
Aromatic-rich hydrocarbon porous networks through alkyne metathesis†
Haishen
Yang‡
 ab,
Youlong
Zhu‡
b,
Ya
Du
b,
Dazhi
Tan
b,
Yinghua
Jin
b and
Wei
Zhang
*b
ab,
Youlong
Zhu‡
b,
Ya
Du
b,
Dazhi
Tan
b,
Yinghua
Jin
b and
Wei
Zhang
*b
aShanghai Key Laboratory of Materials Protection and Advanced Materials in Electric Power, College of Environmental and Chemical Engineering, Shanghai University of Electric Power, Shanghai 200090, P. R. China
bDepartment of Chemistry and Biochemistry, University of Colorado, Boulder, CO 80309, USA. E-mail: wei.zhang@colorado.edu
First published on 9th February 2017
Abstract
Purely hydrocarbon-based porous polymers have generally been prepared through various irreversible transition metal-catalyzed cross-coupling reactions forming C–C bonds. Herein, we report an alternative synthetic approach, namely reversible alkyne metathesis, for the preparation of ethynylene-linked porous polymers. Planar and tetrahedral-shaped monomers were explored to construct poly(aryleneethynylene) (PAE) networks. We systematically varied the size of the monomers and studied the structure–property relationships. The resulting polymers exhibit high Brunauer–Emmett–Teller (BET) surface areas in the range of 736 m2 g−1 to 2294 m2 g−1. The advantages of such aromatic-rich PAE networks are their lightweight, high thermal/chemical stabilities, and superior hydrophobicity, which are beneficial for their application in adsorption/separation of toxic organic pollutants from water. We found that PAEs can adsorb a significant amount of common aromatic solvents, e.g. up to 723 wt% of nitrobenzene. Our study thus demonstrates an encouraging novel approach to prepare purely hydrocarbon-based porous materials.
Introduction
Purely hydrocarbon-based aromatic-rich polymers are interesting light-weight materials which have many potential applications in electrochemical research, development of optoelectronic materials, gas adsorption/separation, and removal of organic pollutants from water.1–4 Various hydrocarbon polymers have been developed to resemble the most well-known naturally occurring carbon allotropes, e.g. graphite and diamonds, and their unique characteristics. For example, chemists have developed conjugated two-dimensional covalent organic frameworks (COFs) through dynamic covalent reactions, mainly imine chemistry and boronic ester formation.5–8 Three-dimensional hydrocarbon polymers closely resembling the tetrahedral structure of diamond have also been developed. One such example is the porous polymer which has one phenyl ring inserted into each C–C bond of the diamond structure. It has been reported that such a polymer exhibits exceptionally high surface area (a Langmuir surface area of 7100 m2 g−1 and a BET surface area of 5600 m2 g−1, the largest reported so far) and excellent thermal and hydrothermal stabilities.9Our group has long-standing interest in developing carbon-rich functional organic materials through alkyne metathesis. Purely hydrocarbon-based porous polymers with high thermal/chemical stability and superhydrophobicity are interesting targets to explore through alkyne metathesis,10–20 which polymerizes monomers by the formation of carbon–carbon triple bonds. Unlike other irreversible C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond forming cross-coupling reactions, alkyne metathesis is reversible, potentially providing an error-correction mechanism. In the past two decades, alkyne metathesis has rapidly emerged as an important dynamic covalent reaction in the synthesis of small molecules,21,22 polymers as well as nanomaterials.23–30 Recently, we have developed a series of highly active and robust alkyne metathesis catalysts containing multidentate ligands.13,31–33 This new class of catalysts completely inhibits the undesired small alkyne polymerization pathway and has excellent functional group compatibility with a broad substrate scope.32,33 The catalyst has enabled metathesis of multiple challenging substrates to provide carbon-rich macrocycles, molecular cages,34,35 conjugated 1D molecular wires (e.g. polydiacetylenes),36 and porous polymers.37
C bond forming cross-coupling reactions, alkyne metathesis is reversible, potentially providing an error-correction mechanism. In the past two decades, alkyne metathesis has rapidly emerged as an important dynamic covalent reaction in the synthesis of small molecules,21,22 polymers as well as nanomaterials.23–30 Recently, we have developed a series of highly active and robust alkyne metathesis catalysts containing multidentate ligands.13,31–33 This new class of catalysts completely inhibits the undesired small alkyne polymerization pathway and has excellent functional group compatibility with a broad substrate scope.32,33 The catalyst has enabled metathesis of multiple challenging substrates to provide carbon-rich macrocycles, molecular cages,34,35 conjugated 1D molecular wires (e.g. polydiacetylenes),36 and porous polymers.37
Herein, we further explored the synthesis of purely hydrocarbon-based porous networks (e.g.PAE 1-1 and PAE 3-3) through alkyne metathesis. We successfully prepared various porous poly(aryleneethynylene) (PAE) networks through polymerization/co-polymerization of planar and tetrahedral-shaped monomers, and systematically investigated their porosities. The effect of monomer structures on the porosity of the resulting PAEs as well as their adsorption capacities toward common organic pollutants is also discussed.
Results and discussion
Planar 1,3,5-tripropynylbenzene (1) and its extended analog 1,3,5-tris(p-propynylphenyl)benzene (2) (Fig. 1) were prepared as monomers to construct 2D conjugated polymers (the ideal structure of PAE A is shown in Fig. 2). Previously, we have reported a porous PAE (the ideal structure of PAE B is shown in Fig. 2) prepared from monomer 3.37 In order to examine the effect of monomer size on the metathesis and properties of the resulting polymer, similar tetrahedral-shaped but enlarged monomer 4 was prepared to construct a 3D polymer with a diamondoid topology. Alkyne metathesis of these monomers produces 2-butyne as a by-product. Since alkyne metathesis is an equilibrium reaction, the removal of 2-butyne is necessary to improve the conversion of monomers and enhance the polymerization degree. Although 2-butyne can be removed under vacuum conditions,10,12,13,38 we found that such conditions require a stringent moisture- and air-free vacuum system and also solvent refill. Therefore, we removed 2-butyne by adding 5 Å molecular sieves (MSs) as scavengers. Pelletized 5 Å MSs were used for the ease of separation. In a typical procedure, the molybdenum catalyst (5) was generated in situ by mixing the triphenolsilane ligand and the molybdenum(VI) trisamide precursor in dry CCl4 (rt, 15 min). Subsequent addition of a monomer solution in CHCl3 and 5 Å MS followed by heating the mixture at 55 °C for 22 h without stirring provided poly(phenylene ethynylene) networks as gel-like materials. After successive washing with acetone, conc. ammonium hydroxide and water, followed by vacuum drying, the PAEs were obtained as beige color solids in excellent yields (82%-quantitative). From a single type of monomer (1–4), we obtained homo-polymerized PAE 1-1, 2-2, 3-3, and 4-4. Alternatively, by using combinations of two different types of monomers (e.g. a 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of monomers 1 and 2), we obtained hetero-polymerized PAE 1-2, 1-3, and 2-3.
:1 molar ratio of monomers 1 and 2), we obtained hetero-polymerized PAE 1-2, 1-3, and 2-3.
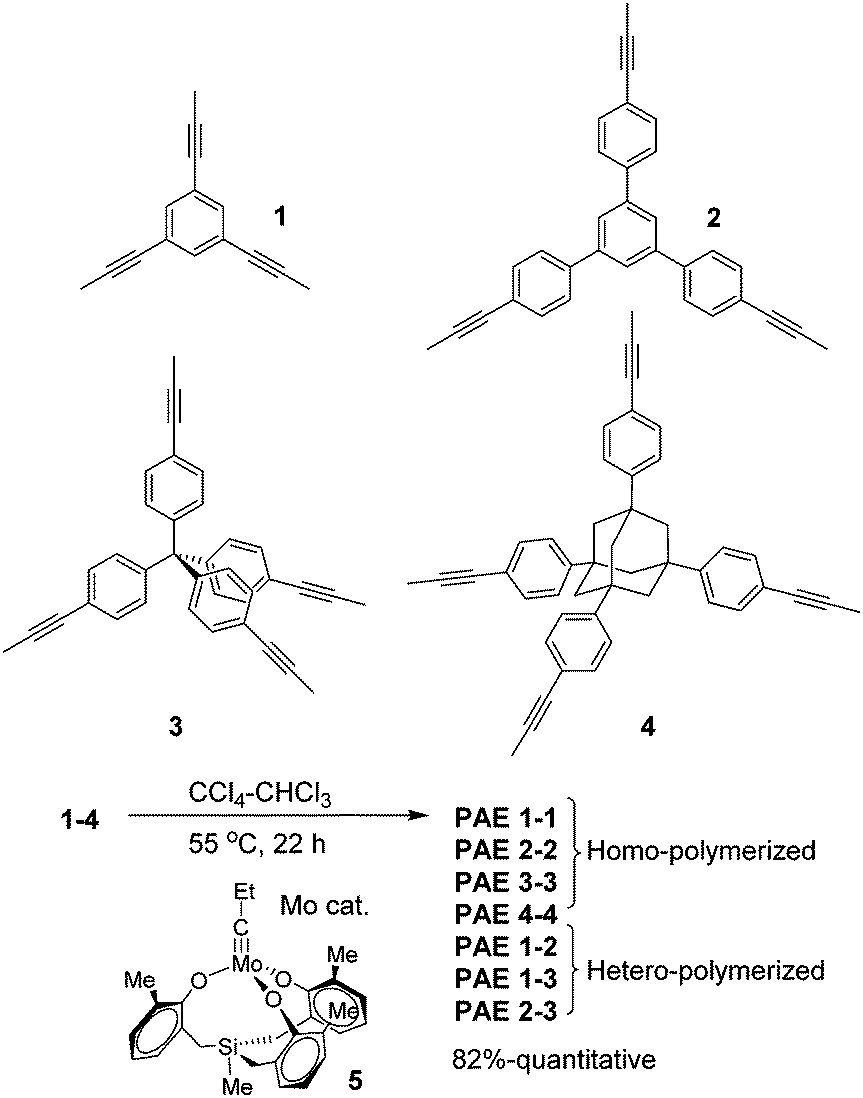 | ||
| Fig. 1 Structures of monomers and synthesis of ethynylene-linked porous polymers. | ||
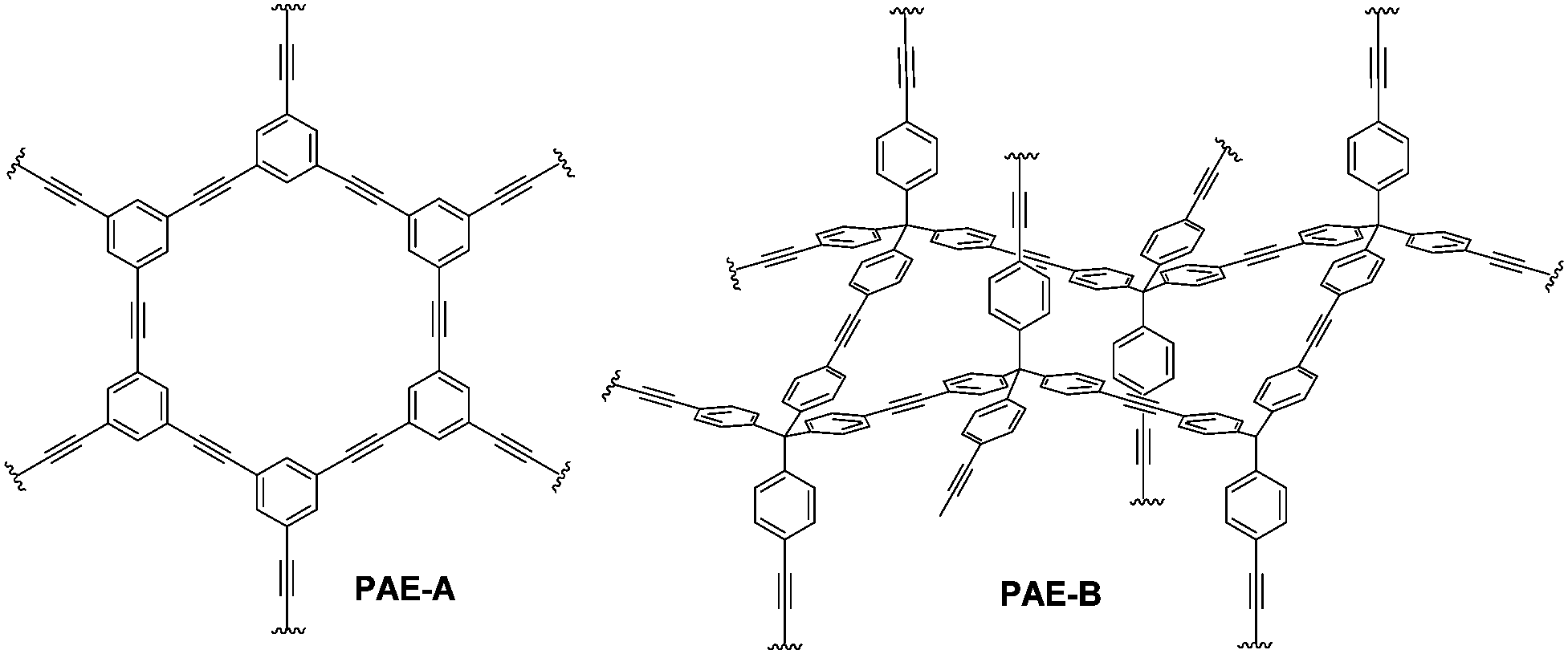 | ||
| Fig. 2 Ideal structures of PAE 1-1 (PAE A) and PAE 3-3 (PAE B). | ||
All PAEs are insoluble in common organic solvents, such as dichloromethane, acetone, ethanol, ethyl acetate, and DMF. The PAEs were characterized by FT-IR, solid state 13C NMR, TGA, SEM, and powder X-ray diffraction (PXRD). The FT-IR spectra (Fig. S2–S8, ESI†) of all the monomers (1–4) show two weak but sharp absorption peaks around 2210–2260 cm−1, which are characteristic of asymmetric CC bond stretching. However, these bands nearly disappear in the spectra of PAEs, indicating the presence of predominantly symmetrical CC bonds in the frameworks. The 13C cross-polarization magic angle spinning (CP-MAS) NMR spectra of PAEs (Fig. S14–S20, ESI†) show ethynylene carbon peaks at 89–91 ppm. We also observed unreacted propyne methyl carbon peaks around 2–4 ppm and a propynyl carbon peak around 79 ppm in some cases, which indicates incomplete conversion of the propynyl groups on the monomers. The thermal stability of PAEs was evaluated through thermogravimetric analyses (TGAs) (Fig. S1, ESI†). Most PAEs lose only <5% weight at 300 °C except hetero-polymerized PAE 1-3 (12% weight loss). A gradual weight loss was observed thereafter until the temperature reaches 800 °C (12–20% total weight loss). PAE 1-3 loses total 38% weight at 800 °C.
Although dynamic alkyne metathesis, which offers reversibility and error correction, has the potential to yield ordered frameworks, we failed to obtain crystalline PAEs. All polymers are amorphous according to PXRD analysis, and featureless agglomerates were observed in SEM images (Fig. S20, ESI†).
Next, we examined the porous properties of these PAEs. Ideally, planar monomers (1–2) can produce 2D networks (e.g.PAE A in Fig. 2) and extension of their layer-structures in the z dimension can produce porous materials. Tetrahedral monomers 3 and 4 would form 3D networks (e.g.PAE B in Fig. 2) with a diamondoid topology in ideal scenarios. We intentionally varied the size of the monomers to study the effect of size variation on the surface area of the resulting polymer networks. Nitrogen adsorption/desorption isotherms of the freshly activated samples at 77 K were measured, and their pore size distributions were calculated by employing non-local density functional theory (NLDFT) (Table 1). As shown in Fig. 3a, all the porous polymers (PAE 1-1, 2-2, and 1-2) consisting of planar monomer units exhibit typical type I nitrogen adsorption isotherms. PAE 1-1 prepared from monomer 1 shows the lowest Brunauer–Emmett–Teller (BET) surface area (736 m2 g−1). When the monomer size increases, the BET surface area of the resulting polymer (PAE 2-2) increases significantly (1419 m2 g−1). The combination of monomer 1 and monomer 2 provided hetero-polymerized PAE 1-2 with a BET surface area of 1088 m2 g−1, which lies in between the surface areas of homo-polymerized PAE 1-1 and PAE 2-2. Pore size distribution profiles (Fig. 3b) show that all three polymers have high proportions of micropores between 1.2 and 1.4 nm. We found that the increase of monomer sizes does not enlarge the pore sizes of porous materials, which is likely due to the incomplete reaction of propynyl end groups, as well as possible framework interpenetration issues.
| PAEs | SABETa | V Total | Pore sizec |
|---|---|---|---|
|
a Surface area (m2 g−1) calculated from the nitrogen adsorption based on the BET models.
b The total pore volumes (cm3 g−1) calculated at P/P0 = 0.99.
c Calculated pore sizes (nm) from nitrogen adsorption isotherms using the NLDFT N2-carbon adsorption branch kernel at 77 K based on a slit/cylindrical pore model.
d The molar ratio of monomers 1 and 2 is 1:1.
e The molar ratio of monomers 1/2 and 3 is 1.3:1.
|
|||
| PAE 1-1 | 736 | 1.20 | 1.3 |
| PAE 2-2 | 1419 | 2.40 | 1.1 |
| PAE 1-2 | 1088 | 2.37 | 1.2 |
| PAE 3-3 | 2294 | 1.54 | 1.2 and 3.4 |
| PAE 4-4 | 1200 | 1.02 | 1.1 and 3.4 |
| PAE 1-3 | 1588 | 1.31 | 1.4 and 3.1 |
| PAE 2-3 | 1188 | 1.14 | 1.4 and 3.4 |
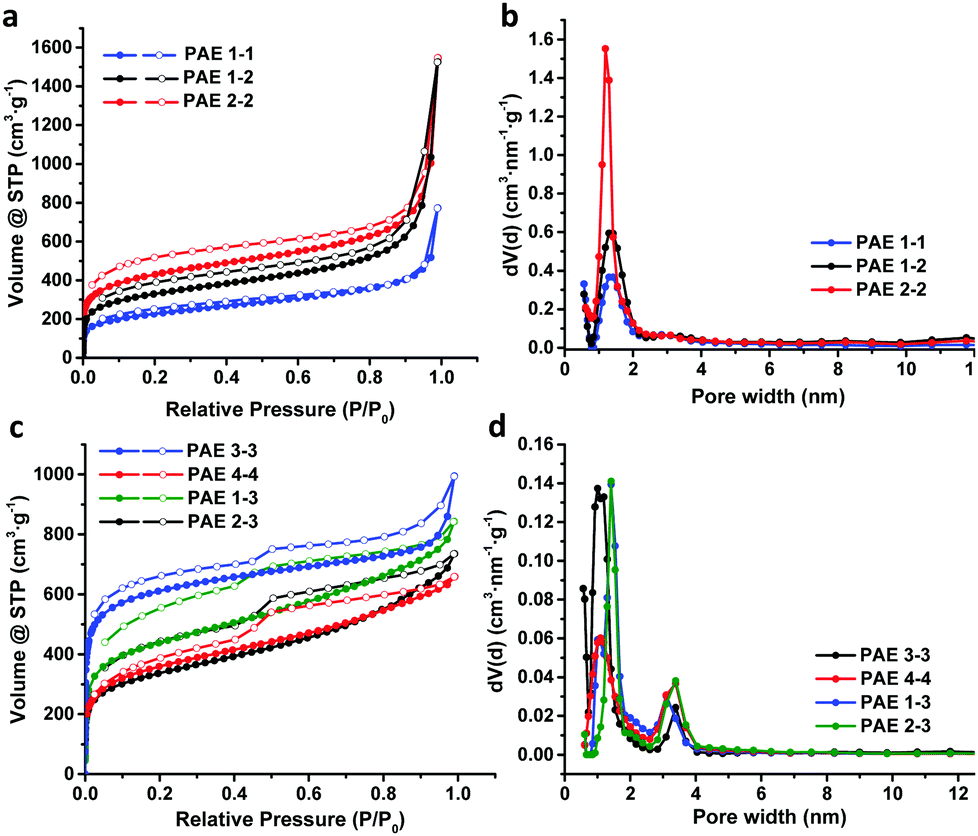 | ||
| Fig. 3 Gas adsorption isotherms and pore size distributions of various PAEs. | ||
When tetrahedral-shaped compound 3 was used as the monomer, the resulting polymer exhibits a BET surface area of 2294 m2 g−1, almost 3-fold that of PAE 1-1. Having one more ethynylene linkage between two phenyl rings compared to the record-holding porous polymer that was prepared from tetrakis(4-bromophenyl)methane through the Yamamoto-type Ullmann cross-coupling reaction,9 the surface area of PAE 3-3 is almost decreased to half, from 5600 m2 g−1 to 2294 m2 g−1. When we further extend the monomer structure by replacing the central carbon of monomer 3 with adamantine moieties, the resulting polymer PAE 4-4 shows a further decreased BET surface area of 1200 m2 g−1, which is in line with the computational prediction reported by Ben et al.9 Nitrogen adsorption isotherms of both PAE 3-3 and 4-4 at 77 K show a rapid uptake at low relative pressure (P/P0 below 0.1) with hysteresis loops at higher pressure, indicating the presence of considerable mesopores. The pore size distribution profiles (Fig. 3d) clearly show the decent proportions of mesopores at ∼3 nm. With the extension of the monomers (PAE 3-3vs.PAE 4-4), the ratio of mesopores is increased. When 2D monomers and 3D monomers were polymerized together through alkyne metathesis, the resulting hetero-polymerized PAE 1-3 and PAE 2-3 exhibit much decreased surface areas compared to the PAE 3-3 consisting of solely 3D monomers. It is interesting to note that introduction of 2D moieties into the 3D polymer networks increased the size of the micropores (1.2 nm in PAE 3-3vs. 1.4 nm in PAE 1-3 and 2-3).
Provided the high surface area and hydrophobicity of PAEs, we next explored their possible application in adsorption of toxic aromatic solvents. We examined the adsorption capacities of PAE 2-2 and PAE 3-3 towards commonly used aromatic solvents: benzene, toluene, o-xylene, mesitylene, aniline, and nitrobenzene. We also examined the adsorption capacity of PAEs toward non-aromatic solvent (decane) and water for comparison purposes. The adsorption capacities were determined by immersing the tips of pipettes loaded with the pre-weighed porous samples until saturation, and calculating the sample weight difference before and after the absorption. Both PAEs adsorb significant amounts of aromatic solvents and adsorption capacities up to 723 wt% were observed for nitrobenzene. As shown in Fig. 4, PAE 3-3 with higher surface area adsorbs considerably larger amounts of organic solvents compared to PAE 2-2. The uptakes of aromatic solvents in PAEs decrease with the increase of structure bulkiness and are less influenced by the polarity of the solvents. The order of adsorption mol% in PAE 3-3 follows the trend of mesitylene < o-xylene < nitrobenzene < aniline < toluene < benzene. In PAE 2-2, bulky mesitylene is also the least adsorbed. In both PAEs, the adsorption mol% of non-aromatic solvent decane was the least, indicating that aromatic-rich PAEs have more favorable interactions with aromatic adsorbates than aliphatic hydrocarbons. Due to the hydrophobicity of PAEs, a negligible amount of water was adsorbed in PAEs (<1%). Therefore, our study suggests the potential application of these PAEs in the removal of organic pollutants from waste water or water streams.
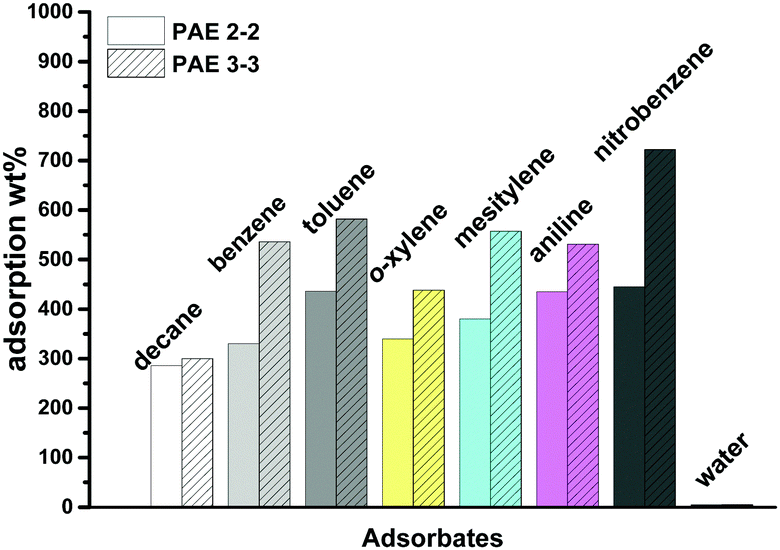 | ||
| Fig. 4 Adsorption capacities of PAE 2-2 and PAE 3-3 toward common organic solvents and water. | ||
Conclusions
In summary, we have successfully synthesized purely hydrocarbon-based polymer networks through alkyne metathesis. The poly(aryleneethynylene)s exhibit porous properties, showing a BET surface area of up to 2294 m2 g−1. We also studied the effect of monomer sizes and geometries on PAE's surface area and pore size distribution. Extension of 2D monomer structures significantly increased the surface area of the resulting polymer, whereas increasing the size of the 3D monomers led to the decrease of the surface area of the resulting polymer. Due to the amorphous nature of PAEs, their pore size distributions do not follow an obvious trend. In all polymers, we observed micropores in the range of 1.2–1.4 nm. When 3D monomers are involved, the PAEs show a considerable portion of mesopores around 3.4 nm. Given their hydrophobicity and porosity, the PAEs adsorb significant amounts of aromatic solvents, e.g. adsorption of 723 wt% of nitrobenzene, thus suggesting their possible applications in absorption and separation of grease and aromatic compounds from an oil–water mixture.Acknowledgements
We thank Dr. Richard Shoemaker for solid state NMR assistance and the National Science Foundation (DMR-1055705) for financial support.References
- E. Stockel, X. F. Wu, A. Trewin, C. D. Wood, R. Clowes, N. L. Campbell, J. T. A. Jones, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Chem. Commun., 2009, 212–214 RSC.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- J. L. Novotney and W. R. Dichtel, ACS Macro Lett., 2013, 2, 423–426 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Q. Ma, D. P. Cao, J. H. Lan, X. F. Jing, W. C. Wang, J. Xu, F. Deng, J. M. Simmons, S. L. Qiu and G. S. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- R. R. Schrock and C. Czekelius, Adv. Synth. Catal., 2007, 349, 55–77 CrossRef CAS.
- R. R. Schrock, Adv. Synth. Catal., 2007, 349, 25 CrossRef CAS.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- K. Jyothish and W. Zhang, Angew. Chem., Int. Ed., 2011, 50, 8478–8480 CrossRef CAS PubMed.
- A. Mortreux and M. Blanchard, Chem. Commun., 1974, 786–787 RSC.
- J. H. Wengrovius, J. Sancho and R. R. Schrock, J. Am. Chem. Soc., 1981, 103, 3932–3934 CrossRef CAS.
- D. Villemin, M. Héroux and V. Blot, Tetrahedron Lett., 2001, 42, 3701–3703 CrossRef CAS.
- V. Huc, R. Weihofen, I. Martin-Jimenez, P. Oulie, C. Lepetit, G. Lavigne and R. Chauvin, New J. Chem., 2003, 27, 1412–1414 RSC.
- A. Fürstner and P. W. Davies, Chem. Commun., 2005, 2307–2320 RSC.
- V. Maraval, C. Lepetit, A.-M. Caminade, J.-P. Majoral and R. Chauvin, Tetrahedron Lett., 2006, 47, 2155–2159 CrossRef CAS.
- S. Lysenko, B. Haberlag, C. G. Daniliuc, P. G. Jones and M. Tamm, ChemCatChem, 2011, 3, 115–118 CrossRef CAS.
- P. Persich, J. Llaveria, R. Lhermet, T. de Haro, R. Stade, A. Kondoh and A. Fürstner, Chem. – Eur. J., 2013, 19, 13047–13058 CrossRef CAS PubMed.
- A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 2794–2819 CrossRef PubMed.
- O. S. Miljanic, K. P. C. Vollhardt and G. D. Whitener, Synlett, 2003, 29–34 CAS.
- W. Zhang and J. S. Moore, J. Am. Chem. Soc., 2004, 126, 12796 CrossRef CAS PubMed.
- W. Zhang and J. S. Moore, Angew. Chem., Int. Ed., 2006, 45, 4416–4439 CrossRef CAS PubMed.
- U. H. F. Bunz, Acc. Chem. Res., 2001, 34, 998–1010 CrossRef CAS PubMed.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605–1644 CrossRef CAS PubMed.
- F. R. Fischer and C. Nuckolls, Angew. Chem., Int. Ed., 2010, 49, 7257–7260 CrossRef CAS PubMed.
- D. F. Sedbrook, D. W. Paley, M. L. Steigerwald, C. Nuckolls and F. R. Fischer, Macromolecules, 2012, 45, 5040–5044 CrossRef CAS.
- H. Yang, Y. Jin, Y. Du and W. Zhang, J. Mater. Chem. A, 2014, 2, 5986–5993 CAS.
- Y. Du, H. Yang, C. Zhu, M. Ortiz, K. D. Okochi, R. Shoemaker, Y. Jin and W. Zhang, Chem. – Eur. J., 2016, 22, 7959–7963 CrossRef CAS PubMed.
- H. Yang, Z. Liu and W. Zhang, Adv. Synth. Catal., 2013, 355, 885–890 CrossRef CAS.
- K. Jyothish, Q. Wang and W. Zhang, Adv. Synth. Catal., 2012, 354, 2073–2078 CrossRef CAS.
- Q. Wang, C. Zhang, B. C. Noll, H. Long, Y. Jin and W. Zhang, Angew. Chem., Int. Ed., 2014, 53, 10663–10667 CrossRef CAS PubMed.
- Q. Wang, C. Yu, H. Long, Y. Du, Y. H. Jin and W. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7550–7554 CrossRef CAS PubMed.
- K. Hu, H. Yang, W. Zhang and Y. Qin, Chem. Sci., 2013, 4, 3649–3653 RSC.
- Y. Zhu, H. Yang, Y. Jin and W. Zhang, Chem. Mater., 2013, 25, 3718–3723 CrossRef CAS.
- W. Zhang and J. S. Moore, Adv. Synth. Catal., 2007, 349, 93–120 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00359a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2017 |