
Time-evolved, far-red, circularly polarised luminescent polymer aggregates endowed with sacrificial helical Si–Si bond polymers†
Michiya
Fujiki
 * and
Shosei
Yoshimoto
* and
Shosei
Yoshimoto
Graduate School of Materials Science, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0192, Japan. E-mail: fujikim@ms.naist.jp
First published on 21st April 2017
Abstract
An enantiomeric pair of non-charged, helical dialkylpolysilanes (PSi-R and PSi-S) were endowed with circularly polarised luminescence (CPL) at 700 nm and circular dichroism (CD) at 660 nm when non-helical poly{[dioctylfluorene]-alt-[bis(thiophenyl)-benzothiazole]} (PF8DBT) formed a hetero-aggregate with PSi-R and PSi-S in a 1-to-1 ratio as repeating units. The initial ultraweak CPL (gem) and CD (gabs) amplitudes of the as-prepared aggregates were magnified up to −0.019 at 706 nm (induced by PSi-S) and +0.017 at 695 nm (induced by PSi-R) after prolonged ageing at ambient temperature for 24 h. These CPL amplitudes were maintained after the selective photoscissoring of PSi-S and PSi-R at 313 nm for 60 s. A time-dependent growth behaviour in the hetero-aggregate size (initially ≈400 nm; reached ≈2000 nm in one day) was observed and characterised using dynamic light scattering (DLS). To investigate the origin of the time-evolution characteristics, the energy potential, dipole moments and CD/UV-Vis-near-infrared (NIR) spectra were simulated upon altering the dihedral angle sets of several model oligomers of PF8DBT using density functional theory (DFT)/time-dependent DFT (TD-DFT) (B3LYP/6-31G(d,p)). An additional Zerner's Intermediate Neglect of Differential Overlap (ZINDO) calculation qualitatively established that the two hypothetical, chirally dipole–dipole-cancelled, π–π-stacked, slipped dimers cause red shifts in the CD and UV-Vis-NIR spectra, which were associated with an enhanced |gabs|, ≈1 × 10−3 at 715 nm and 0.5 × 10−3 at 660 nm, compared to the corresponding monomeric value (|gabs| ≈ 0.1 × 10−3 at 620 nm) obtained using TD-DFT.
1. Introduction
Time-dependent mirror symmetry breaking (TD-MSB) under the far-from-equilibrium, open-flow conditions of external chemical influences and physical sources has become a hotly debated topic in the fields of chirality and chiral materials.1–4 Optically active scaffolds efficiently build-up and maintain helically assorted architectures when optically inactive organic, polymeric and inorganic resources are used as building blocks.4 One of the most sophisticated chiral organisations in natural-occurring systems is the light-harvesting system of helically organised, multiple loops in purple bacteria, which are reinforced via helical α- and β-polypeptides.5Collagens, DNA, polysaccharides, synthetic helical polymers and chiral molecules are candidates for helix-inducible scaffolds and templates.6–8 These scaffolds and templates exhibit chiroptical and other elaborate functions when circularly polarised luminescent (CPL)-silent and circular dichroism (CD)-silent substances are used as the source materials. These systems can be designed based on the transition of the non-restricted, rotational freedom of C–X (X = C, O, N, S) single bonds to a restricted molecular motion upon complexation.6,9 Note that a scaffold is a temporary platform used to construct a building and is removable after the architecture is complete;10 whereas, a template is a long-lived platform used to make copies with other materials.11
To obtain scaffolding capabilities, non-covalent intermolecular attractive interactions, including C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/HN, YH/π (Y = O, N), cation/π, CH/X (X = F, O, N, H, C), π/π, C–F/Si, electrostatic and solvophobic interactions,12 and London dispersion and Casimir forces13 help organise and disorganise the architecture. If scaffolding forces are too strong chemically and thermally, the scaffold instead acts as a template.
O/HN, YH/π (Y = O, N), cation/π, CH/X (X = F, O, N, H, C), π/π, C–F/Si, electrostatic and solvophobic interactions,12 and London dispersion and Casimir forces13 help organise and disorganise the architecture. If scaffolding forces are too strong chemically and thermally, the scaffold instead acts as a template.
For example, chiral organic gelators generate helical inorganic motifs via a sol–gel process with alkoxysilanes, and the organic moieties are removable at high temperatures.8b–d Enantiopure amines and amino alcohols cause CD-silent π-conjugated polymers to become CD-active helical polymers via acid–base interactions; whereas, the enantiopure sources can be replaced with other achiral molecules.8a
Aggregation-induced CPL substances containing luminogens14 and inherent luminophores15 can efficiently generate CPL- and CD-active aggregates and chiral nano-structures. L-/D-Glutamides, as chiral gelators, generate CPL- and CD-active polyfluorene derivatives, and the glutamide can be removed by adding MeOH, which breaks the organisation.16 Non-charged, helical polysaccharides transform CPL-/CD-silent cationic polythiophenes and anionic/non-charged polyfluorenes into their corresponding CPL-/CD-active compounds.7c–e,hD-/L-Methionine on a quartz surface leads to helically grown CPL-/CD-active aggregates of ZnO nanoparticles.17 These polysaccharides and methionines are chiral templates.
Recently, we showed that the use of non-charged poly(n-hexyl-(S)-2-methylbutylsilane) (PSi-S) and poly(n-hexyl-(R)-2-methyl-butylsilane) (PSi-R) as photoscissorable helical scaffolds (Fig. 1) generates CPL-/CD-active poly(di-n-octylfluorene) (PF8).18 To resolve the multiple non-covalent interactions between PSi and PF8, irradiation via unpolarised light at 313 nm was used to achieve the production of CPL-/CD-active PF8 homo-aggregates. To further verify the scaffolding capability of PSi-S and PSi-R, we tested the non-charged, non-helical poly{[9,9-di-n-octyl-fluorene]-alt-[4,7-bis(thiophen-2-yl)-2,1,3-benzothia-diazole]} (PF8DBT). PF8DBT and its derivatives are low-band-gap (LBG) polymers19 that emit in the red-to-far-red (FR) region (Fig. 1).20 Herein, we report the TD-MSB characteristics of the CPL-/CD-active PF8DBT hetero-aggregates with PSi-S and PSi-R under optimised conditions. The hetero-aggregates reached gem ≈ −0.019 at 706 nm with PSi-S and ≈+0.017 with PSi-R at 695 nm in 24 h when the generated aggregate was allowed to stand at room temperature. The CPL-/CD-amplitudes of the PF8DBT aggregates were maintained after photoscissoring with PSi-S and PSi-R at 313 nm.
 | ||
| Fig. 1 Chemical structures of PF8DBT, PSi-S and PSi-R used in the experiments and DBT and FDBTF used in the theoretical study. | ||
2. Experimental section
2.1 Materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400, a weight-average molecular, (Mw) = 24900, and a polydispersity index, PDI = Mw/Mn = 1.62. The Mn values corresponded to an average repeating unit of 22.4 and an extended polymer length of ≈43 nm.
:1 (v/v) (refractive index [nD] of 1.405), and the total volume of mixed toluene and MeOH was fixed at 3.0 mL. The desired molar ratio of the polymers dissolved in chloroform was tuned according to the experimental requirements. The dissolved PF8DBT was added to the cuvette and followed by the rapid addition of fractionated PSi-S (Mn = 20400, Mw = 26900, PDI = 1.31, polymer length for an ideal rod ≈ 16 nm) or PSi-R (Mn = 23900, Mw = 39200, PDI = 1.64, polymer length for an ideal rod ≈ 19 nm) to produce a well-mixed homogeneous solution. Subsequently, MeOH was slowly added to produce the hetero-aggregates. The resulting aggregate-containing solvents were transferred to an SQ cuvette with a path length of 5 mm to simultaneously measure the CD/UV-Vis and CPL/photoluminescence (PL) spectra as a function of the ageing time.
400, a weight-average molecular, (Mw) = 24900, and a polydispersity index, PDI = Mw/Mn = 1.62. The Mn values corresponded to an average repeating unit of 22.4 and an extended polymer length of ≈43 nm.
:1 (v/v) (refractive index [nD] of 1.405), and the total volume of mixed toluene and MeOH was fixed at 3.0 mL. The desired molar ratio of the polymers dissolved in chloroform was tuned according to the experimental requirements. The dissolved PF8DBT was added to the cuvette and followed by the rapid addition of fractionated PSi-S (Mn = 20400, Mw = 26900, PDI = 1.31, polymer length for an ideal rod ≈ 16 nm) or PSi-R (Mn = 23900, Mw = 39200, PDI = 1.64, polymer length for an ideal rod ≈ 19 nm) to produce a well-mixed homogeneous solution. Subsequently, MeOH was slowly added to produce the hetero-aggregates. The resulting aggregate-containing solvents were transferred to an SQ cuvette with a path length of 5 mm to simultaneously measure the CD/UV-Vis and CPL/photoluminescence (PL) spectra as a function of the ageing time.
2.2 Characterisation
980]/absorbance at the CD extremum of the hetero-aggregates. The absorbance often increased because of the Rayleigh scattering of the aggregates in the heterogeneous suspension; all CD, PL and CPL signals were insensitive to the aggregate scattering. An unfavourable increment in the apparent absorbance that resulted from the scattering was subtracted from the scattering term to obtain the plausible absorbance of the aggregate sample. The degree of circular polarisation in the photoexcited state was defined as: gem = 2 × (IL − IR)/(IL + IR), at the CPL extremum of the aggregates, where IL and IR are the photomultiplier signals for the left- and right-circularly polarised emissions under the excitation of monochromated unpolarised light, respectively.
2.3 Instrumentation
The CD/UV-Vis spectra of the aggregates were simultaneously obtained using JASCO (Hachioji-Tokyo, Japan) J-820/J-725 spectropolarimeters with Peltier-controlled equipment to control the sample temperature. The scanning rate was 100 nm min−1, the bandwidth was 2 nm, and the response time was 2 s at 20 °C. We used a cuvette with a path length of 5 mm to satisfy the requirements of a maximum absorbance less than 2.0 and a maximum ellipticity of ±2000 mdeg, which were specified by the instrument manufacturer, with the exception of several preliminary evaluations of the as-prepared aggregate samples in which a cuvette with a 10 mm path length was used. The CPL/PL spectra were simultaneously recorded at room temperature (23–25 °C) on a JASCO CPL-200 spectrofluoropolarimeter with focusing lenses in the absence of Peltier-controlled equipment. A path length of 5 mm, scanning rate of 50 nm min−1, excitation at 420 nm, excitation bandwidth of 1200 μm, monitoring bandwidth of 1500 μm, photomultiplier tube (PMT) response time of 4 s and single accumulation were applied for all measurements.The values of Mw, Mn and PDI were evaluated using a GPC (Shimadzu [Kyoto, Japan] A10 chromatograph) with a PL Gel mixed B column (25 cm in length, 4.6 mm ID, Varian-Agilent) and special-grade tetrahydrofuran (Wako Chemical [Osaka, Japan]) as an eluent at 40 °C based on the calibration with polystyrene standards (Varian-Agilent).
The time-dependent hydrodynamic diameters of the hetero-aggregates were analysed using an Otsuka Electronics (Hirakata-Osaka, Japan) DLS-6000 dynamic light scattering (DLS) apparatus with a detector angle of 90°, and 30 accumulations using the solution viscosity data obtained with a Sekonic (Tokyo, Japan) VM-100 viscometer at 25 °C and the nD value of MeOH-toluene (1:1 (v/v)) at 589 nm at 25 °C using an Atago (Tokyo, Japan) thermo-controlled DR-M2 refractometer at 589 nm (Tokyo, Japan) at 20 °C.
Several computer-generated rotamers of DBT and FDBTF were optimised using PM3MM (universal force field [UFF], Gaussian09 rev. D.01, Gaussian, Inc., Wallingford CT, 2013).22 These DBT and FDBTF models were further optimised using a density functional theory (DFT) program with B3LYP and the 6-31G(d,p) basis set as a function of dihedral angle sets and followed by time-dependent (TD)-DFT calculations to obtain the UV-Vis and CD spectra (10 singlet transitions) with a full-width-at-half-maximum (FWHM) of 0.1 eV. The twisted, slipped π–π stack dimer models of FDBTF were optimised using the PM3MM program and followed by Zerner's Intermediate Neglect of Differential Overlap (ZINDO) calculations to obtain the UV-Vis and CD spectra (20 singlet transitions) with a FWHM of 0.05 eV. These simulated UV-Vis and CD spectra were saved as text data sets and re-plotted using KaleidaGraph ver. 4.5 (Synergy Software, Reading, PA 19606, USA). An Apple PowerMac (2.67 GHz clock, 8-core and 32 GB memory) was used to perform the calculations.
To measure the PL lifetime of PF8DBT in chloroform and the suspension states, we performed time-correlated single-photon counting (TCSPC). The second harmonic (SHG = 400 nm) of a tuneable Ti:sapphire laser (Mira 900, Coherent) with an ≈150 fs pulse width and 76 MHz repetition rate was used as an excitation source. The PL emission intensities at 680 nm (aggregates) and 580 nm (non-aggregates) were spectrally resolved using a collection of optics and a monochromator (Acton SP-2150i, Princeton Instrument [Trenton, NJ, USA]). A TCSPC module (PicoHarp, PicoQuant [Berlin, Germany]) with a microchannel plate (MCP)-PMT (C4780, Hamamatsu Photonics [Hamamatsu, Japan]) was used for ultrafast detection. The total instrument response function (IRF) for the PL decay was less than 100 ps, which provided a temporal resolution of less than 10 ps. The deconvolution of the actual PL decay and IRF was performed using fitting software (FlouFit, PicoQuant) to deduce the time constant associated with each exponential decay.
The quantum yield of PL (ΦPL) was evaluated using a rigid 5,5′′′-bis(N,N-diphenylamino)-4′-dimesitylboryl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene as a secondary reference for a green-red emitter (ΦPL 0.38 in CHCl3).23
To photochemically degrade PSi-S (and PSi-R) in the PSi-PF8DBT hetero-aggregate as a suspension in a cuvette at room temperature, an ultra-high-pressure 500 W Hg lamp (Optiplex BA-H501 and USH-500SC2 Ushio [Tokyo, Japan]) with a narrow band-pass filter (FWHM: 10 nm) of 313 nm (Asahi Spectra [Tokyo, Japan]) was used. The photon flux at 313 nm was 3.65 μW cm−2 for 60 s determined using an Si photodetector (Nova PD300-UV, Ophir-Japan [Tokyo, Japan]). The detailed experimental set-up was reported previously.4d,18
3. Results and discussion
3.1 Generation, amplification and retention from optically active PF8DBT endowed with PSi-S and-R
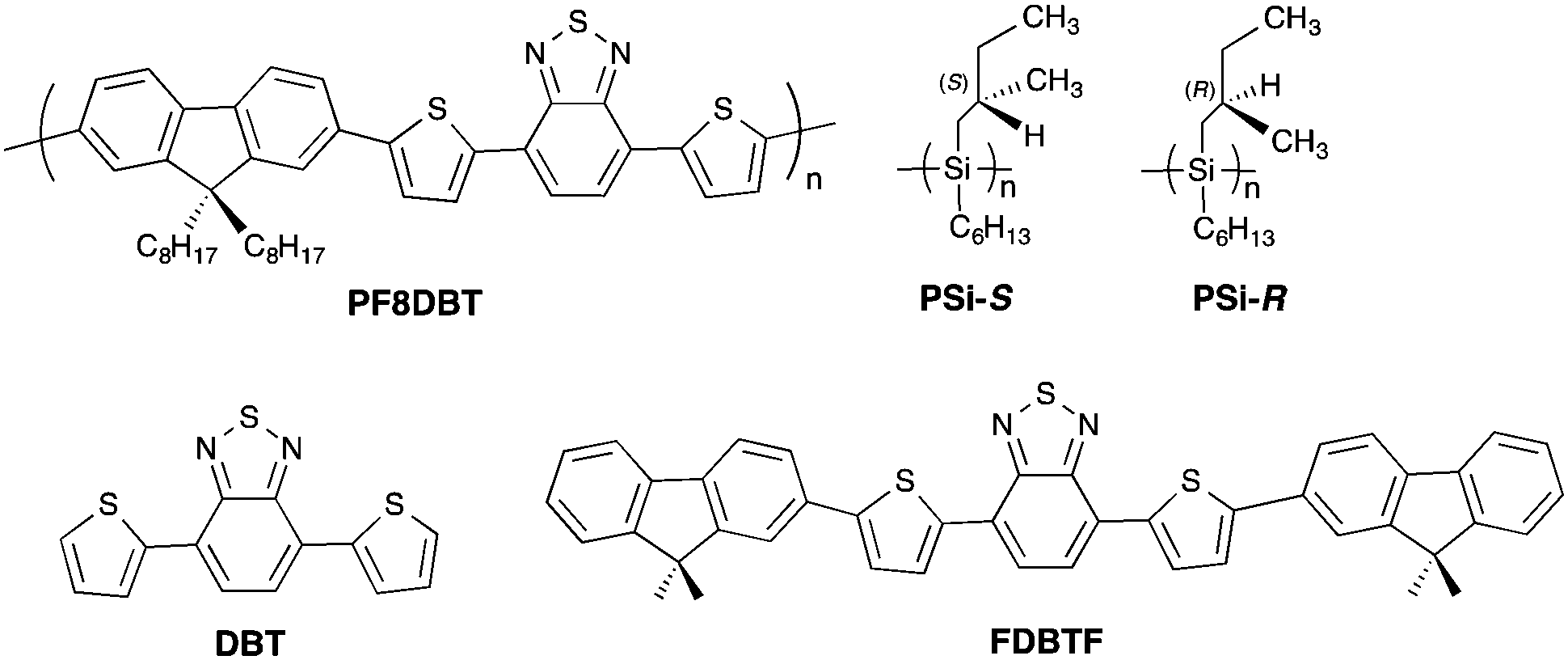
Fig. 2a and c show the CD/UV-Vis-NIR spectra arising from hetero-aggregates of PF8DBT-and-PSi-S and PF8DBT-and-PSi-R, respectively. For comparison, the CD/UV-Vis-NIR spectra of PF8DBT in dilute CHCl3 at 20 °C and its homo-aggregate dispersed in chloroform-MeOH (2/1 (v/v)) at 20 °C are given in the ESI† (Fig. S1 and S2).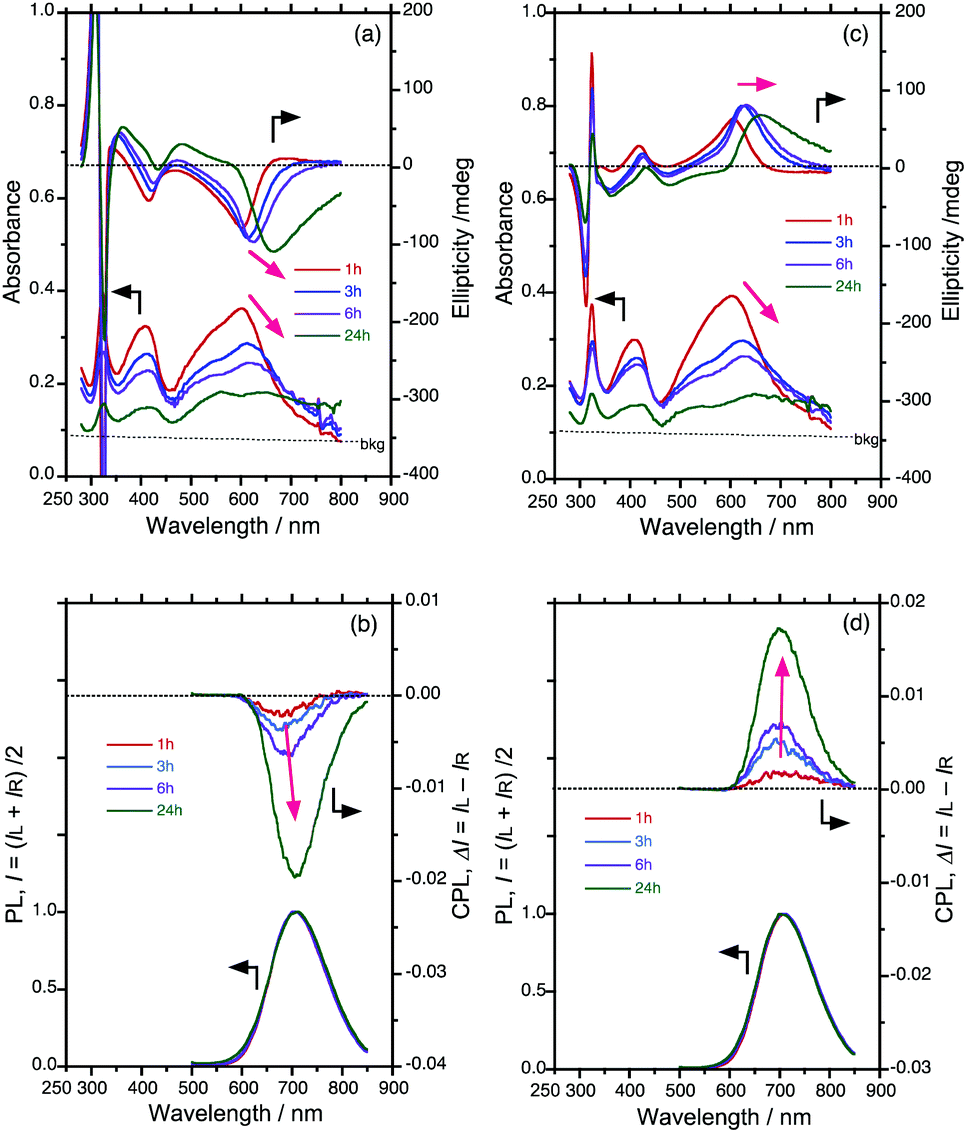 | ||
| Fig. 2 Changes in the (a) CD and UV-Vis-NIR spectra and (b) CPL and PL spectra excited at 400 nm for the PSi-S-and-PF8DBT hetero-aggregates (1-to-1 molar ratio). Changes in (c) the CD and UV-Vis-NIR spectra and (d) the CPL and PL spectra excited at 400 nm for the PSi-R-and-PF8DBT hetero-aggregates (1-to-1 molar ratio). These aggregates were produced in chloroform-MeOH (2/1 (v/v)). [PF8DBT] = 2.5 × 10−5 M, [PSi-S] = 2.5 × 10−5 M and [PSi-R] = 2.5 × 10−5 M. | ||
The intense bisignate CD bands at 305 and 322 nm, which are associated with a narrow UV band at 320 nm, are attributed to the exciton couplet of the Siσ–Siσ* transition arising from helically assorted PSi-S and PSi-R aggregates, respectively.15c,18 The intense negative and positive CD bands associated with two major Vis-NIR bands at ≈400 nm and ≈600 nm are attributable to the S0–S2 and S0–S1 π–π* transitions of the PF8DBT main chain, respectively.20 Evidently, the two λmax values at 400 nm and 600 nm frequently red shift, and their UV-Vis intensities weaken as the ageing time increases (1, 3, 6 and 24 h). In contrast, the 320 nm band of PSi remained nearly constant with the ageing time.
For comparison, PF8DBT in the absence and presence of PSi-S and PSi-R in a homogeneous CHCl3 solution and PF8DBT homo-aggregates in a CHCl3–MeOH cosolvent (2:1, v/v) did not exhibit any CD signals in the range of 250–800 nm because PF8DBT does not exhibit a helical preference in a fluidic solution and the homo-aggregates form an isotropic suspension state.
These time-evolved CD/UV-Vis-NIR characteristics are synchronised with the corresponding CPL/PL characteristics. Fig. 2b and d show the CPL/PL spectra of the PF8DBT-and-PSi-S and PF8DBT-and-PSi-R hetero-aggregates excited at 400 nm, respectively. Evidently, the initial CPL-/PL-bands at approximately 680–700 nm, arising from the S0–S1 band of the PF8DBT-and-PSi-S and PF8DBT-and-PSi-R hetero-aggregates, progressively increased with the ageing time. Nearly 24 h was required to boost the CD/CPL amplitudes and reach constant CD/CPL magnitudes. Although significant time-evolved alterations on the order of several hours were reported in several molecular aggregates and molecular stacks, which was confirmed by CD and Raman optical activity (ROA), the marked time-evolved changes in the CPL and CD characteristics may be the first examples among the aggregation-induced chiroptical studies reported so far.
Fig. 3 summarises the alterations of all the gabs, gem, λmax and λem values of the S0–S1 Cotton bands at approximately 600–700 nm for the four ageing times tested. Here, gabs and gem are dimensionless chiroptical parameters used to quantitatively discuss the degrees of molecular and macromolecular chirality in the ground (S0) and photoexcited (S1) states.21 The initially very weak gem amplitudes of the as-prepared aggregates increased to ≈−0.019 at 706 nm (with PSi-S) and ≈+0.017 at 695 nm (with PSi-R) after ageing under ambient temperature for 24 h. Similarly, the initially weak gabs values obtained after ageing for 1 h increased to ≈−0.033 at 666 nm (with PSi-S) and ≈+0.033 at 659 nm (with PSi-R) after 24 h. However, the interpretation of these gabs values is somewhat questionable because of an unavoidable baseline increment in the absorbance, which is a result of scattering from the aggregates.
 | ||
| Fig. 3 Time-dependent gabs-λext and gem-λem relationships of the PSi-S-and-PF8DBT and PSi-R-and-PF8DBT hetero-aggregates (1-to-1 ratio) in the CHCl3–MeOH cosolvent (2/1 (v/v)). All data were taken from Fig. 2. | ||
The CPL/PL transitions occur from the lowest S1-state with ν = 0′ to the S0-state with ν = 0, 1, 2… according to Kasha's rule.24 The Stokes shift in the hetero-aggregates after ageing for 24 h was estimated from the CD and CPL spectra and was as small as 960 cm−1 (with PSi-S) and 860 cm−1 (with PSi-R) (Fig. 3). This small Stokes shift results in the highly emissive state in the hetero-aggregates, ΦPL = 22% and ⋯ΦPL ≈ 49% for PF8DBT in the CHCl3 solution. It should be mentioned that vibronic 0–0′ and 0–1′ bands with a spacing of ≈2000 cm−1 can be seen in the hetero-aggregates near 500 nm and 700 nm, indicating highly restricted rotational freedom. In contrast, the vibronic bands in the CHCl3 solution were not obvious because of the non-restricted rotational freedom (ESI,† Fig. S1).
3.2 Photochemical removal of the PSi-S and PSi-R scaffolds
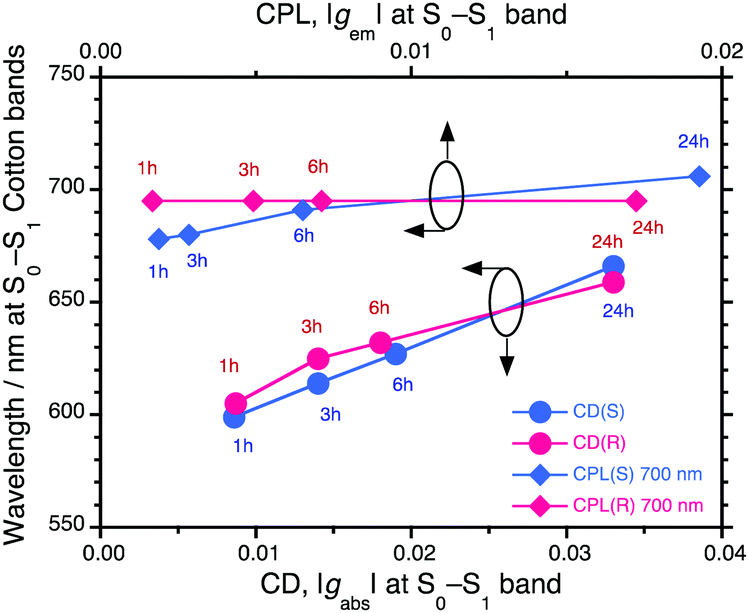
To chiroptically elucidate the helix scaffolding capability of PSi-S and PSi-R towards PF8DBT, we performed an Si–Si bond selective photoscissoring experiment at 313 nm on the PF8DBT-and-PSi aggregates after ageing for 24 h.Fig. 4a and b show the CD/UV-Vis-NIR and CPL/PL spectra (excited at 400 nm) of the photoscissored PF8DBT-and-PSi-S aggregates, respectively, and Fig. 4c and d display the same for the photoscissored PF8DBT-and-PSi-R aggregates, respectively. Evidently, for both PF8DBT-and-PSi-S and PF8DBT-and-PSi-R, all the CD/UV-Vis-NIR and CPL/PL spectral characteristics associated with the gem values are maintained, even after the photoscissoring reactions, except for the complete disappearance of the Siσ–Siσ* bands. The gem values were −0.022 at 703 nm for PSi-S and +0.015 at 698 nm for PSi-R, and they were nearly unchanged after the removal of the photoscissible helical Si–Si bonds.
 | ||
| Fig. 4 (a) CD/UV-Vis-NIR and (b) CPL/PL spectra of PF8DBT-and-PSi-S hetero-aggregates excited at 400 nm after ageing for 24 h, followed by photoscissoring at 313 nm. (c) CD/UV-Vis-NIR spectra and (d) CPL/PL spectra excited at 400 nm of PF8DBT-and-PSi-R hetero-aggregates after ageing for 24 h, followed by 313 nm photoscissoring. (e and f) CD/CPL spectra as a function of the photon energy (in eV) of the photoscissored PF8DBT-and-PSi-S and PF8DBT-and-PSi-R hetero-aggregates, respectively. These data were taken from (a–d). | ||
Fig. 4e and f plot the CPL and CD spectral amplitudes as a function of the photon energy in electron volts; the data presented in these figures were taken from the scattering free CPL and CD spectra in Fig. 4a–d. Eventually, when the PF8DBT endowed with PSi-S or PSi-R was used as an achiral building block, we obtained a pair of chiral PF8DBT semiconducting emitters with optical band gaps (Eg,opt) of 1.76 eV and 1.78 eV and small Stokes shifts of 0.11 eV (880 cm−1) and 0.14 eV (1130 cm−1), respectively. These small Stokes shifts indicate that minimal structural reorganisation of the chiral PF8DBT aggregates occurs in the S1 state because of the limited rotational freedom.
3.3 Time evolution of the hetero-aggregate size
To determine the origin of the time-dependent gem and gabs amplitudes associated with the marked red shift in the corresponding S0–S1 Cotton bands in the photoexcited and ground states, we measured the hydrodynamic diameters of the aggregates as a function of the ageing time, followed by a photoscissoring reaction at 313 nm of the 24 h aged samples.Fig. 5a and b show the size distribution curves of the PF8DBT-and-PSi-S (1-to-1) and PF8DBT-and-PSi-R (1-to-1) aggregates and the photoscissored aggregates. Although a mixture of PF8DBT and PSi-S (1-to-1) initially forms a ca. 390 nm hetero-aggregate (0 h), the size gradually increased to 430 nm (3 h) and 1810 nm (24 h). The photoscissoring reaction of the 24 h sample led to a reduced aggregate volume (≈790 nm or (790/1810)3 = 0.08) because the PSi-S was removed from the hetero-aggregate. A similar tendency was observed for a mixture of PF8DBT and PSi-R (1-to-1). In this case, the sizes were 550 nm (0 h), 820 nm (1 h), 940 nm (3 h), and 2070 nm (24 h), and after the photoscissoring reaction of the 24 h sample, the values 1340 nm and 390 nm were obtained.
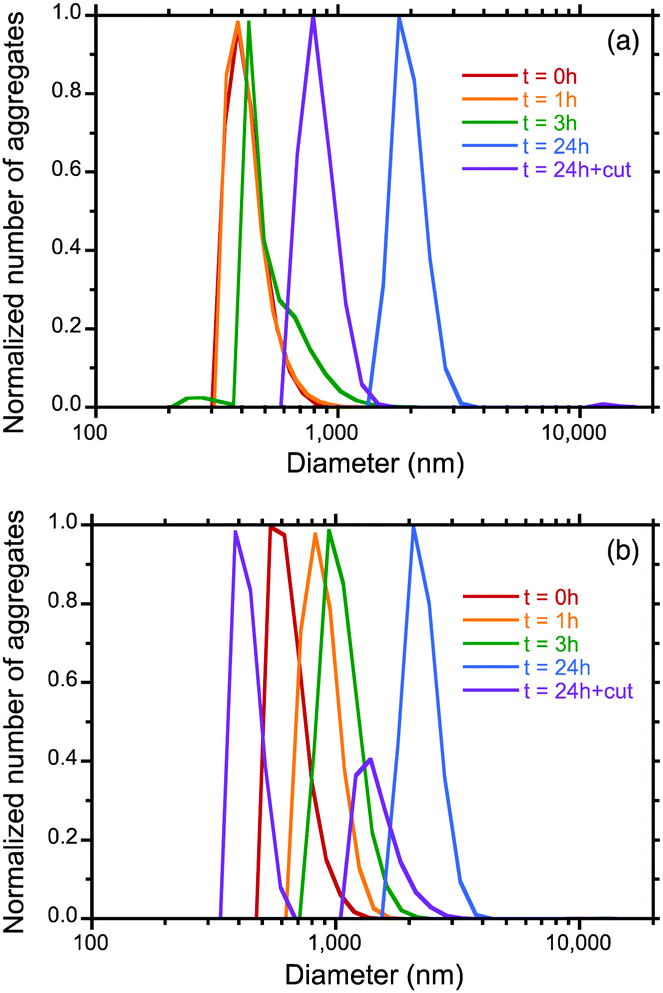 | ||
| Fig. 5 Time-dependent hydrodynamic diameters of (a) PSi-S-and-PF8DBT hetero-aggregates and (b) PSi-R-and-PF8DBT hetero-aggregates as a function of the ageing time (0 h, 1 h, 3 h, 24 h) and 24 h after irradiation at 313 nm (3.65 μW cm−2) for 60 s. | ||
The time-evolved growth of the aggregate size is accounted for by the phenomenon of Ostwald ripening. If smaller aggregates and larger aggregates co-exist in solution, the smaller aggregates re-deposit onto the larger aggregates. The larger aggregates become larger over time, and this is associated with the disappearance of the smaller aggregates. Similar increase in the aggregation size of non-charged polymers over time has been reported by us, but the reasons for the increases were unresolved at that time.4e,18
In contrast, the photoscissoring reaction of the larger aggregates in solution should promote the backward reaction of Ostwald ripening.25 The cleaved fragments of PSis help generate smaller aggregates (Fig. 5b). Some of the smaller aggregates more easily dissolve into the solution, which is followed by the disappearance of the smaller aggregates (Fig. 5a). Generally, low molecular weight species have better solubility than high molecular weight species. This concept is used in a fractional dissolution method to isolate a lower molecular weight polymer from a broader molecular weight polymer and in a lithography solvent-based rinsing process for fine pattering using a positive-type photoresist polymer deposited onto a solid surface.
These progressive increments in the aggregate size may be responsible for the time evolution of the gem and gabs amplitudes associated with the marked red shift in the S0–S1 transitions. A larger aggregate size can resist the thermal fluctuations and rotational freedom of the single bonds and Brownian motion. We assume that an aggregate larger than ≈1000 nm would exhibit larger gem and gabs amplitudes than smaller aggregates of 300–500 nm. Indeed, the larger aggregate should act as an efficient optical cavity to confine external and internal light according to a modern photonic concept, so-called optofluidics.26 We should note that in a hypothetical optical cavity with a refractive index of 2.0, the light wavelength of 700 nm under vacuum (and air) decreases by half to 350 nm, and the speed of light slows to 1.5 × 108 m s−1.
Fig. 6 compares four optical microscopic images of PF8DBT-and-PSi-S (1-to-1) hetero-aggregates associated with bright-field images under white-light illumination and their corresponding dark-field fluorescence images upon 510–550 nm excitation using an appropriate filter block set with an Hg-lamp light source. Bright- and dark-field fluorescence images (Fig. 6a and b) without ageing tend to exhibit many small red dots and 1–2 μm aggregates. In contrast, a sample aged for 24 h contains more large aggregates, and the smaller red dots nearly disappear. This tendency is consistent with the time-evolved aggregate size alteration observed via DLS measurements and is accountable for by the phenomenon of Ostwald ripening.
 | ||
| Fig. 6 Optical microscopic images of PF8DBT-and-PSi-S (1-to-1) hetero-aggregates without ageing and after ageing for 24 h. (a and c) Bright-field images under white-light illumination. (b and d) Dark-field fluorescence images upon 510–550 nm excitation. Hg-lamp light source filter block set: (G-2A): EX 510–550 nm, DM 575 nm, BA 590 nm. | ||
3.4 Time-evolved photodynamics and quantum yield
One question that remains relates to the alteration of the photodynamics and quantum yields of the time-evolved PF8DBT-and-PSi-S and PF8DBT-and-PSi-R aggregates. Fig. 7 compares three PL decay curves: one monitored at 580 nm for PF8DBT in a homogeneous CHCl3 solution and two monitored at 680 nm for a PF8DBT-and-PSi-S hetero-aggregate (1:1) obtained with ageing times of 30 min and 24 h in CHCl3/MeOH (2/1 (v/v)).
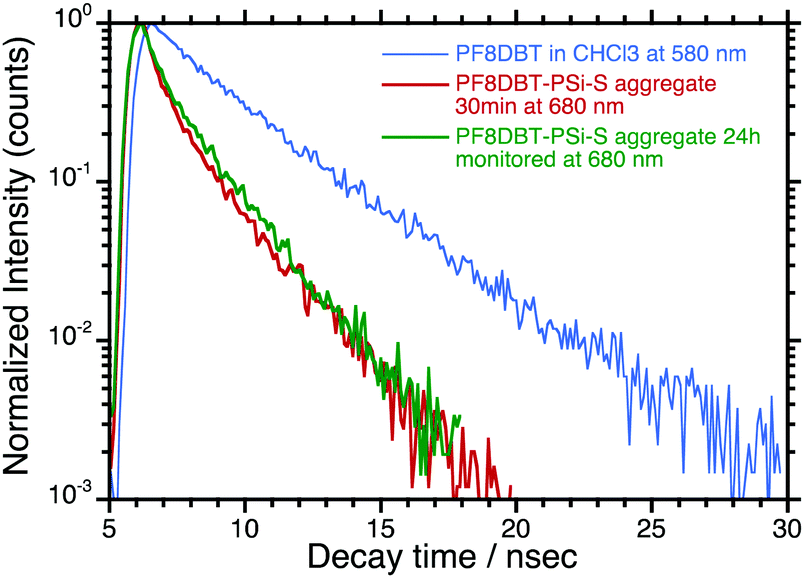 | ||
| Fig. 7 (a) PL decay curve at 580 nm of PF8DBT in a CHCl3 solution and, for comparison, (b) PL decay curves at 680 nm of the aged PF8DBT/PSi-S hetero-aggregates (1:1) (30 min and 24 h) in CHCl3/MeOH (2/1 (v/v)). | ||
The photoexcited PF8DBT in the CHCl3 solution decayed exponentially into two longer-lived radiative species with τ1/2(1) ≈ 2.34 ns (49%) and τ1/2(2) ≈ 4.01 ns (51%) with χ = 1.236. In contrast, the photoexcited PF8DBT-and-PSi-S aggregates subjected to 30 min of ageing in a CHCl3–MeOH (2:1) solvent had two shorter-lived species with τ1/2 ≈ 0.64 ns (75%) and 2.23 ns (25%) with χ = 2.81, whereas the photoexcited PF8DBT-and-PSi-S aggregates subjected to 24 h of ageing decayed with τ1/2 ≈ 0.54 ns (64%) and 2.10 ns (36%) with an accuracy χ = 2.57. A population of two species (1 and 2) was slightly altered upon aggregation but it was only weakly dependent on the ageing time. Since τ1/2 = 1/(kr + knr) (kr and knr are the radiative and non-radiative rate constants, respectively) and ΦPL = kr/(kr + knr) ≈ τ1/2, species 1 (2.34 ns) and 2 (4.01 ns) become two shorter-lived species 1 (0.54–0.64 ns) and 2 (2.10–2.23 ns) in the aggregates. Furthermore, because the ΦPL and τ1/2 values of the aggregate are nearly half those in CHCl3, we assumed that the non-radiative component (knr) of the aggregate increases.
3.5 The optofluidic effect26,27
The cosolvent was optimised by optofluidically tuning the nD value and resonantly enhancing the gabs value of the hetero-aggregate at a very specific nD of ≈1.405 (Fig. 8a), where the ratio of good to poor solvent (CHCl3:MeOH) was 2 to 1 (v/v). This tendency is commonly observed in a series of CPL-/CD-active polymer aggregates existing in a heterogeneous suspension state.4d,e,15b,18,28 In terms of the optofluidics, an efficient confinement of circularly polarised light in a polymeric optical cavity with a high refractive index is realised by finely tuning the lower refractive index of the surrounding solvents.26–28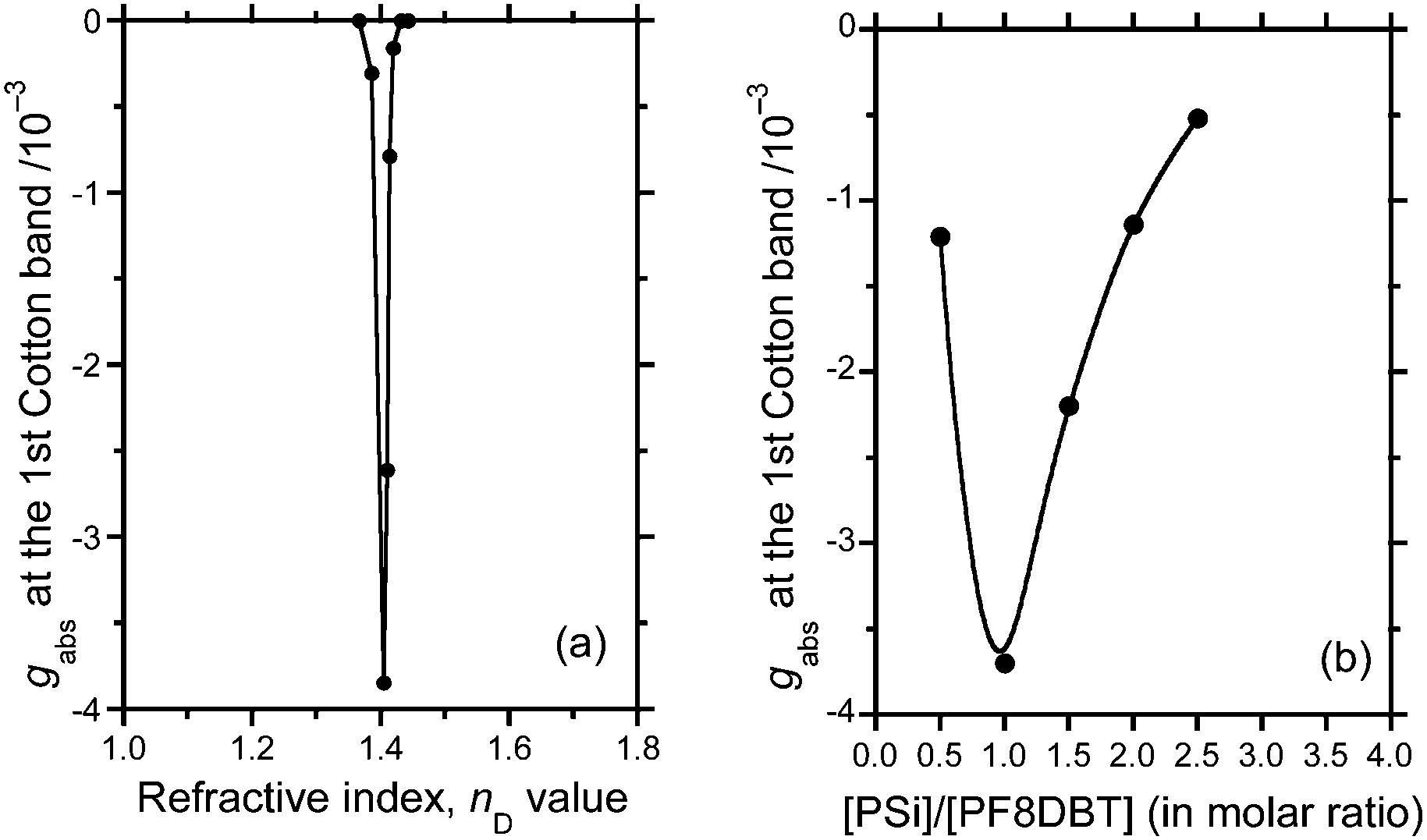 | ||
| Fig. 8 (a) gabs value at 580 nm of the PF8DBT-PSi-S hetero-aggregate (1:1) as a function of the average refractive index (nD) of the CHCl3–MeOH cosolvent. (b) gabs value of the PF8DBT-and-PSi-S hetero-aggregates as a function of the PSi-S-to-PF8DBT ratio generated in the CHCl3–MeOH (2:1) cosolvent. [PF8DBT]0 = 6.6 × 10−6 mol L−1, path length 5 mm. It should be noted that the hetero-aggregates were formed under all conditions, as proven by an increment of the baseline signal of the UV-Vis absorption due to the slight turbidity of the sample that can be recognised by the naked eye. | ||
Alternatively, although we tested several combinations of good and poor solvents in a 2 to 1 ratio (e.g., CHCl3 and limonene, CHCl3 and IPA, CHCl3 and diethyl ether, CHCl3 and n-hexane, tetrahydrofuran and MeOH, and toluene and MeOH), CHCl3 and MeOH in a 2 to 1 ratio resulted in the most intense gabs value (ESI,† Fig. S5–S11). Choosing good and poor cosolvents with a tuned refractive index is vital for the design of aggregation-induced enhancement in chiroptical responses.
3.6 PSi-to-PF8DBT molar ratio dependency
To obtain additional insight into whether the intermolecular interactions between PSi-S and PF8DBT are stoichiometric, the gabs value at the first Cotton band (≈600 nm) as a function of the PSi-S-to-PF8DBT ratio in the hetero-aggregates after 1 h of ageing is plotted in Fig. 8b. The PSi-S-and-PF8DBT hetero-aggregate prefers a 1-to-1 stoichiometry because the |gabs| value is maximised at [PSi-S]/[PF8DBT] = 1.0. The hetero-aggregates do not obey the sergeant-and-soldier scenario that is observed in the π–π-stacked supramolecules and stiff helical copolymers.29 Thus, finding the best macromolecular complexation ratio between PSi-S and PF8DBT is critical.3.7 Potential energy surface revealed via DFT calculations
To computationally discuss the origin of the TD-MSB characteristics as evident in the marked red shift in the CD and CPL associated with the enhanced gabs and gem magnitudes, we generated several hypothetical rotamers of DBT and FDBTF as molecular models of PF8DBT optimised using PM3MM and DFT (B3LYP, 6-31G(d,p) basis set) (Fig. 9 and 109,10). DBT and FDBTF are molecular models of PF8DBT.20f,30DBT has two rotational degrees of freedom between the benzodithiazole and the two thiophene rings,30 and FDBTF has two additional rotational degrees of freedom between the two fluorenes and the thiophene rings.20f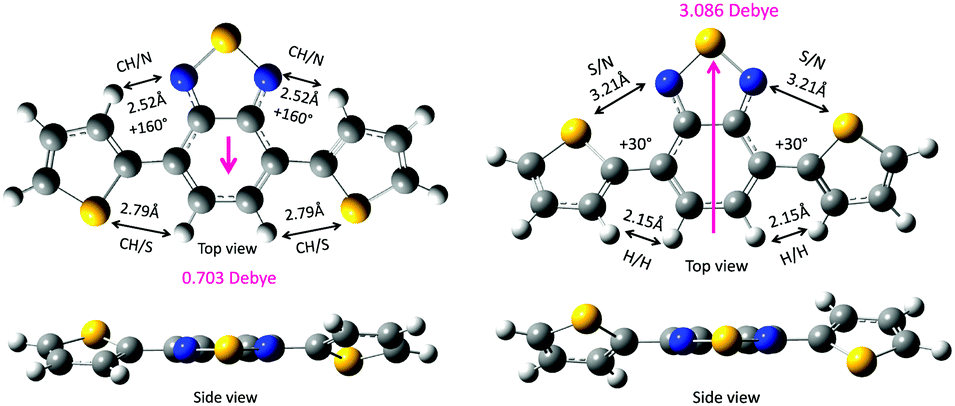 | ||
| Fig. 9 Two representative twist (non-coplanar) rotamers of DBT (left: anti–anti and right: syn–syn) optimised using DFT (B3LYP, 6-31G(d,p) basis set). | ||
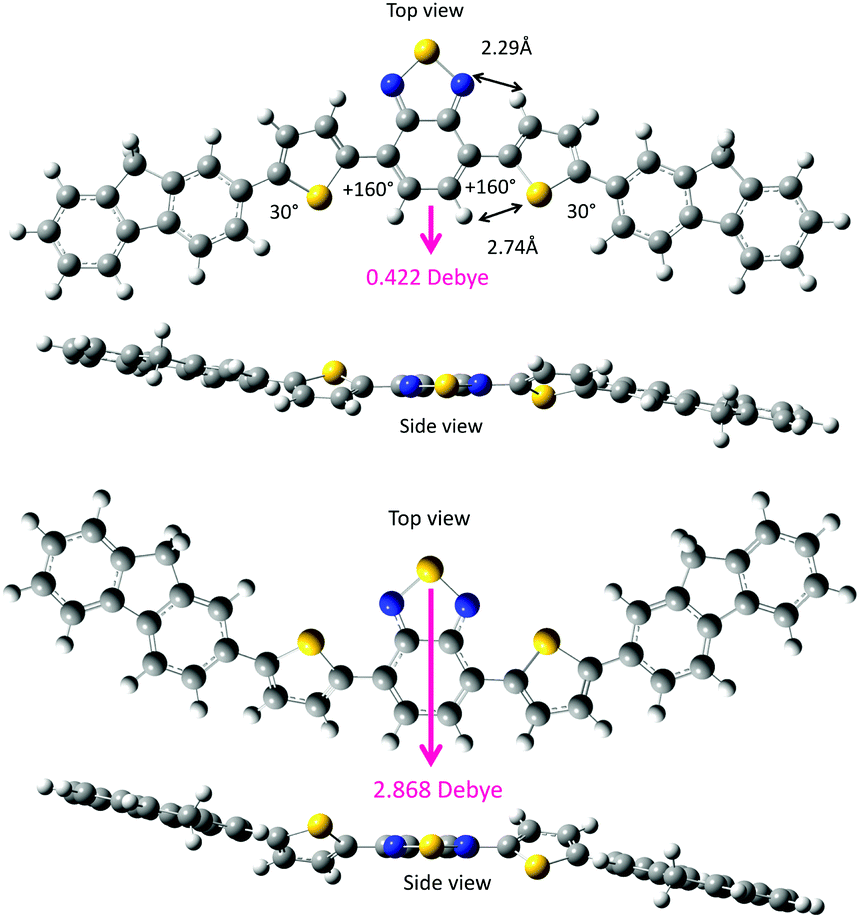 | ||
| Fig. 10 Two representative twist rotamers of the less-polar FDBTF (top: anti–anti (or 160°–160°) of DBT) and polar FDBTF (bottom: syn–syn (or 30°–30°) of DBT) that were optimised using DFT (B3LYP, 6-31G(d,p) basis set). | ||
First, the two DBT rotamers were generated in twist geometries at 10° intervals. For example, the anti–anti (160°–160°) form is stabilised via several weak intramolecular CH/N (2.52 Å) and CH/S (2.79 Å) interactions, and the syn–syn (30°–30°) form is stabilised via several weak intramolecular S/N (2.52 Å) and CH/HC (2.15 Å) interactions. According to the van der Waals radii compiled by Bondi,31 the van der Waals contacts are estimated to be CH/N (2.75 Å), CH/S (3.00 Å), S/N (3.35 Å) and CH/HC (2.40 Å). The distances that were theoretically optimised via the DFT calculations are considerably shorter than the corresponding Bondi van der Waals contact distances. This indicates the existence of multiple attractive intramolecular CH/N, CH/S, S/N and CH/HC interactions within DBT. The less-polar anti–anti form (0.703 Debye) is slightly more stable (by 0.075 kcal mol−1) than the polar syn–syn form (3.086 Debye).
Recently, McCulloch et al. revealed that in a vacuum, the anti–anti form with a small dipole moment (0.14 Debye) is more stable than the syn–synDBT with a large dipole moment (2.21 Debye).30 However, a disordered crystal of DBT consists of 71% of a relatively planarised trans–syn form (dihedral angles: 4.26° and 2.03°) and 29% of a syn–syn form because of the dipole–dipole cancellation.30 In the condensed phases, such as the solvent-free crystal and aggregates surrounded by solvent molecules, an intermolecular dipole–dipole cancellation, rather than an intramolecular dipole–dipole cancellation, may be crucial beyond a theoretical prediction of the molecular rotamers in a vacuum.
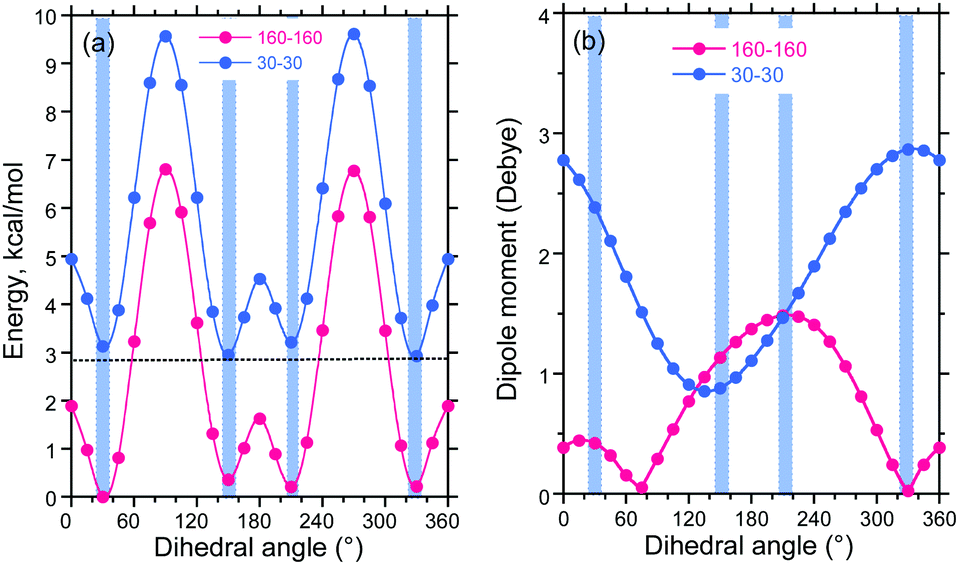
Based on our knowledge and understanding of DBT, we generated syn–syn and anti–antiFDBTF rotamers with two fluorenes as molecular models of the PF8DBT high polymer (Fig. 10). Here, two sets of the dihedral angles between the fluorene and thiophene ring altered the disrotatory with 15° intervals. Fig. 11a and b depict the potential energy (in kcal mol−1) and permanent dipole moments (in Debye) in a vacuum as a function of the dihedral angles between the fluorene and thiophene rings.
Fig. 11a shows that the syn–syn and anti–antiFDBTF rotamers commonly have four energy minima at dihedral angles of +30°, +150°, +210° (−150°), and +330° (−30°), but the four syn–syn rotamers are destabilised by ≈2.9–3.1 kcal mol−1 relative to the corresponding anti–anti rotamers. Among the four anti–antiFDBTF rotamers, global minima exist at dihedral angles of +30°, but the energy difference among the four minima is as small as 0.36 kcal mol−1. The barrier heights among these local minima are quite small (e.g., 1.8 and 6.8 kcal mol−1). Conversely, among the four syn–synFDBTF rotamers, global minima exist at dihedral angles of +330°, but the energy difference among the four minima can be as small as 0.28 kcal mol−1. The barrier heights among these local minima are as small as ≈2 and ≈7 kcal mol−1.
 | ||
| Fig. 11 (a) Potential energy and dipole moments of two representative FDBTF sets as a function of the dihedral angle between the fluorene and thiophene rings. Here, 160–160 stands for the anti–anti twist FDBTF rings, and 30–30 refers to the syn–syn twist FDBTF rings. | ||
Based on the eight energy minima of the anti–anti and syn–synFDBTF rotamers for the relative population in a vacuum at 0 K, the following estimations were obtained based on the Boltzmann distribution using a global minimum of the 30°–30° anti–antiFDBTF rotamers. In the anti–antiFDBTF rotamers, the dihedral angles are (30°–30°) 34.9%, (150°–150°) 18.8%, (330°–330°) 22.8% and (210°–210°) 21.6%, and in the syn–syn rotamers, the dihedral angles are (30°–30°) 0.16%, (150°–150°) 0.22%, (330°–330°) 0.14% and (210°–210°) 0.23%. These estimations led us to conclude that in a fluidic solution at ambient temperature, all anti–antiFDBTF rotamers exist equally and that all syn–synFDBTF rotamers coexist in small proportions. For the PF8DBT high polymer, most of the possible rotational isomeric structures (RISs) statistically coexist in the same main chain. This is the inherent nature of floppy chain-like polymers. The population of these RISs may be somewhat altered by the polarity of the surrounding solvent.
Fig. 11b shows that the anti–anti rotamers have nearly null dipole moments at the two local minima of +75° (0.054 Debye) and 330° (0.025 Debye), which is similar to the syn–syn rotamer at a local (global) minimum of +135° (0.855 Debye). Polar solvents and poorer cosolvents, including polar solvent molecules such as MeOH, may minimise the large dipole moment of PF8DBT in solution. Given that the dipole moments are perpendicular to the main chain axis, the dipole moments being aligned in the same direction reveals the ferroelectricity, the moments being aligned in an alternating pattern shows anti-ferroelectricity, and the moments being aligned randomly shows paraelectricity.
The CD and UV-Vis-NIR spectra (20 singlet transition states) of the polar syn–synFDBTF rotamer (dihedral angles 210°–30°–30°–210°) and less-polar anti–antiFDBTF rotamer (dihedral angles 30°–160°–160°–30°) simulated with TD-DFT (B3LYP, 6-31G(d,p), FWHM: 0.1 eV) are shown in Fig. 12. The polar syn–syn rotamer has gabs ≈ +1.4 × 10−4 at 618 nm, whereas the less-polar anti–anti rotamer has gabs ≈ −1.1 × 10−4 at 636 nm.
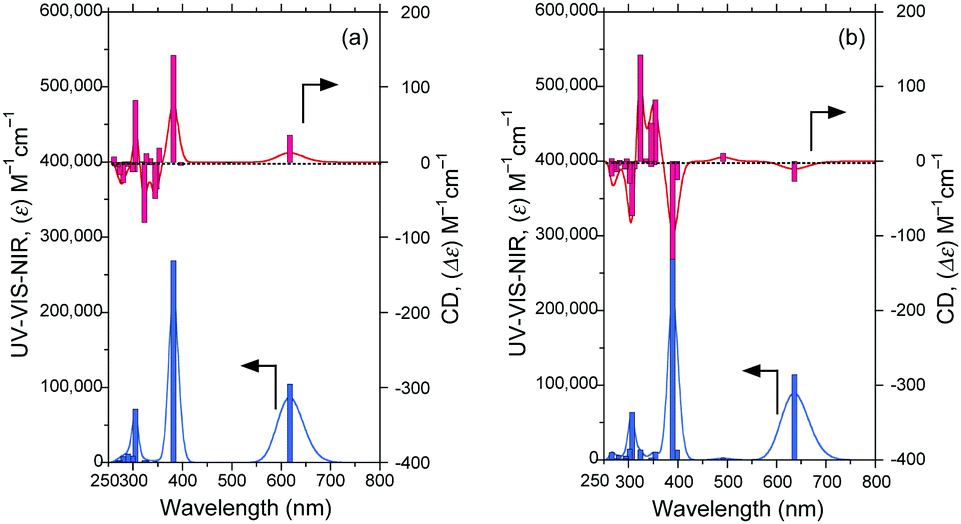 | ||
| Fig. 12 CD and UV-Vis-NIR spectra (20 singlet transition states) of (a) the polar syn–synFDBTF rotamer (dihedral angles 210°–30°–30°–210°) and (b) the less-polar anti–antiFDBTF rotamer (dihedral angles 30°–160°–160°–30°) simulated using TD-DFT (B3LYP, 6-31G(d,p), FWHM: 0.1 eV). Structures were taken from Fig. 10. | ||
During the course of the aggregation associated with the desolvation process from the aggregates, the inter-macromolecular dipole–dipole cancellations between the PF8DBT chains should substitute for the intermolecular dipole–dipole cancellation between PF8DBT and the solvent molecules. This process requires substantial energy and a prolonged annealing time for the ill-organised metastable structures in the aggregates to become the energetically most-stable state by overcoming the many energy potential barriers among the metastable structures. In this work, the spontaneous annealing process occurring at ambient temperature is detected because the time evolution of the chiroptical data is on the order of hours.
3.8 Simulated CD/UV-Vis-NIR spectra of the slip π–π stack FDBTF dimers
For (chir)optical visualisation, we simulated the CD and UV-Vis-NIR spectra of the four isolated syn–syn and four anti–antiFDBTF rotamers (ESI,† Fig. S19–S26) and the corresponding dipole-dipole-cancelled, slip, J-like, π–π stack dimers with the help of the ZINDO program (20 singlet transition states) using the optimised individual syn–syn and anti–anti molecules (Fig. 13a and b). The dimer structures were optimised in advance using PM3MM-level calculations. The reason for the slip π–π stack dimers is that the slip-type, J-aggregate affords an emissive state using the additive effect between the two electric dipole-allowed transition moments, whereas a cofacial type H-aggregate is non-emissive because of the cancellation between the two electric transition dipole moments according to Kasha's theory.32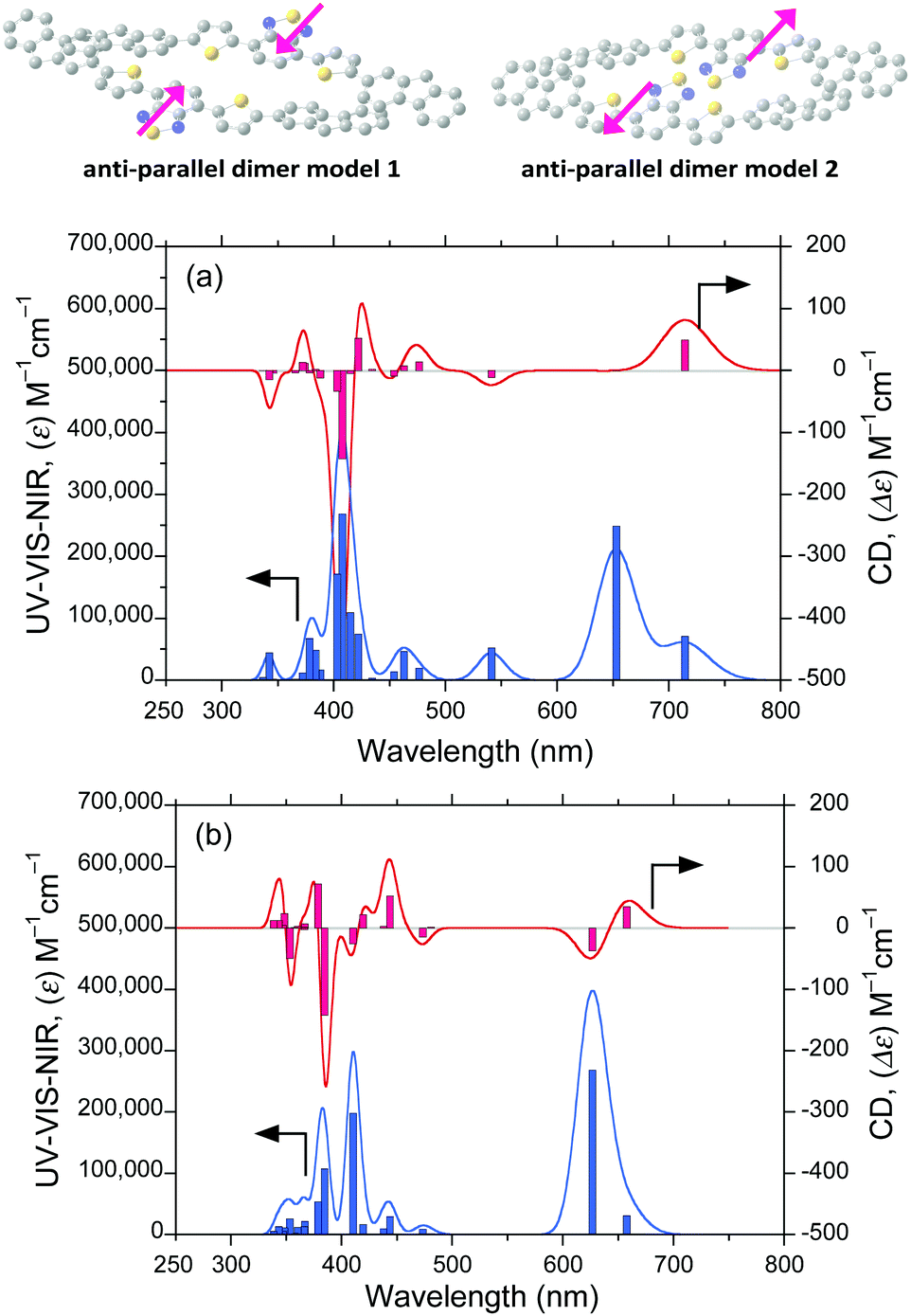 | ||
| Fig. 13 (a) CD and UV-Vis-NIR spectra (20 singlet transition states) of an optimised anti-parallel dimer 1 initialised using the less-polar FL(30°)-DBT(160°-and-160°)-FL(30°) and (b) another anti-parallel dimer 2 simulated using the ZINDO program (FWHM: 0.05 eV) and initialised using the polar FL(330°)-DBT(30°-and-30°)-FL(330°). These dimers were optimised via PM3MM-level calculations. | ||
3.9 Scenario of the time-evolved hetero-aggregates
According to the Flory–Huggins theory,33 two different non-charged polymers are inherently immiscible, which causes micro-segregation and/or microphase separation that results in turbidity. This is visible to the naked eye and an increase in the UV-Vis-NIR spectral signals is associated with an increment of the wavelength in the background spectra (notes as bkg in Fig. 1). The explanations for the time-dependent gabs and gem characteristics associated with their signs at the S0–S1 Cotton bands are as follows.The as-prepared hetero-aggregates that formed instantly via the rapid addition of the poor solvent (MeOH) involve many metastable, kinetically formed, poorly organised, associated structures with random orientations of the DBT moieties. The structures include some species emitting at ≈700 nm because of the huge RISs of the PF8DBT main chains and side chains. The poorly organised structures are PF8DBT chains with less-polar DBT groups due to intramolecular dipole–dipole cancellation (Fig. 5, 10 and 14). The fraction of the 700 nm species in the metastable aggregates is not large, and we primarily observed an intense, broadened visible band (λmax) at ≈600 nm, which was followed by the 700 nm species because of an energy migration scheme that exists in the aggregates upon excitation at 400 nm.
The metastable hetero-aggregate structures spontaneously become the most stable helically ordered hetero-aggregate structures over time (Fig. 15) via the given scheme. The rotatable C–C bonds are indicated by the red bold lines, and the arrows induce a swapping in the orientation of the DBT rings from up to down or vice versa (Fig. 14). The presence of two n-octyl groups at the 9,9-position of the fluorene rings associated with the intramolecular CH/π interactions34 should prevent cofacial π–π stacking of the fluorene rings, which is responsible for the aggregation-caused quenching of the luminescence.9,14 This situation should spontaneously lead to an emissive J-aggregation32,35 associated with a marked red shift in the Vis-NIR spectra. The structural reorganisation processes involving the swapping of the DBT rings and the slipped translational association of the PF8DBT chains (Fig. 14 and 15) are responsible for the larger gabs and gem values, and this should take a long time in the liquid suspension state.
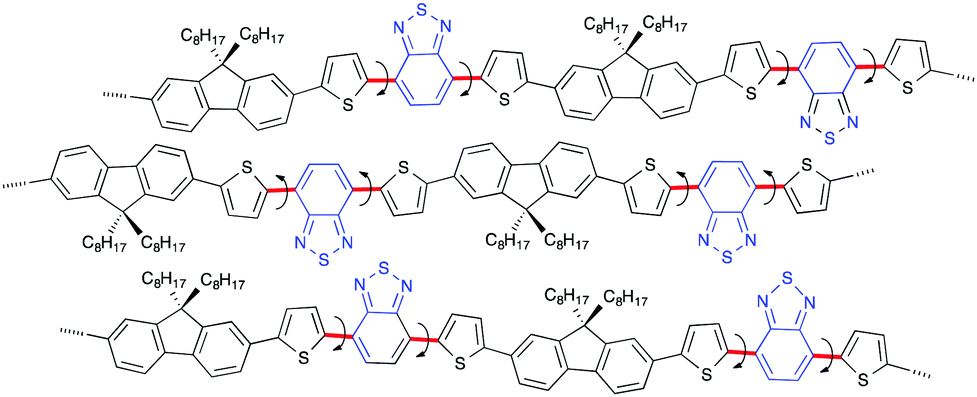 | ||
| Fig. 14 Poorly ordered metastable structure of the PF8DBT chains with less polar DBT groups due to intramolecular dipole–dipole cancellation. The rotatable C–C bonds indicated by red bold lines with turned arrows induce the swapping orientation of DBT rings from up to down or vice versa over time. | ||
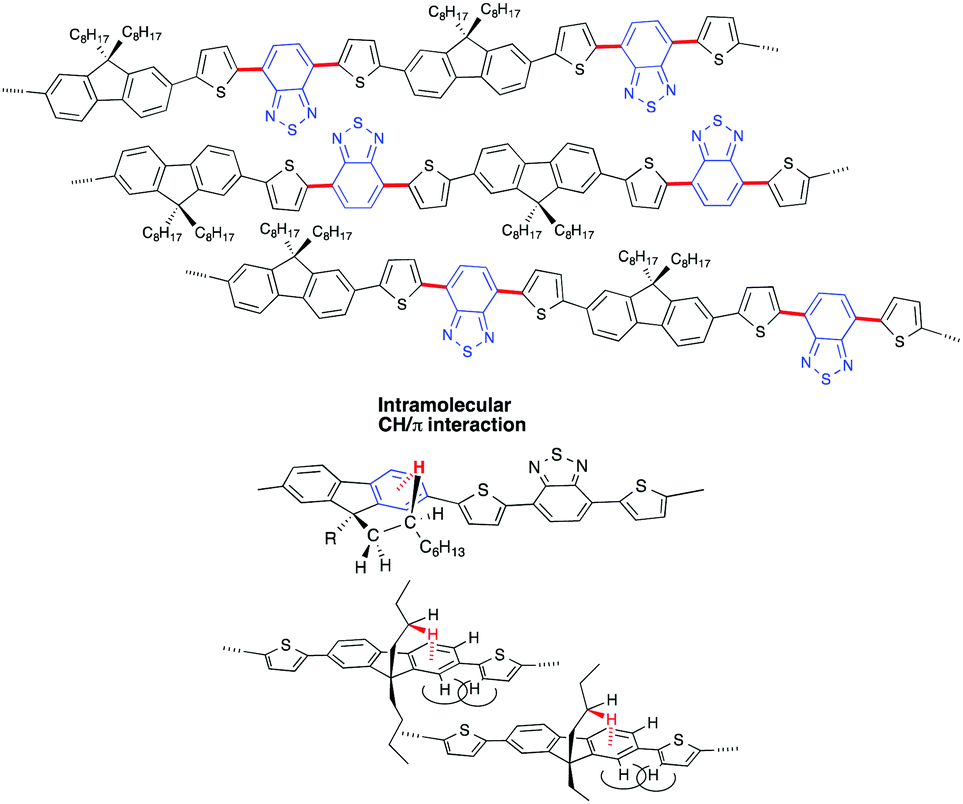 | ||
| Fig. 15 (top) Helically twisted and ordered structure of PF8DBT chains with polar DBT groups led by swapping the orientation of the DBT rings, leading to interchain dipole–dipole cancellation over time. (middle) The presence of the two octyl groups at the 9,9-position of the fluorene rings prevents the cofacial π–π stacked fluorene rings responsible for the aggregation-caused PL quenching. (bottom) This geometry should lead to an efficient J-aggregation associated with a marked red shift in the π–π* transition energy. | ||
The transfer of the macromolecular helicity from the non-charged PSi to the non-charged PF8DBT can occur during the course of the hetero-aggregation process in the poorer CHCl3–MeOH cosolvent regardless of any specific inter-macromolecular interactions. Only the chiral directional London dispersion force is responsible for the chirality-inducing capability. The helix scaffolds consisting of PSi-S and PSi-R with PF8DBT exhibit nearly ideal mirror-image CD-/CPL-spectral profiles relative to the PF8DBT-and-PSi hetero-aggregates, which is in agreement with the facts previously reported for several chiral polymer homo-aggregates.15,18,28
3.10 Scope and limitations of hetero-aggregation with helical PSi
Various supramolecular polymer complexes using single- and two-component molecular and oligomeric building blocks are promising for the design of (chir)optical, semiconducting, and biological functions and well-defined, higher-order fibrous and gel structures.36 Relatively directional and intense hydrogen, π–π stacking and electrostatic interactions are the driving forces for these structures.Dipolar-dipolar cancellation, which is a chirality-inducing driving force, is crucial for CPL-/CD-active supra-macromolecular complexation between non-charged π-/π-, π-/σ- and σ-/σ-conjugated polymers, although the supra-macromolecular complexes increased the difficulty of characterising the detailed structures of the resultant hetero-aggregates. This process provides a versatile approach in which two or more types of luminophores (and possibly PL-silent luminogens)14 can be chosen relatively freely to obtain a scaffold-free chiroptical substance with a desired wavelength and chiroptical sign with the help of a chirality-inducing scaffold. This process is in addition to a homo-aggregation process using a single luminophore and/or luminogen component.14
In principle, a mixture consisting of two or more types of polymers will eventually undergo micro-segregation and multi-phase separation after a prolonged time if Flory–Huggins theory33 is obeyed because of the minimal entropic gain upon mixing two different macromolecular chains.
As a preliminary experiment, we confirmed that although the aggregation-induced scaffold with PSi-S and PSi-R can generate poly[(9,9-di-n-octylfluoren-2,7-diyl)-alt-bithiophene] (PF8T2), which has large gem and gabs amplitudes, the time-evolved characteristics are not obvious.18 The lack of an intramolecular permanent dipole moment perpendicular to the main chain axis of PF8T2 is assumed to be responsible for the lack of marked time-evolved gem and gabs characteristics.
Recently, highly emissive and semiconducting materials in the red, FR and NIR regions have attracted interest for important applications.37–44 Such red/FR/NIR emitters are applicable to crypto-inks, boosting the efficiency of solar cells, improved charge carrier mobility and medical imaging. However, chiral red/FR/NIR emitters and their candidates, including J-aggregates,35,37 are rarely observed because the building sources are limited to 4f–4f transition lanthanides and Pt(II) complexes,38 inorganic network polymers and quantum dots,39 boron-dipyrromethane (BODIPY),40 perylene,37,41 helicene,42 dibenzoterrylene (DBT),20 polydiacetylene,43 twisted aromatics and polymers44 and electric-field driven thin-film devices.45 In addition, these building sources often require expensive chiral sources and multiple synthetic processes. The present hetero-aggregation-induced chiroptical emissive substance, which can be produced using any achiral π-conjugated polymers with the help of photoscissable PSi-S and PSi-R,46 facilitates efficient production of chiral emitters as a sacrificial helix at any chosen wavelength under mild conditions and at ambient temperature via a chiral catalyst-free preparation and loss-less aggregation process.
4. Conclusions
In this study, we demonstrated the time-evolved characteristics of the enhanced FR-CPL and -CD signals on the order of gem = ±0.02 at ≈700 nm starting from a non-helical LBG PF8DBT aggregate endowed with photoscissible, rigid, rod-like, helical PSi-S and PSi-R. This protocol allowed the successful production of ambidextrous CPL- and CD-active PF8DBT aggregates through the complete removal of the PSi used as the sacrificial scaffold. We discussed the origin of the time-evolved chiroptical characteristics based on the CD- and CPL spectra, DLS measurements and fluorescence optical microscopic (FOM) observations as a function of the ageing time and theoretical calculations of the various model rotamers of PF8DBT using DFT and TD-DFT (B3LYP, 6-31G(d,p)) and ZINDO. Although the individual model rotamers are stabilised in the preferred twist form via multiple intramolecular CH/N, CH/S, S/N and CH/HC interactions, dipole–dipole cancellation among the large dipole moments of the DBT moiety perpendicular to the main chain, followed by the slip stack, J-type aggregation, was assumed to be responsible for the time-evolved characteristics. Indeed, to generate optically active PF8DBT aggregates, our aggregation process, in which a poor solvent (MeOH) is rapidly added to a homogeneous CHCl3 solution of PF8DBT, kinetically produced thermodynamically metastable stacking structures. This metastable state spontaneously transformed to the thermodynamically stable, slip stack, J-type structures in one day.Acknowledgements
MF acknowledges financial support from JSPS KAKENHI (16H04155). SY wishes to thank Profs Hiroharu Ajiro, Hiroko Yamada, Fumihiko Matsui, Shun Hirota and Tsuyoshi Kawai for discussion and comments. We also thank Drs Nor Azura Abdul Rahim and Sibo Guo for stimulating discussions. Yasuo Okajima (PL lifetime), Yoshiko Nishikawa (DLS), Toshiki Nagai (FOM), Kazuki Yamazaki (GPC) and Sang Thi Duong (GPC) at NAIST contributed to this work by providing technical assistance.Notes and references
- For reviews and a book: (a) I. Prigogine, G. Nicolis and A. Babloyants, Phys. Today, 1972, 25(Nov), 23–28 CrossRef CAS; (b) I. Prigogine, G. Nicolis and A. Babloyants, Phys. Today, 1972, 25(Dec), 38–44 CrossRef; (c) D. Kondepudi and I. Prigogine, Modern Thermodynamics: From Heat Engines to Dissipative Structures, John Wiley & Sons, Chichester, UK, 1998 Search PubMed; (d) R. Plasson, D. P. Kondepudi, H. Bersini, A. Commeyras and K. Asakura, Chirality, 2007, 19, 589–600 CrossRef CAS PubMed.
- For a review and a book: (a) E. Schrodinger, What is life? With Mind and Matter and Autobiographical Sketches, Cambridge University Press, 1944 Search PubMed; (b) V. Avetisov and V. Goldanskii, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11435–11442 CrossRef CAS PubMed.
- For reviews: (a) S. F. Mason, Nature, 1984, 311, 19–23 CrossRef CAS PubMed; (b) J. K. Barton, Science, 1986, 233, 727–733 CrossRef CAS PubMed; (c) W. A. Bonner, Origins Life Evol. Biospheres, 1992, 21, 407–420 CrossRef; (d) Y. Inoue, Chem. Rev., 1992, 92, 741–770 CrossRef CAS; (e) B. L. Feringa and R. A. van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418–3438 CrossRef; (f) K. Soai, I. Sato and T. Shibata, Chem. Rec., 2001, 1, 321–332 CrossRef CAS PubMed; (g) M. Fujiki, Chem. Rec., 2009, 9, 271–298 CrossRef CAS PubMed.
- (a) I. Sato, H. Urabe, S. Ishiguro, T. Shibata and K. Soai, Angew. Chem., Int. Ed., 2003, 42, 315–317 CrossRef CAS PubMed; (b) T. Kawasaki, M. Sato, S. Ishiguro, T. Saito, Y. Morishita, I. Sato, H. Nishino, Y. Inoue and K. Soai, J. Am. Chem. Soc., 2005, 127, 3274–3275 CrossRef CAS PubMed; (c) C. Blanco and D. Hochberg, Phys. Chem. Chem. Phys., 2011, 13, 839–849 RSC; (d) M. Fujiki, Y. Donguri, Y. Zhao, A. Nakao, N. Suzuki, K. Yoshida and W. Zhang, Polym. Chem., 2015, 6, 1627–1638 RSC; (e) M. Fujiki, K. Yoshida, N. Suzuki, N. A. A. Rahim and J. A. Jalil, J. Photochem. Photobiol., A, 2016, 331, 120–129 CrossRef CAS.
- For reviews: (a) W. Kühlbrandt, Structure, 1995, 3, 521–525 CrossRef; (b) C. N. Hunter, Curr. Biol., 1995, 5, 826–828 CrossRef CAS PubMed; (c) X. Hu and K. Schulten, Phys. Today, 1997, 50(8), 28–34 CrossRef CAS; (d) X. Hu, A. Damjanovic, T. Ritz and K. Schulten, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5935–5941 CrossRef CAS PubMed; (e) V. Sundström, T. Pullerits and R. van Grondelle, J. Phys. Chem. B, 1999, 103, 2327–2346 CrossRef; (f) for a recent time-revolutional analysis, Y. Yoneda, T. Noji, T. Katayama, N. Mizutani, D. Komori, M. Nango, H. Miyasaka, S. Itoh, Y. Nagasawa and T. Dewa, J. Am. Chem. Soc., 2015, 137, 13121–13129 CrossRef CAS PubMed.
- For reviews. (a) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS PubMed; (b) J. Glowacki and S. Mizuno, Biopolymers, 2008, 89, 338–344 CrossRef CAS PubMed; (c) E. Schwartz, L. G. Stephane, J. J. L. M. Cornelissen, R. J. M. Nolte and A. E. Rowan, Chem. Soc. Rev., 2010, 39, 1576–1599 RSC.
- (a) J. D. Le, Y. Pinto, N. C. Seeman, K. Musier-Forsyth, T. A. Taton and R. A. Kiehl, Nano Lett., 2004, 4, 2343–2347 CrossRef CAS; (b) Y.-M. Guo, H. Oike and T. Aida, J. Am. Chem. Soc., 2004, 126, 716–717 CrossRef CAS PubMed; (c) S. Haraguchi, M. Numata, C. Li, Y. Nakano, M. Fujiki and S. Shinkai, Chem. Lett., 2009, 38, 254 CrossRef CAS; (d) T. Shiraki, Y. Tsuchiya, T. Noguchi, S.-I. Tamaru, N. Suzuki, M. Taguchi, M. Fujiki and S. Shinkai, Chem. – Asian J., 2014, 9, 218–222 CrossRef CAS PubMed; (e) T. Shiraki, A. Dawn, Y. Tsuchiya and S. Shinkai, J. Am. Chem. Soc., 2010, 132, 13928–13935 CrossRef CAS PubMed; (f) S. Le Gac, E. Schwartz, M. Koeph, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. – Eur. J., 2010, 16, 6176–6186 CrossRef CAS PubMed; (g) G. Fukuhara, K. Iida, Y. Kawanami, H. Tanaka, T. Mori and Y. Inoue, J. Am. Chem. Soc., 2015, 137, 15007–15014 CrossRef CAS PubMed; (h) S. Guo, N. Suzuki and M. Fujiki, Macromolecules, 2017, 50, 1778–1789 CrossRef CAS.
- (a) E. Yashima, K. Maeda and Y. Okamoto, Nature, 1999, 399, 449 CrossRef CAS; (b) Y. Ono, K. Nakashima, M. Sano, Y. Kanekiyo, K. Inoue, J. Hojo and S. Shinkai, Chem. Commun., 1998, 1477–1478 RSC; (c) Y. Yang, M. Suzuki, S. Owa, H. Shirai and K. Hanabusa, J. Mater. Chem., 2006, 16, 1644–1650 RSC; (d) D. Liu, B. Li, Y. Guo, Y. Li and Y. Yang, Chirality, 2015, 27, 809–815 CrossRef CAS PubMed; (e) D. Yang, Y. Zhao, K. Lv, X. Wang, W. Zhang, L. Zhang and M. Liu, Soft Matter, 2016, 12, 1170–1175 RSC; (f) R. Tamoto, N. Daugey, T. Buffeteau, B. Kauffmann, M. Takafuji, H. Ihara and R. Oda, Chem. Commun., 2015, 51, 3518–3521 RSC; (g) T. Goto, Y. Okazaki, M. Ueki, Y. Kuwahara, M. Takafuji, R. Oda and H. Ihara, Angew. Chem., Int. Ed., 2017, 56, 2989–2993 CrossRef CAS PubMed; (h) J. Han, J. You, X. Li, P. Duan and M. Liu, Adv. Mater., 2017 DOI:10.1002/adma.201606503.
- For reviews, see. (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- http://https://en.oxforddictionaries.com/definition/scaffolding .
- http://https://en.oxforddictionaries.com/definition/template .
- For a book and reviews: (a) G. Desiraju and T. Steiner, The Weak Hydrogen Bond: In Structural Chemistry and Biology, Oxford Science Pub, Oxford, UK, 1998 Search PubMed; (b) O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049–6076 CrossRef CAS PubMed; (c) M. Fujiki and A. Saxena, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4637–4650 CrossRef CAS.
- (a) S. K. Lamoreaux, Phys. Rev. Lett., 1997, 78, 5–8 CrossRef CAS; (b) A. Meurk, P. F. Luckham and L. Bergström, Langmuir, 1997, 13, 3896–3899 CrossRef CAS; (c) J. N. Munday and F. Capasso, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 75, 060102 CrossRef; (d) J. N. Munday, F. Capasso and V. A. Parsegian, Nature, 2009, 457, 170–173 CrossRef CAS PubMed; (e) E. Shahmoon, I. Mazets and G. Kurizki, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10485–10490 CrossRef CAS PubMed.
- (a) J. Liu, H. Su, L. Meng, Y. Zhao, C. Deng, J. C. Y. Ng, P. Lu, M. Faisal, J. W. Y. Lam, X. Huang, H. Wu, K. S. Wong and B. Z. Tang, Chem. Sci., 2012, 3, 2737–2747 RSC; (b) J. C. Y. Ng, J. Liu, H. Su, Y. Hong, H. Li, J. W. Y. Lam, K. S. Wong and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 78–83 RSC ; For a review, ; (c) J. Roose, B. Z. Tang and K. S. Wong, Small, 2016, 12, 6495–6512 CrossRef CAS PubMed.
- (a) K. Kawagoe, M. Fujiki and Y. Nakano, New J. Chem., 2010, 34, 637–647 RSC; (b) Y. Nakano, Y. Liu and M. Fujiki, Polym. Chem., 2010, 1, 460–469 RSC; (c) Y. Nakano, F. Ichiyanagi, M. Naito, Y.-G. Yang and M. Fujiki, Chem. Commun., 2012, 48, 6636–6638 RSC; (d) L. Wang, N. Suzuki, J. Liu, T. Matsuda, N. A. A. Rahim, W. Zhang, M. Fujiki, Z. Zhang, N. Zhou and X. Zhu, Polym. Chem., 2014, 5, 5920–5927 RSC; (e) Y. Zhao, N. A. A. Rahim, Y. Xia, M. Fujiki, B. Song, Z. Zhang, W. Zhang and X. Zhu, Macromolecules, 2016, 49, 3214–3221 CrossRef CAS.
- D. Yang, Y. Zhao, K. Lv, X. Wang, W. Zhang, L. Zhang and M. Liu, Soft Matter, 2016, 12, 1170–1175 RSC.
- (a) Y. Duan, X. Liu, L. Han, S. Asahina, D. Xu, Y. Cao, Y. Yao and S. Che, J. Am. Chem. Soc., 2014, 136, 7193–7196 CrossRef CAS PubMed; (b) Y. Duan, L. Han, J. Zhang, S. Asahina, Z. Huang, L. Shi, B. Wang, Y. Cao, Y. Yao, L. Ma, C. Wang, R. K. Dukor, L. Sun, C. Jiang, Z. Tang, L. A. Nafie and S. Che, Angew. Chem., Int. Ed., 2015, 54, 15170–15175 CrossRef CAS PubMed.
- N. A. A. Rahim and M. Fujiki, Polym. Chem., 2016, 7, 4618–4629 RSC.
- (a) J. Roncali, Chem. Rev., 1997, 97, 173–206 CrossRef CAS PubMed; (b) E. Bundgaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2007, 91, 954–985 CrossRef CAS; (c) Y. Li and Y. Zou, Adv. Mater., 2008, 20, 2952–2958 CrossRef CAS.
- (a) Q. Hou, Y. Xu, W. Yang, M. Yuan, J. Peng and Y. Cao, J. Mater. Chem., 2002, 12, 2887–2892 RSC; (b) M. Svensson, F. Zhang, S. C. Veenstra, W. J. H. Verhees, J. C. Hummelen, J. M. Kroon, O. Inganäs and M. R. Andersson, Adv. Mater., 2003, 15, 988–991 CrossRef CAS; (c) Q. Zhou, Q. Hou, L. Zheng, X. Deng, G. Yu and Y. Cao, Appl. Phys. Lett., 2004, 84, 1653–1655 CrossRef CAS; (d) K. G. Jesperson, W. J. D. Beenken, Y. Zaushitsyn, A. Yartsev, M. Anderson, T. Pullerits and V. Sundström, J. Chem. Phys., 2004, 121, 12613–12617 CrossRef PubMed; (e) H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed; (f) J. Liu, G. Feng, D. Ding and B. Liu, Polym. Chem., 2013, 4, 4326–4334 RSC; (g) H. Bronstein, J. M. Frost, A. Hadipour, Y. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Chem. Mater., 2013, 25, 277–285 CrossRef CAS; (h) C. Dai, D. Yang, W. Zhang, B. Bao, Y. Cheng and L. Wang, Polym. Chem., 2015, 6, 3962–3969 RSC; (i) C. Yang, H. Liu, Y. Zhang, Z. Xu, X. Wang, B. Cao and M. Wang, Biomacromolecules, 2016, 17, 1673–1683 CrossRef CAS PubMed.
- E. L. Eliel, S. H. Wilen and L. N. Mander, Chiroptical Properties, in Stereochemistry of Organic Compounds, Wiley-Interscience, New York, NY, 1994, ch. 13 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- A. Wakamiya, K. Mori and S. Yamaguchi, Angew. Chem., Int. Ed., 2007, 23, 4273–4276 CrossRef PubMed.
- (a) J. G. Calvert and J. N. Pitts, Jr., Photochemistry, John Wiley and Sons, New York, NY, US, 1966 Search PubMed; (b) N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, CA, US, 1978 Search PubMed.
- (a) R. Zott, Angew. Chem., Int. Ed., 2003, 42, 3990–3995 CrossRef CAS PubMed; (b) B. Liu and H. C. Zeng, Small, 2005, 1, 566–571 CrossRef CAS PubMed; (c) H. Ham, N.-H. Park, S. S. Kim and H. W. Kim, Sci. Rep., 2014, 4, 3579 CrossRef PubMed.
- For reviews and a book: (a) D. Psaltis, S. R. Quake and C. Yang, Nature, 2006, 442, 381–386 CrossRef CAS PubMed; (b) C. Monat, P. Domachuk and B. J. Eggleton, Nat. Photonics, 2006, 1, 106–114 CrossRef; (c) Optofluidics–Fundamentals, Devices, and Applications, ed. Y. Fainman, L. P. Lee, D. Psaltis and C. Yang, McGraw-Hill, 2010 Search PubMed.
- (a) J. A. Sherwin and A. Lakhtakia, Opt. Commun., 2002, 214, 231–245 CrossRef CAS; (b) J. Pedersen and N. A. Mortensen, Appl. Phys. Lett., 2007, 91, 213501 CrossRef; (c) N. A. Mortensen and S. Xiao, Appl. Phys. Lett., 2007, 90, 141108 CrossRef; (d) F. B. Arango, M. B. Christiansen, M. Gersborg-Hansen and A. Kristensen, Appl. Phys. Lett., 2007, 91, 223503 CrossRef; (e) H. C. Hunt and J. S. Wilkinson, Microfluid. Nanofluid., 2008, 4, 53–79 CrossRef CAS.
- (a) Y. Nakano and M. Fujiki, Macromolecules, 2011, 44, 7511–7519 CrossRef CAS; (b) M. Fujiki, K. Yoshida, N. Suzuki, J. Zhang, W. Zhang and X. Zhu, RSC Adv., 2013, 3, 5213–5219 RSC; (c) M. Fujiki, A. J. Jalilah, N. Suzuki, M. Taguchi, W. Zhang, M. M. Abdellatif and K. Nomura, RSC Adv., 2012, 2, 6663–6671 RSC.
- M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CAS.
- C. B. Nielsen, A. J. P. White and I. McCulloch, J. Org. Chem., 2015, 80, 5045–5048 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- For early reviews, see: (a) M. Kasha, Radiat. Res., 1963, 20, 55–71 CrossRef CAS PubMed; (b) M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- (a) P. J. Flory, J. Chem. Phys., 1942, 10, 51–61 CrossRef CAS; (b) M. L. Huggins, J. Phys. Chem., 1942, 46, 151–158 CrossRef CAS.
- (a) M. Taguchi, N. Suzuki and M. Fujiki, Polym. J., 2013, 45, 1047–1057 CrossRef CAS; (b) N. Suzuki, T. Matsuda, T. Nagai, K. Yamazaki and M. Fujiki, Cryst. Growth Des., 2016, 16, 6593–6599 CrossRef CAS.
- For a recent review, F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- For recent reviews: (a) T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171–173 CrossRef CAS PubMed; (b) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (c) L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed; (d) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed; (e) E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed; (f) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- For a review: E. M. Sμnchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem. – Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
- (a) S. D. Bonsall, M. Houcheime, D. A. Strausa and G. Muller, Chem. Commun., 2007, 3676–3678 RSC; (b) S. Petoud, G. Muller, E. G. Moore, J. Xu, J. Sokolnicki, J. P. Riehl, U. N. Le, S. M. Cohen and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 77–83 CrossRef CAS PubMed; (c) J. L. Lunkley, D. Shirotani, K. Yamanari, S. Kaizaki and G. Muller, J. Am. Chem. Soc., 2008, 130, 13814–13815 CrossRef CAS PubMed; (d) F. Song, G. Wei, X. Jiang, F. Li, C. Zhu and Y. Cheng, Chem. Commun., 2013, 49, 5772–5774 RSC; (e) J. D. Routledge, M. W. Jones, S. Faulkner and M. Tropiano, Inorg. Chem., 2015, 54, 3337–3345 CrossRef CAS PubMed; (f) N. Shi, Y. Guan, J. Zhang and X. Wan, RSC Adv., 2015, 5, 34900–34907 RSC; (g) L. Dai, W.-S. Lo, I. D. Coates, R. Pa and G.-L. Law, Inorg. Chem., 2016, 55, 9065–9070 CrossRef CAS PubMed; (h) Y. Kono, K. Nakabayashi, S. Kitamura, M. Shizuma, M. Fujiki and Y. Imai, RSC Adv., 2016, 6, 40219–40224 RSC; (i) For a recent review, R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC; (j) For a recent book, G. Muller, In Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials, ed. A. Bettencourt-Dias, Wiley, Chichester, West Sussex, UK, 2014, ch. 3, pp. 77–123 Search PubMed; (k) T. Ikeda, M. Takayama, J. Kumar, T. Kawai and T. Haino, Dalton Trans., 2015, 44, 13156–13162 RSC.
- (a) M. Fujiki, Y. Kawamoto, M. Kato, Y. Fujimoto, T. Saito, S. Hososhima and G. Kwak, Chem. Mater., 2009, 42, 3443–3447 Search PubMed; (b) S. Fukao and M. Fujiki, Chem. Mater., 2009, 42, 8062–8067 CAS; (c) M. Fujiki, M. Kato, Y. Kawamoto and G. Kwak, Polym. Chem., 2011, 2, 914–922 RSC; (d) I. Russier-Antoine, F. Bertorelle, R. Hamouda, D. Rayane, P. Dugourd, Z. Sanader, V. Bonacic-Koutecky, P.-F. Brevet and R. Antoine, Nanoscale, 2016, 8, 2892–2898 RSC; (e) J. Kumar, T. Kawai and T. Nakashima, Chem. Commun., 2017, 53, 1269–1272 RSC.
- (a) R. Donuru, S. Zhu, S. Green and H. Liu, Polymer, 2010, 51, 5359–5368 CrossRef; (b) E. M. Sańchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. de la Moya, J. Am. Chem. Soc., 2014, 136, 3346–3349 CrossRef PubMed; (c) C. Ray, E. M. Sμnchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, Í. López-Arbeloa, J. Bañuelos, K. D. Cohovi, J. L. Lunkley, G. Muller and S. de la Moya, Chem. – Eur. J., 2016, 22, 8805–8808 CrossRef CAS PubMed; (d) R. B. Alnoman, S. Rihn, D. C. O'Connor, F. A. Black, B. Costello, P. G. Waddell, W. Clegg, R. D. Peacock, W. Herrebout, J. G. Knight and M. J. Hall, Chem. – Eur. J., 2016, 22, 93–96 CrossRef CAS PubMed; (e) Y. Wang, Y. Li, S. Liu, F. Li, C. Zhu, S. Li and Y. Cheng, Macromolecules, 2016, 49, 5444–5451 CrossRef CAS.
- J. Kumar, H. Tsumatori, J. Yuasa, T. Kawai and T. Nakashima, Angew. Chem., Int. Ed., 2015, 54, 5943–5947 CrossRef CAS PubMed.
- (a) I. H. Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guénéé, C. Nançoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC; (b) J. Bosson, G. M. Labrador, S. Pascal, F.-A. Miannay, O. Yushchenko, H. Li, L. Bouffier, N. Sojic, R. C. Tovar, G. Muller, D. Jacquemin, A. D. Laurent, B. Le Guennic, E. Vauthey and J. Lacour, Chem. – Eur. J., 2016, 22, 18394–18403 CrossRef CAS PubMed; (c) H. Sakai, T. Kubota, J. Yuasa, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, T. Kawai and T. Hasobe, Org. Biomol. Chem., 2016, 14, 6738–6743 RSC.
- F. Li, Y. Wang, Z. Wang, Y. Cheng and C. Zhu, Polym. Chem., 2015, 6, 6802–6805 RSC.
- (a) K. Watanabe, I. Osaka, S. Yorozuya and K. Akagi, Chem. Mater., 2012, 24, 1011–1024 CrossRef CAS; (b) M. Li, H.-Y. Lu, C. Zhang, L. Shi, Z. Tang and C.-F. Chen, Chem. Commun., 2016, 52, 9921–9924 RSC; (c) C. Chen, J. Chen, T. Wang and M. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 30608–30615 CrossRef CAS PubMed.
- (a) S. Iba, S. Koh, K. Ikeda and H. Kawaguchi, Appl. Phys. Lett., 2011, 98, 081113 CrossRef; (b) N. Nishizawa, K. Nishibayashi and H. Munekata, Appl. Phys. Lett., 2014, 104, 111102 CrossRef; (c) J. Xiang, Y. Li, Q. Li, D. A. Paterson, J. M. D. Storey, C. T. Imrie and O. D. Lavrentovich, Adv. Mater., 2015, 27, 3014–3018 CrossRef CAS PubMed; (d) F. Zinna, U. Giovanella and L. Di Bari, Adv. Mater., 2015, 27, 1791–1795 CrossRef CAS PubMed; (e) N. Nishizawa, K. Nishibayashi and H. Munekata, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1783–1788 CrossRef CAS PubMed.
- For reviews. (a) R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS; (b) M. Fujiki, J. R. Koe, K. Terao, T. Sato, A. Teramoto and J. Watanabe, Polym. J., 2003, 35, 297–344 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, CPL/PL/CD/UV-Vis spectra, DFT/TD-DFT-calculations, photographs of the colour change and characterisation of F8DBT by 1H-NMR. See DOI: 10.1039/c7qm00096k |
| This journal is © the Partner Organisations 2017 |