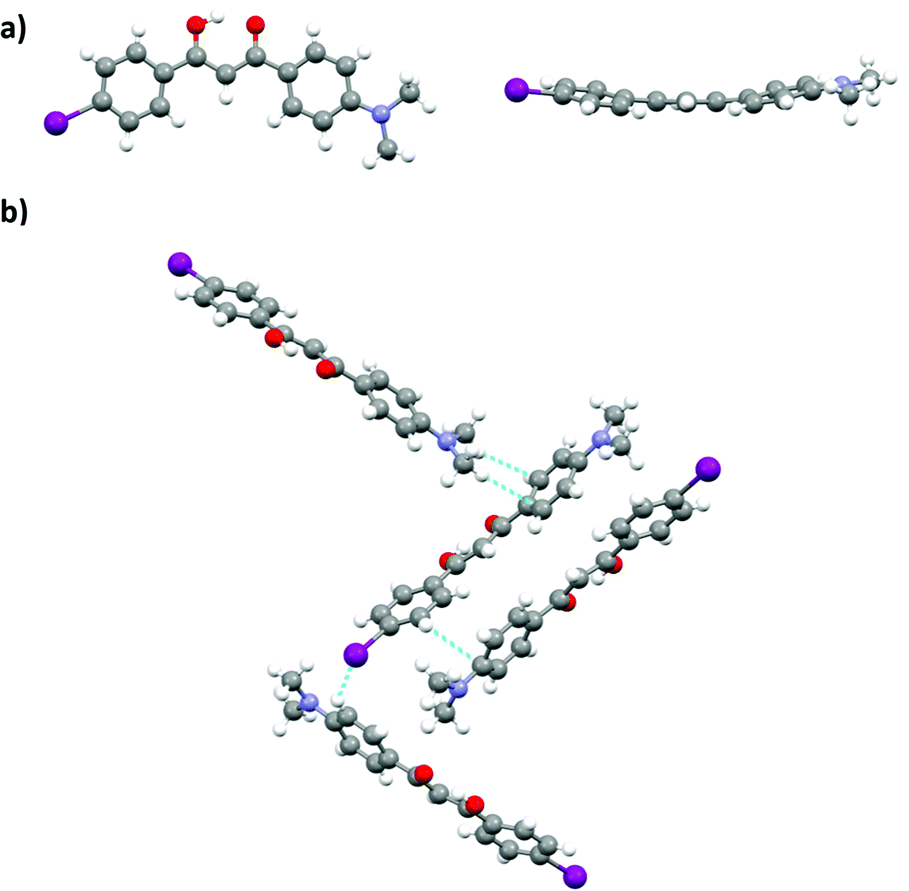 | ||
| Fig. 10 Crystal structure of DMA-I from two perspectives (a). Unit cell of DMA-I showing packing interactions (b). | ||
Conclusion
In summary, the electron withdrawing ability of dye substituents had a large effect on the solution and solid-state optical properties of DMA dyes. According to DFT calculations, all dyes showed evidence of ICT in their HOMO and LUMO molecular orbitals which correlated with a positive solvatochromic shift. Dye sensitivity toward solvent polarity depended on the electron withdrawing strength of the para substituent, however the cyano substituted dye, DMA-CN, exhibited evidence of ESIPT. The AIE properties of DMA-I and DMA-CN were tested in THF/H2O solutions with increasing H2O fractions. Both dyes showed AIE, however longer wavelength emission and a larger intensity increase was observed for DMA-CN. Polystyrene thin films of DMA-I and DMA-CN with increasing concentrations of dye and camphoric anhydride, an external polar dopant exhibited red-shifted emission, which indicated that they are sensitive to matrix polarity in addition to exhibiting solvatochromism. Both smearing and melting dye films on weigh paper resulted in emission responses, however the maximally red-shifted emission was observed after melt quenching. Like other ML active BF2bdk and bdk systems, powder XRD patterns indicate that the change in emission is linked with a phase change from crystalline in the TA state, to amorphous for SM and MT films. As with their emission in solution, a strong correlation between the strength of the electron withdrawing group and the wavelength of solid-state emission, especially in the amorphous MT phase. Based on the sensitivity of DMA-I and DMA-CN emission toward matrix polarity in PS films, it is possible that neighboring dye molecules stabilize the excited state of homogenous dye films as well, leading to red shifted emission. While there are likely many effects that determine emission in stimuli responsive materials (e.g. aggregation), engineering dyes with large molecular dipoles in order to utilize matrix polarity effects is a design strategy for future stimuli responsive luminescent materials. Furthermore, this work represents an important step forward in the design of boron-free bdk materials, given compounds with wide ranging colors and thermochromic properties were achieved.Acknowledgements
We thank the National Science Foundation (NSF CHE-1213915) and University of Virginia Department of Chemistry for support for this research. Christopher DeRosa is acknowledged for thoughtful discussions.References
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- K. Chung, M. S. Kwon, B. M. Leung, A. G. Wong-Foy, M. S. Kim, J. J. Kim, S. Takayama, J. Gierschner, A. J. Matzger and J. J. Kim, ACS Cent. Sci., 2015, 1, 94–102 CrossRef CAS PubMed.
- Y. Jiang, Mater. Sci. Eng., C, 2014, 45, 682–689 CrossRef CAS PubMed.
- F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 RSC.
- Y. Sagara, T. Komatsu, T. Ueno, K. Hanaoka, T. Kato and T. Nagano, J. Am. Chem. Soc., 2014, 136, 4273–4280 CrossRef CAS PubMed.
- S. K. Park, I. Cho, J. Gierschner, J. H. Kim, J. H. Kim, J. E. Kwon, O. K. Kwon, D. R. Whang, J.-H. Park, B.-K. An and S. Y. Park, Angew. Chem., 2016, 128, 211–215 CrossRef.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- G. Liang, J. Wu, H. Gao, Q. Wu, J. Lu, F. Zhu and B. Z. Tang, ACS Macro Lett., 2016, 5, 909–914 CrossRef CAS.
- H. Mao, P. Pan, G. Shan and Y. Bao, J. Phys. Chem. B, 2015, 119, 6471–6480 CrossRef CAS PubMed.
- D. G. Abebe and T. Fujiwara, Biomacromolecules, 2012, 13, 1828–1836 CrossRef CAS PubMed.
- Y. Fang, Y. Ni, S.-Y. Leo, B. Wang, V. Basile, C. Taylor and P. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 23650–23659 CAS.
- A. L. Black, J. M. Lenhardt and S. L. Craig, J. Mater. Chem., 2011, 21, 1655–1663 RSC.
- M. Moßhammer, M. Strobl, M. Kühl, I. Klimant, S. M. Borisov and K. Koren, ACS Sensors, 2016, 1, 681–687 CrossRef.
- X. Wang, R. J. Meier and O. S. Wolfbeis, Angew. Chem., 2013, 125, 424–427 CrossRef.
- X. Zhang, X. Liu, R. Lu, H. Zhang and P. Gong, J. Mater. Chem. C, 2012, 22, 1167–1172 RSC.
- S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 4757–4764 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- J. R. Hemmer, S. O. Poelma, N. Treat, Z. A. Page, N. D. Dolinski, Y. J. Diaz, W. Tomlinson, K. D. Clark, J. P. Hooper, C. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2016, 138, 13960–13966 CrossRef CAS PubMed.
- S. Varughese, J. Mater. Chem. B, 2014, 2, 3499–3516 CAS.
- C. A. DeRosa, C. Kerr, Z. Fan, M. Kolpaczynska, A. S. Mathew, R. E. Evans, G. Zhang and C. L. Fraser, ACS Appl. Mater. Interfaces, 2015, 7, 23633–23643 CAS.
- R. Yoshii, A. Nagai, K. Tanaka and Y. Chujo, Macromol. Rapid Commun., 2014, 35, 1315–1319 CrossRef CAS PubMed.
- W. Z. Yuan, P. Lu, S. Chen, J. W. Y. Lam, Z. Wang, Y. Liu, H. S. Kwok, M. Yuguang and B. Z. Tang, Adv. Mater., 2010, 22, 2159–2163 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. Sing Wong, E. Peñ a-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 1845–15853 Search PubMed.
- H. Tong, D. Yongqiang, Y. Hong, M. Häussler, J. W. Y. Lam, H. H.-Y. Sung, X. Yu, J. Sun, I. D. Williams, H. S. Kwok and B. T. Zhong, J. Phys. Chem. C, 2007, 111, 2287–2294 CAS.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- R. G. Gri and M. Dinca, J. Am. Chem. Soc., 2012, 134, 15061–15070 CrossRef PubMed.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed.
- Z. Li, Y. Q. Dong, J. W. Y. Lam, J. Sun, A. Qin, M. Häußler, Y. P. Dong, H. H. Y. Sung, I. D. Williams, H. S. Kwok and B. Z. Tang, Adv. Funct. Mater., 2009, 19, 905–917 CrossRef CAS.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 1–4 Search PubMed.
- C. Li, M. Liu, N. G. Pschirer, M. Baumgarten and K. Muellen, Chem. Rev., 2010, 110, 6817–6855 CrossRef CAS PubMed.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed.
- G. Zhang, S. H. Kim, R. E. Evans, B. H. Kim, J. N. Demas and C. L. Fraser, J. Fluoresc., 2009, 19, 881–889 CrossRef CAS PubMed.
- C. M. Ip and A. Troisi, J. Phys. Chem. Lett., 2016, 7, 2989–2993 CrossRef PubMed.
- C.-T. Poon, D. Wu, W. H. Lam and V. W.-W. Yam, Angew. Chem., 2015, 127, 10715–10719 CrossRef.
- L. Zöphel, V. Enkelmann and K. Müllen, Org. Lett., 2013, 15, 804–807 CrossRef PubMed.
- A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS PubMed.
- E. J. Choi, E. Kim, Y. Lee, A. Jo and S. B. Park, Angew. Chem., Int. Ed., 2014, 53, 1346–1350 CrossRef CAS PubMed.
- J. Wu, Y. Cheng, J. Lan, D. Wu, S. Qian, L. Yan, Z. He, X. Li, K. Wang, B. Zou and J. You, J. Am. Chem. Soc., 2016, 138, 12803–12812 CrossRef CAS PubMed.
- X. Y. Shen, Y. J. Wang, E. Zhao, W. Z. Yuan, Y. Liu, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2013, 117, 7334–7347 CAS.
- E. Lippert, Z. Naturforsch., A: Phys. Sci., 1955, 10, 541–545 Search PubMed.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- V. Bulović, R. Deshpande, M. E. Thompson and S. R. Forrest, Chem. Phys. Lett., 1999, 308, 317–322 CrossRef.
- C. F. Madigan and V. Bulović, Phys. Rev. Lett., 2003, 91, 247403 CrossRef PubMed.
- W. Chang, G. M. Akselrod and V. Bulović, ACS Nano, 2015, 9, 4412–4418 CrossRef CAS PubMed.
- A. P. Green, K. T. Butler and A. R. Buckley, Appl. Phys. Lett., 2013, 102, 133501 CrossRef.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- G. Zhang, T. L. S. Clair and C. L. Fraser, Macromolecules, 2009, 42, 3092–3097 CrossRef CAS.
- T. Liu, A. D. Chien, J. Lu, G. Zhang and C. L. Fraser, J. Mater. Chem., 2011, 21, 8401–8408 RSC.
- W. A. Morris, M. Kolpaczynska and C. L. Fraser, J. Phys. Chem. C, 2016, 120, 22539–22548 CAS.
- N. D. Nguyen, G. Zhang, J. Lu, A. E. Sherman and C. L. Fraser, J. Mater. Chem., 2011, 21, 8409–8415 RSC.
- G. Zhang, J. Lu and C. L. Fraser, Inorg. Chem., 2010, 49, 10747–10749 CrossRef CAS PubMed.
- W. A. Morris, M. Sabat, T. Butler, C. A. DeRosa and C. L. Fraser, J. Phys. Chem. C, 2016, 120, 14289–14300 CAS.
- W. A. Morris, T. Liu and C. L. Fraser, J. Mater. Chem. C, 2015, 3, 352–363 RSC.
- W. A. Morris, T. Butler, M. Kolpaczynska and C. L. Fraser, Mater. Chem. Front., 2017, 1, 158–166 RSC.
- X. Sun, X. Zhang, X. Li, S. Liu and G. Zhang, J. Mater. Chem., 2012, 22, 17332–17339 RSC.
- T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, Chem. Commun., 2015, 51, 3359–3362 RSC.
- T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, ACS Appl. Mater. Interfaces, 2016, 8, 1242–1251 CAS.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- J. Zhao and Y. Yang, J. Mol. Liq., 2016, 220, 735–741 CrossRef CAS.
- A. P. Demchenko, K.-C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC.
- J. E. Kwon, S. S. Y. Park and S. S. Y. Park, J. Am. Chem. Soc., 2013, 135, 11239–11246 CrossRef CAS PubMed.
- K.-C. Tang, M.-J. Chang, T.-Y. Lin, H.-A. Pan, T.-C. Fang, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Am. Chem. Soc., 2011, 133, 17738–17745 CrossRef CAS PubMed.
- H. Shi, R. Liu, S. Zhu, Q. Gong, H. Shi, X. Zhu and H. Zhu, J. Fluoresc., 2016, 26, 2005–2013 CrossRef CAS PubMed.
- R. Ghosh and D. K. Palit, Photochem. Photobiol. Sci., 2013, 12, 987–995 CAS.
- H. Fujii, S. Hirao, H. Sakurai and K. Tani, JP 2005035902, 2005.
- Y. Sato, M. Morimoto, H. Segawa and T. Shimidzu, J. Phys. Chem., 1995, 99, 35–39 CrossRef CAS.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 5118–5120 RSC.
- Y. L. Chow, C. I. Johansson, Y.-H. Zhang, R. Gautron, L. Yang, A. Rassat and S.-Z. Yang, J. Phys. Org. Chem., 1996, 9, 7–16 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09 Revis. A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. Xu, R. E. Evans, L. Tiandong, G. Zhang, J. N. Demas, C. O. Trindle and C. L. Fraser, Inorg. Chem., 2013, 52, 3597–3610 CrossRef CAS PubMed.
- Y. Kubota, Y. Sakuma, K. Funabiki and M. Matsui, J. Phys. Chem. A, 2014, 118, 8717–8729 CrossRef CAS PubMed.
- X. Y. Shen, W. Z. Yuan, Y. Liu, Q. Zhao, P. Lu, Y. Ma, I. D. Williams, A. Qin, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2012, 116, 10541–10547 CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- Y. Kubota, Y. Ozaki, K. Funabiki and M. Matsui, J. Org. Chem., 2013, 78, 7058–7067 CrossRef CAS PubMed.
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS PubMed.
- T. Butler, A. S. Mathew, M. Sabat and C. L. Fraser, ACS Appl. Mater. Interfaces, 2017 DOI:10.1021/acsami.7b01985.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1542425. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00157f |
| This journal is © the Partner Organisations 2017 |