
Green light-emitting 2-(1H-indol-3-yl)acetonitrile-based D–A fluorophores – a combined theoretical and experimental study†
Subramanian
Muruganantham
a,
Natarajan
Nagarajan
a,
Gunasekaran
Velmurugan
ab,
Asit
Prakash
c,
Monica
Katiyar
*c,
Ponnambalam
Venuvanalingam
*a and
Rajalingam
Renganathan
*a
aSchool of Chemistry, Bharathidasan University, Tiruchirappalli-620 024, Tamil Nadu, India. E-mail: rrengas@gmail.com; Fax: +91-431-2407045; Tel: +91-431-2407053
bDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai, Maharashtra-400 076, India
cDepartment of Materials Science & Engineering and Samtel Centre for Display Technologies Indian Institute of Technology Kanpur, Kanpur-208016, India
First published on 6th February 2017
Abstract
A series of new fluorophores based on 2-(1H-indol-3-yl)acetonitrile incorporated with various polycyclic aromatic hydrocarbons, carbazole, pyridine, and triphenylamine was designed and synthesised. The optical, electrochemical, thermal, and morphological properties of the fluorophores were studied using various spectroscopic techniques. The demonstration of electroluminescence behaviour was carried out by fabricating a single layer device with the device structure of indium tin oxide (ITO – 150 nm)/poly(ethylenedioxythiophene)–polystyrene sulfonate (PEDOT:PSS – 40 nm)/emissive layer (65 nm)/lithium fluoride (LiF – 0.5 nm)/Al (120 nm), which exhibited a turn-on voltage of 6.5 V, maximum brightness of 110 cd m−2 and 133 cd m−2, maximum current efficiency of 0.34 cd A−1 and 0.73 cd A−1, and power efficiency of 0.10 lm W−1 and 0.28 lm W−1 for TIN and T2IN, respectively. Density functional theory (DFT) results revealed that the highest occupied molecular orbital (HOMO) is mainly localized on the donor part, whereas the lowest unoccupied molecular orbital (LUMO) is spread over the entire molecule i.e. over the donor, π–spacer, and acceptor units. The higher HOMO and lower LUMO energies of fluorophores reduce the energy barrier for hole creation and electron acceptance processes, respectively. All the fluorophores show a narrow band gap, and the charge injection ability increases with suitable substitution, which leads to improved organic light-emitting diode (OLED) performance.
Introduction
Organic molecules with a fluorescent nature have been attracting the attention of scientists for more than a century. They have good sensitivity, excellent specificity, and a large linear range for analysis1,2 both in solid form and in liquid solution. Current scientific researchers have extensively used these materials in different optoelectronic applications,3–5 such as organic light-emitting diodes (OLEDs),6 and in the wide range of full-color flat-panel display as novel organic donor–acceptor compounds, which are enlarge newer emitters with thermally activated delayed fluorescence (TADF).7 The extensive emitter should have lower ΔE(S − T) such that the process leads to a reversible intersystem crossing and the excitons harvest the singlet state and the excited state. Moreover, the lifetime should be longer in contrast to the normal excited sate fluorescence lifetime of organic fluorescent sensors,8 organic light-emitting field effect transistors (OLEFETs),9 organic solid-state lasers,10etc. The materials that display efficient optical response are required for all these applications.11 Often, materials that are strongly fluorescing in liquid solution become non-fluorescent in the solid form and vice versa. Therefore, materials that fluoresce in both forms have a wide range of applications in all the abovementioned fields. Recently, new developments in the highly emissive organic light-emitting diodes have provoked concerted research in the scientific and industrial communities. In the past two decades, active research is ongoing owing to the wide applicability of these materials in flexible full-color flat-panel displays, solid-light emitters, and low-cost high-potential light applications.12 The essential criteria for a good organic emitter are its morphology, thermal stability, and emission in the solid state.13To date, appreciable functionalized materials, such as dendrimers,14 starbursts,15,16 nanocomposites,17 and spiro compounds,18 have been well-established as light-emitting materials. Moreover, organic π-conjugated molecules with donor–acceptor architecture,19 such as triphenylamine,20,21 carbazole,22,23 naphthalene, anthracene,24 and pyrene25,26 with a non-planar and twisted framework, have attracted a new strategy for optical and optoelectronics materials. These molecules possess strong fluorescence and π–π interactions that are favourable to enhance the charge carrier/hole transport ability, which is required for the ingenious application in OLEDs.27,28 In this regard, donor–acceptor π-conjugated small molecules with good structure and optical properties are favourable for efficient light-emitting devices.29,30
As part of a broader project aimed at the development of fluorophores for OLEDs, we investigated the 2-(1H-indol-3-yl)acetonitrile scaffold for the construction of donor–acceptor (D–A) systems. N-Heterocycles are commonly found as structural motifs in many materials such as biologically active natural products and pharmaceutical intermediates. In particular, 3-functionalised indoles are versatile intermediates in the synthesis of alkaloids and several heterocycles. They have been found to exhibit various pharmaceutical activities, such as anticancer, antidiabetic, and antinociceptive, and inhibit HIV-1 integrase. Marketed drugs include pravadoline (analgesic) and ramosetron (antiemetic). Recently, Chowdhury et al.31 studied white-light generation due to proton transfer and the sensing efficiency of some substituted indole groups of molecules such as indole, indole-3-carboxaldehyde, and indole-7-carboxaldehyde in the solid (powder) form and in liquid solution. However, a negligible amount of optical, electrochemical, and device data is available for 2-(1H-indol-3-yl) acetonitrile, and this encouraged us to study the synthesis and photophysical investigation of substituted 2-(1H-indol-3-yl) acetonitrile derivatives. Since the optoelectronic applications of arylamine, carbazole, polyaromatic hydrocarbons, and pyridine moieties are already well-known, we used these groups as donor moieties to design new fluorophores. Considering all these precedents, we designed and synthesized seven D–A type fluorophores based on 2-(1H-indol-3-yl)acetonitrile as the acceptor and pyrene, anthracene, naphthalene, carbazole, pyridine, and triphenylamine as the donor moieties. The photophysical, electrochemical, thermal, and morphological properties were explored. To the best of our knowledge, this is the first report on the sequential screening of this type of D–A fluorophores. Theoretical calculations were also performed using density functional theory (DFT) to obtain information about the geometric and electronic properties of these new fluorophores. In addition, the electroluminescence (EL) properties were explored using these compounds as the emitting layer in OLEDs.
General information
Experimental methods
| Φf = ΦRISODRηS2/IRODSηR2 | (1) |
Fabrication of the OLED device
Pre-coated indium tin oxide (ITO) on glass substrates with a sheet resistance of 15–20 Ω cm−2 was used to pre-pattern the glass substrates by a photolithography technique to provide an effective device area of 4 mm × 4 mm. The pretreatment of the ITO glass substrates included cleaning using ethanol, acetone, and detergent via ultrasonication for 0.5 h, spraying with deionised water for 10 min, and drying with nitrogen. Finally, the substrates were treated with UV ozone for 10 min. All the organic layers were sublimated by thermal evaporation in an oven at 1.0 Å s−1 at a pressure of ca. 3.5 × 10−4 Pa. The rate of deposition and thickness were measured using a quartz control monitor placed in a substrate holder. (For the device structure, the substrate ITO acted as an anode (150 nm), poly(3,4-ethylene-dioxythiophene)–poly(styrenesulfonate) PEDOT:PSS as a hole-injecting layer (HIL) (about 40 nm), the emissive layer (EML, 65 nm), LiF (0.5 nm) as a electron injection layer (EIL), and Al layer (120 nm) as the cathode). We fabricated the device structure as ITO/HIL (40 nm)/EML (65 nm)/Li/F. The current density–voltage–luminance (J–V–L) and current density–voltage (I–V) characteristics were measured. The optical performance, such as EL spectra and CIE color coordinates, and the progression of the fabricated devices were measured using a CS-1000 spectroradiometer (Konica Minolta) and a Keithley 2400 source meter interfaced with a computer in a dark place. All the measurements were carried out at room temperature under ambient conditions.Computational details
All the calculations on the synthesized molecules were performed using Gaussian 09 code.33 The ground-state geometries of the molecules were fully optimized at the DFT level at B3LYP34–40 with the 6-311+G(d,p) basis set. The vibrational frequency analysis was performed to ensure that all the optimized geometries were minima on the potential energy surface.Synthesis
Synthesis of (E)-2-(1H-indol-3-yl)-3-(alkenyl) acrylonitrile (NIN). A mixture of the aromatic aldehyde 1a (1 mmol) and 2-(1H-indol-3-yl)acetonitrile 2 (1.1 mmol) was dissolved in 10 mL of 10% ethanolic NaOH. The mixture was stirred at room temperature for 1–2 h under nitrogen atmosphere. The progress of the reaction was monitored by TLC. After the completion of the reaction, the solvent was evaporated under reduced pressure. The residue was poured onto crushed ice under vigorous stirring. The obtained precipitate was filtered and dried in a vacuum oven. The crude product was purified by column chromatography (SiO2; EtOAc: hexane, 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 v/v) to afford 3.
:9 v/v) to afford 3.
Yield: 80.12%; m.p. = 167–168 °C; 1H NMR (DMSO-d6, Me4Si, 400 MHz) δ 7.36–7.29 (m, 2H), 7.48 (d, J = 8 Hz, 1H), 7.61–7.54 (m, 3H), 7.68 (d, J = 2.8Hz, 1H), 7.92 (d, J = 8.8 Hz, 2H), 8.10–8.05 (m, 3H), 8.32 (s), 8.49 (brs, 1H), ppm. 13C NMR (100 MHz, DMSO-d6) δ 115.1, 116.3, 117.5, 123.1, 124.3, 125.8, 127.6, 128.6, 129.0, 130.3, 131.2, 131.4, 131.6, 131.7, 133.6, 134.4, 136.2, 137.0, 138.3, 138.9, 142.5 ppm. HRMS (ESI) m/z calcd for C21H14N2: 294.1157 [M + Na+]; found 317.1054.
(E)-2-(1H-Indol-3-yl)-3-(alkenyl) acrylonitrile (AIN). Yield: 78.5%; m.p. = 209–210 °C; 1H NMR (DMSO-d6, Me4Si, 400 MHz) δ 7.34–7.15 (m, 2H), 7.54–7.5 (m, 5H), 7.89 (d, J = 2.8 Hz 1H), 8.15–8.09 (m, 5H), 8.51 (s, 1H), 8.66 (s, 1H), 11.78 (brs, 1H), ppm. 13C NMR (100 MHz, DMSO-d6) δ 103.6, 109.9, 111.7, 112.5, 114.9, 117.2, 119.0, 120.0, 121.0, 122.0, 123.0, 124.0, 125.0, 125.4, 125.6, 126.0, 126.5, 127.0, 127.03, 128.0, 129.0, 129.1, 129.4, 131.0, 134.07, 137.3 ppm. HRMS (ESI) m/z calcd for C25H16N2: 345.1392 [M + H+]; found 345.1391.
(E)-2-(1H-Indol-3-yl)-3-(alkenyl) acrylonitrile (PY2IN). Yield: 72.3%; m.p. = 281–283 °C; 1H NMR (DMSO-d6, Me4Si, 400 MHz) δ 7.22 (t, J = 9.2 4H), 7.52 (d, J = 6.8 Hz 2H), 7.86 (s, 4H), 8.10 (s, 9H), 8.42 (d, J = 8.0 Hz 4H), 11.80 (brs, 2H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 106. 2, 110.8, 112.5, 118.5, 119.4, 119.6, 120.6, 122.5, 123.7, 126.9, 126.9, 129.0, 135.4, 135.7, 137.2, 138.4, 139.0, 154.8 ppm. HRMS (ESI) m/z calcd for C39H25N5: 563.211 [M + H+]; found 564.2189.
(E)-2-(1H-Indol-3-yl)-3-(alkenyl) acrylonitrile (TIN). Yield: 69.2%; m.p. = 174–176 °C; 1H NMR (DMSO-d6, Me4Si, 400 MHz) δ 6.97 (d, J = 8.4 Hz 3H), 7.10–7.06 (m, 5H), 7.29–7.13 (m, 2H), 7.36–7.32 (4H), 7.49 (J = 8.0 Hz 1H), 7.63 (s, 1H), 7.73 (s, 1H), 7.81 (s, J = 8.4 Hz, 2H), 8.01 (d, J = 7.6 Hz, 1H), 11.80 (brs, 1H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 79.14, 102.7, 111.0, 112.4, 119.0, 119.4, 120.3, 121.1, 122.3, 123.7, 124.0, 124.8, 125.0, 125.8, 127.7, 129.7, 129.8, 136.4, 137.1, 146.4, 148.2 ppm. HRMS (ESI) m/z calcd for C29H21N3: 412.1816 [M + H+]; found 412.1813.
(E)-2-(1H-Indol-3-yl)-3-(alkenyl) acrylonitrile (PYRIN). Yield: 77.5% m.p. = 214–215 °C; 1H NMR (DMSO-d6, Me4Si, 400 MHz) δ 7.27 (s, 2H), 7.56 (d, J = 7.4 1H), 8.0 (s, 1H), 8.55–8.13 (m, 10H), 8.7 (s, 1H), 11.8 (brs, 1H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 106.2, 110.8, 112.5, 118.5, 119.4, 119.6, 120.6, 122.5, 123.7, 126.9, 127.0, 129.0, 135.4, 135.7, 137.2, 138.4, 139.0, 154.8 ppm. HRMS (ESI) m/z calcd for C27H16N2: 369.1391 [M + H+]; found 369.1392.
(E)-2-(1H-Indol-3-yl)-3-(alkenyl) acrylonitrile (CIN). Yield: 72.0%; m.p. = 115–116 °C; 1H NMR (DMSO-d6, Me4Si, 400 MHz) δ 1.3 (s, 3H), 4.5 (d, J = 6.4 2H) 7.26–7.16 (m, 3H), 7.54–7.5 (m, 3H), 7.62 (d, J = 8.0 Hz 1H), 7.71 (d, J = 8.0 Hz 1H), 7.8 (s, 1H), 7.9 (s, 1H), 8.20–8.12 (m, 3H), 8.7 (s, 1H), 11.7 (brs, 1H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 13.7, 37.15, 102.11, 109.42, 109.51, 111.18, 112.4, 119.4, 120.2, 120.3, 121.6, 122.1, 122.3, 122.4, 123.8, 125.5, 125.6, 126.0, 126.3, 137.1, 138.4, 140.0, 140.07 ppm. HRMS (ESI) m/z calcd for C25H19N3: 362.1657 [M + H+]; found 362.1657.
(E)-2-(1H-Indol-3-yl)-3-(alkenyl) acrylonitrile (T2IN). Yield: 68.0%; m.p. = 174.2–176 °C; 1H NMR (CDCl3, Me4Si, 400 MHz) δ 6.77 (J = 8.0 Hz, d, 1H), 6.95–7.03 (m, 4H), 7.05–7.12 (m, 5H), 7.15–7.25 (m, 5H), 7.27–7.40 (m, 4H), 7.45–7.48 (m, 2H), 7.64–7.73 (m, 3H), 7.86–7.91 (m, 1H), 8.46 (br, 2H) ppm. 13C NMR (CDCl3, 100 MHz) δ 102.7, 111.0, 112.3, 118.9, 119.4, 120.2, 121.0, 122.3, 123.7, 124.0, 124.8, 125.0, 125.8, 127.7, 129.6, 129.7, 129.8, 136.4, 137.1, 146.4, 148.2 ppm. HRMS (ESI) m/z calcd for C40H27N5: 578.2344 [M + H+]; found 578.2343.
Results and discussion
Synthesis and characterisation
The chemical structures of the series of (2-(1H-indol-3-yl)acetonitrile)-based fluorophores synthesised herein are shown in Scheme 1, and their corresponding synthetic routes are shown in Scheme 2. Carbazole, triphenylamine, and pyridine aldehydes were prepared via Vilsmeier–Haack formylation,41 and they were isolated and purified by column chromatography. Finally, the desired product was synthesised via the Knoevenagel condensation42 of various aldehydes with the active methylene (2-(1H-indol-3-yl)acetonitrile) compound as per the literature procedure. The synthesized compounds were obtained as yellowish green solids and were characterised via1H NMR, 13C NMR, and mass spectroscopies. The synthetic procedures and characterisation data are summarized in the Experimental section.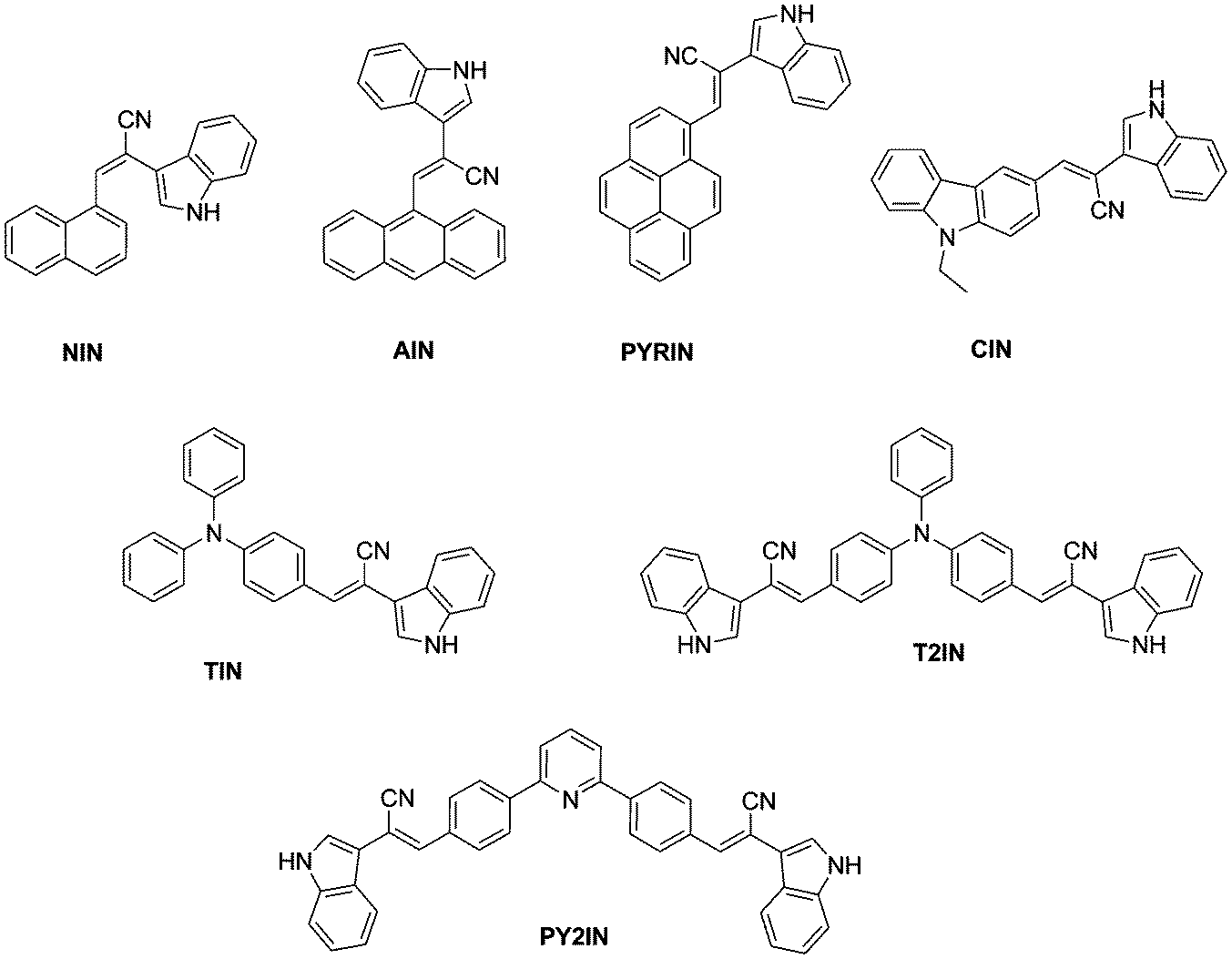 | ||
| Scheme 1 Chemical structures of the compounds synthesized herein. | ||
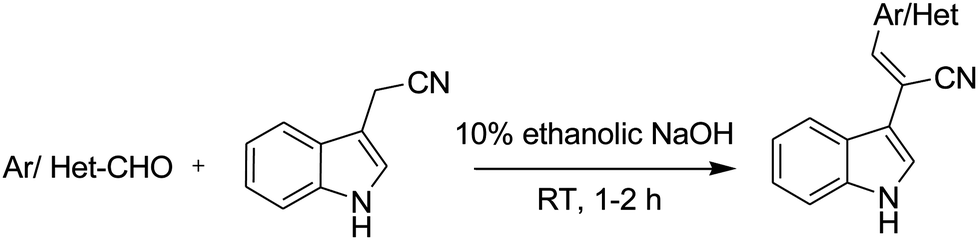 | ||
| Scheme 2 Synthetic route for all the derivatives. | ||
Electronic spectra
All the fluorophores were yellowish green solids and were readily soluble in common organic solvents. More or less similar absorption spectral patterns were observed for all the compounds studied herein (Fig. 1). A representative absorption spectrum of all the derivatives in a THF solution is depicted in Fig. S1 and S3–S9 (ESI†) illustrate the spectra of the derivatives in solvents with different polarities. The detailed photophysical data are presented in Table 1. The optical band-gaps of PY2IN, NIN, PYRIN, TIN, T2IN, CIN, and AIN in the THF solution were evaluated to be 2.78, 2.66, 2.58, 2.57, and 2.56 eV, respectively, from the onset of absorption. The optical band gaps of CIN, T2IN, and AIN are less compared to those of other derivatives.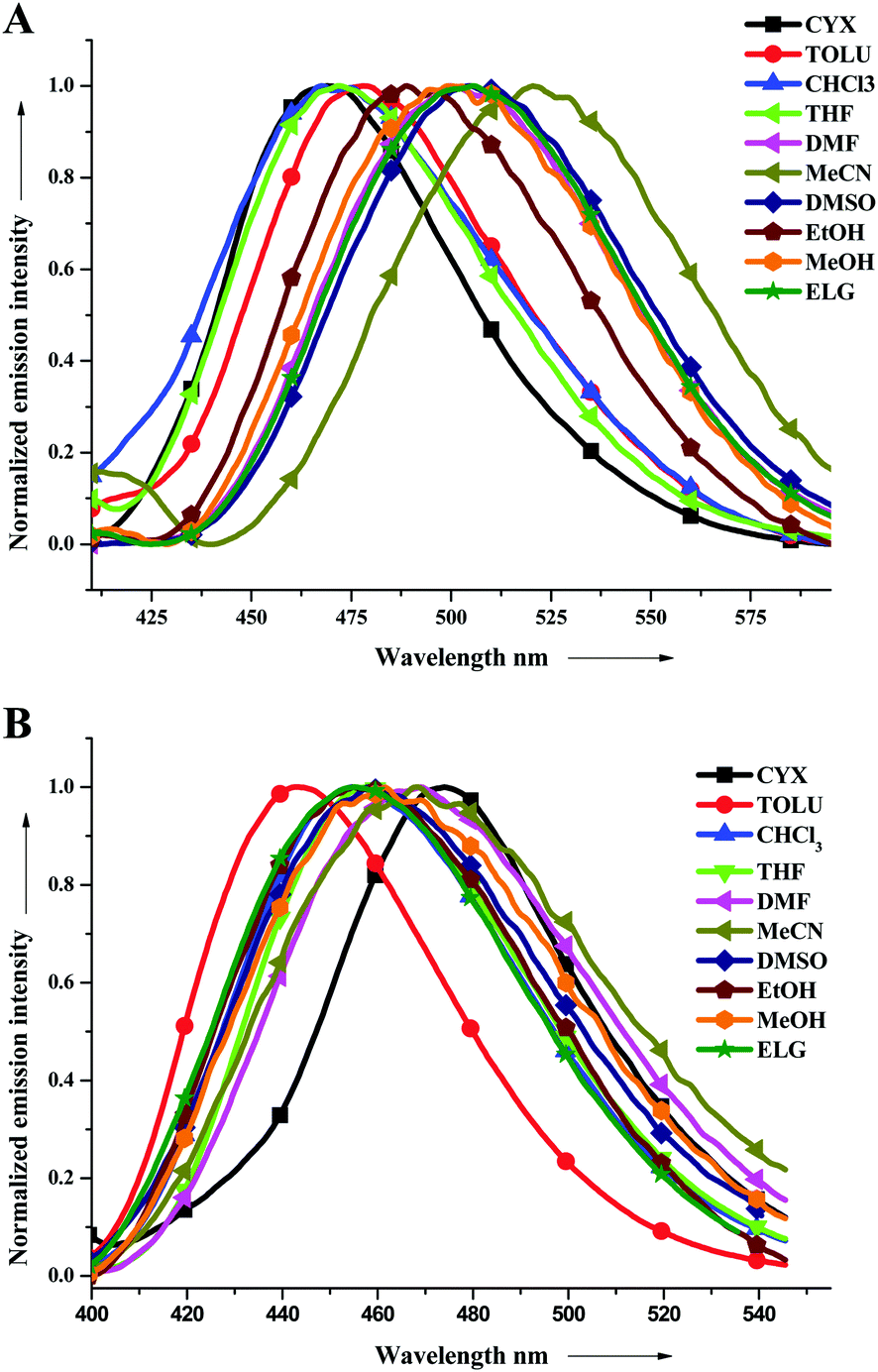 | ||
| Fig. 1 Emission spectra of TIN (A) and T2IN (B) in all the solvents studied herein. | ||
| Solvent | λ abs [nm] | λ emi/Φfc [nm/%] | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AIN | NIN | PYRIN | CIN | TIN | T2IN | PY2IN | AIN | NIN | PYRIN | CIN | TIN | T2IN | PY2IN | |
| a Absorption and emission measured for various solvents. b In thin films for TINT2IN, and PY2IN. c Fluorescence quantum yield (Φf) | ||||||||||||||
| CYX | 390 | 349 | 389 | 369 | 391 | 370 | 370 | 515 | 454 | 470 | 455 | 469 | 474 | 450 |
| TOLU | 392 | 359 | 389 | 395 | 400 | 419 | 379 | 517 | 464 | 484 | 468 | 478 | 443 | 462 |
| CHCl3 | 389 | 354 | 396 | 400 | 401 | 422 | 378 | 441 | 469 | 464/4.0 | 510/22.9 | 468/10.8 | 454/13.9 | 472/49.5 |
| THF | 389 | 359 | 394 | 396 | 397 | 418 | 382 | — | 474 | 494 | 479 | 480 | 457 | 479 |
| DMF | 391 | 360 | 400 | 400 | 402 | 423 | 386 | 555 | 462 | 472 | 514 | 500 | 468 | 504 |
| MeCN | 388 | 352 | 390 | 395 | 395 | 416 | 377 | 440 | 486 | 499/3.7 | 524/3.5 | 510/2.6 | 468/1.1 | 497/7.2 |
| DMSO | 392 | 364 | 401 | 404 | 404 | 426 | 390 | 436 | 435 | 484/17.5 | 520 | 505/8.4 | 458/2.7 | 494/20.3 |
| EtOH | 390 | 360 | 399 | 400 | 399 | 421 | 381 | — | 485 | 499 | 560 | 486 | 455 | 494 |
| MeOH | 387 | 357 | 394 | 400 | 398 | 418 | 380 | 403 | 495 | 499/3.8 | 572/14.8 | 495/2.5 | 461/1.4 | 502/17.8 |
| ELG | 390 | 365 | 402 | 404 | 405 | 426 | 388 | 518 | 489 | 510/16.0 | 566/34.8 | 504/34.8 | 456/18.3 | 500/34.9 |
| Thin filmb | — | — | — | — | 410 | 430 | 381 | — | — | — | — | 532 | 528 | 497 |
From the spectra, the shorter wavelength band at 354–380 nm is attributed to the π–π* transition from the conjugated aromatic ring systems, and the longer wavelength band at 382–426 nm is attributed to the intramolecular charge transfer between the donor and acceptor units. Compared to the shorter wavelength band, the longer wavelength band is very sensitive to the solvent environment. This has been exemplified in the case of T2IN (Table 1) that exhibited a significant bathochromic shift of 56 nm while moving from the less polar cyclohexane to more polar DMSO and ELG. The effects of solvent polarity in the polyaromatic systems, such as AIN, NIN, and PYRIN, are very less. i.e., the shift is 2 nm, 18 nm, and 13 nm, respectively. However, a strong donor group, such as a carbazole or triphenylamine group containing fluorophores (CIN, T2IN), has a very high solvent effect, leading to 35 nm or 56 nm red shifts, respectively.
Photoluminescence spectra
Photoluminescence (PL) spectroscopy was performed to examine the emission behaviour of electronically excited molecules in various solvents, and the emission spectra are displayed in Fig. 1 (for TIN and T2IN) and the spectra of other related derivatives are shown in Fig. S10–S14 (ESI†). The observed emission behaviour is independent of the excitation wavelength. All the compounds showed emissions in the range of 403–570 nm in various solvent environments. As observed in the ground state electronic spectra, the excited state was also very sensitive to solvent polarity. The emission spectra obtained in less polar solvents showed a blue emission, whereas on tuning the solvent polarity, a higher bathochromic shift (towards red wavelength) was observed (see Fig. 1 and Table 1). The shift from 30 to 70 nm in the emission wavelength observed on tuning the solvent from nonpolar (cyclohexane) to highly polar (DMSO) suggests that the excited state is more polar in nature and very sensitive to different solvent environments. This is due to the presence of various hetero-donor and acceptor groups, leading to a strong intramolecular charge transfer, which is very sensitive to solvent polarity.The fluorescence quantum yields (Φf) for PYRIN, CIN, TIN, T2IN, and PY2IN obtained in various solvents are presented in Table 1 along with their absorption (λmax) and emission (λmax) maxima. The Φf values for PYRIN, CIN, TIN, T2IN, and PY2IN were found in the range of 0.2–0.56 relative to that of perylene (ΦR = 0.94) in cyclohexane, proving that these are better candidates for electroluminescence applications.
Lifetime measurement
The excited state behaviour of the compounds was further analysed via a time-resolved single photon counting technique, and the results are shown in Tables S1–S5 (ESI†) and Fig. 2. The decays of TIN, T2IN, CIN, PYRIN, and PY2IN show good fits with a biexponential function, which deduces the existence of different lifetime components in their excited state. In general, all the compounds exhibit two lifetimes with the major component (60–75%) of a short-lived lifetime that ranged from 1.33 to 3.81 ns. For TIN, the lifetime of the short-lived component ranged from 2.62 to 6.12 ns, whereas that of the long-lived component ranged from 5.0 to 5.76 ns. For T2IN, the corresponding parameters ranged from 2.91 to 5.23 and 5.03 to 5.90 ns, respectively. The long-lived components indicate stabilisation of the excited state due to solvent–solute interaction. The occurrence of two lifetimes reveals different conformations of the excited state with different spin multiplicities. As reported earlier,43 the existence of a quinoidal excited state resonance structure or twisted intermolecular charge-transfer structure due to the electron-withdrawing nitrile moiety leads to different conformations, resulting in two different decay lifetime components.44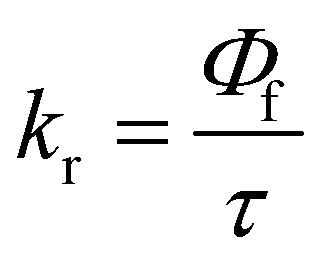 | (2) |
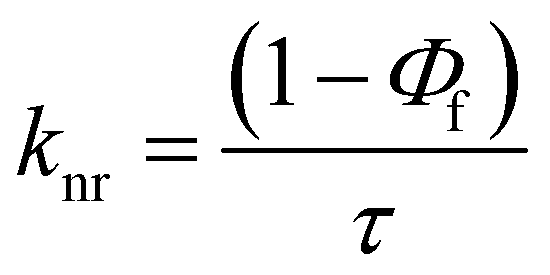 | (3) |
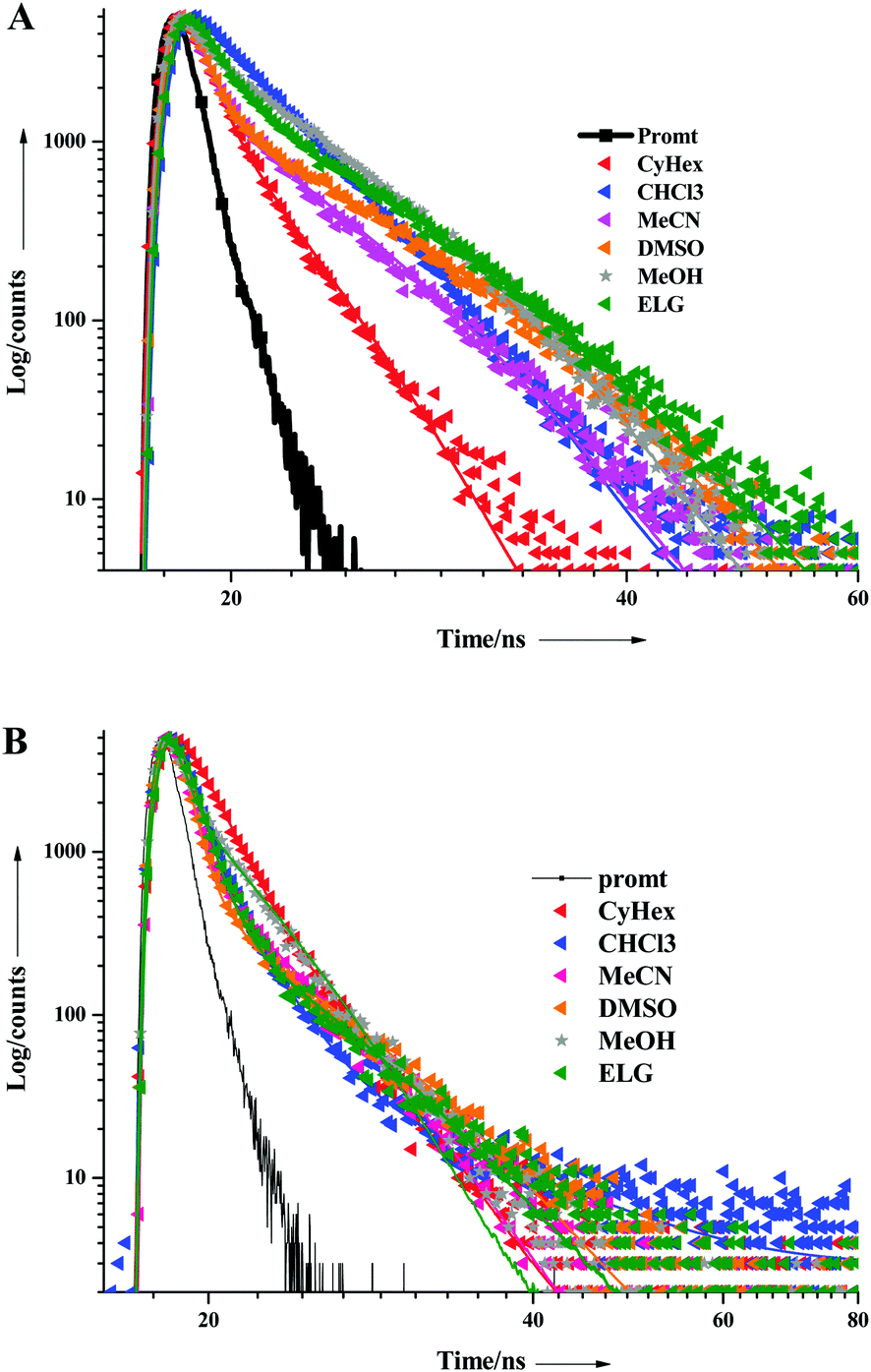 | ||
| Fig. 2 Fluorescence decay curves of (A) TIN and (B) T2IN in all the solvents studied herein. | ||
Electrochemical properties
The electrochemical behaviour of the new fascinating D–π–A molecules was investigated via cyclic voltammetry. Cyclic voltammetry was performed using a three electrode cell setup at the scan rate of 100 mV s−1, as shown in Fig. 3, in a 0.1 M DMF solution with TBAPF6 as the supporting electrolyte, ferrocene (4.8 eV) as the internal standard, and Ag/Ag+ as the reference electrode. For each compound, the band-gap energy values (ΔEg = HOMO–LUMO)46 were determined from the UV-visible absorption onset wavelength. The HOMO energy level of the studied molecules ranges from −5.32 to −5.43 eV and the LUMO energy levels range from −2.65 to −2.85 eV; the pertinent data are displayed in Table 2. The band-gap energy levels of TIN and T2IN are close to each other and to the value obtained from a similar photoluminescence emission study.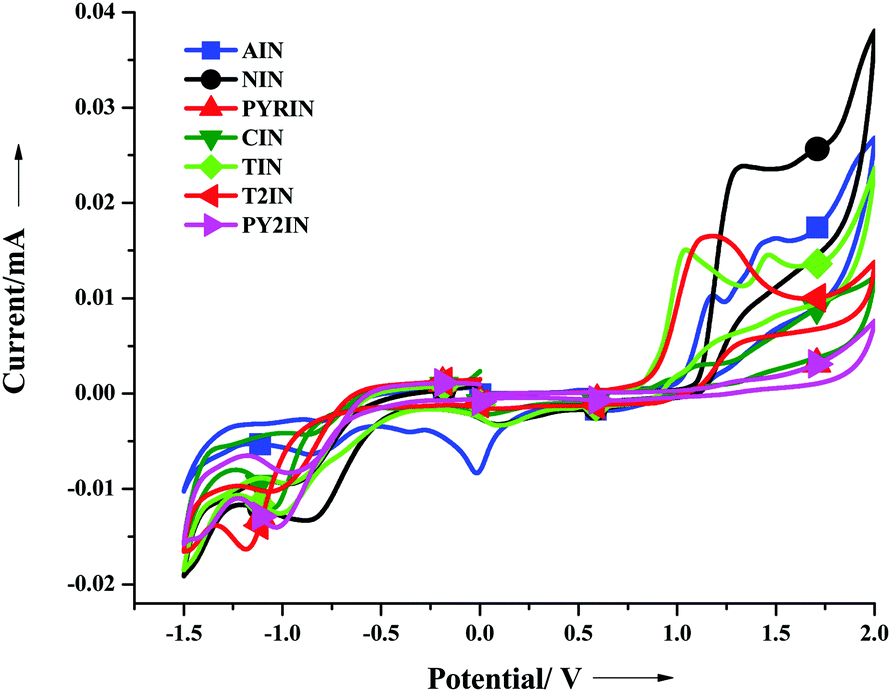 | ||
| Fig. 3 Cyclic voltammograms of all the derivatives. | ||
| Entry | T m | T d [°C] | T g | (εmax, M−1 cm−1 × 10−4) | E Ox (V) | HOMOc [eV] | LUMO [eV] | E o–o [eV] |
|---|---|---|---|---|---|---|---|---|
| a Measured for Tg & Td. b Obtained from CV measurements in DMF solution. c The HOMO and LUMO energies were determined from CV and the absorption onset. Ferrocene (Fc; 4.8 eV) was used as the internal standard in each experiment. The Fc oxidation potential was located at +410 mV, relative to that of the saturated Ag/AgCl reference electrode. Estimated from the onset of the absorption spectra, Eg = (1240/λ onset). | ||||||||
| NIN | 2225.10 | 1.31 | −5.61 | −2.82 | 2.78 | |||
| AIN | 525.22 | 1.18 | −5.48 | −2.91 | 2.56 | |||
| CIN | 2967.35 | 1.04 | −5.34 | −2.75 | 2.58 | |||
| PYRIN | 4134.51 | 1.17 | −5.47 | −2.80 | 2.66 | |||
| TIN | 173 | 240 | 76 | 3525.90 | 1.02 | −5.32 | −2.65 | 2.66 |
| T2IN | 218 | 243 | 77 | 4826.82 | 1.13 | −5.43 | −2.85 | 2.57 |
| PY2IN | 5312.11 | 1.17 | −5.47 | −2.80 | 2.78 | |||
All the compounds exhibit quasi-reversible oxidation and reduction processes. For TIN, we observed two oxidation and reduction peak potentials (see Fig. S19–S24, ESI†). We observed a stable peak for the entire scan range, which indicated considerable stability of the compounds for use in optoelectronic devices.
Thermal properties
The thermal features of the TIN and T2IN derivatives were evaluated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) under nitrogen atmosphere, and the data are depicted in Table 2. Both the derivatives possess good thermal stability. The melting temperatures (Tm) obtained using DSC experiments were 173 and 218 °C for TIN and T2IN, respectively. Moreover, the thermal decomposition temperatures (Td) were 240 and 243 °C for TIN and T2IN, respectively. The onsets of the glass transition temperatures were (Tg) 76 and 77 °C for TIN and T2IN, respectively.47AFM analysis
The morphological properties of the active layer of the thin film were examined by atomic force microscopy (AFM) analysis. The thin films of PY2IN, TIN, and T2IN were fabricated on a quartz substrate via a thermal evaporation method. To assess their thermal and amorphous stability, the film surface was exposed to high thermal stress conditions from room temperature to 100 °C. As seen in Fig. S25 (ESI†), the film morphology was smooth, and the surface-root-mean-square (rms) values were ∼0.7, 1.061, and 2.92 nm for PY2IN, TIN, and T2IN, respectively, and even after an exposure to 100 °C, the RMS values of these compounds remained unchanged (from 1.04 to 3.08 nm), which implied that the thin films are very stable, which is important for their application in electroluminescence devices.Density functional theory analysis
The optimized structures of the eight fluorophores are shown in Fig. S26 (ESI†). Herein, the molecules containing different donor substituents were chosen, and their structural and electronic properties have been discussed in detail. Fig. S27 (ESI†) reveals that the C–C bond lengths in all the fluorophores fall in between their respective single and double bond limits. This confirms that all the C–C bonds present in the molecules are no longer pure single bonds, and the extensive delocalization of the π-electrons is enhanced due to the co-planarity of the benzene rings. Interestingly, the C–N bond lengths were predicted to be ∼1.15 Å, and this typical length suggests that the C–N bond is also involved in the extensive delocalization. Among the studied molecules, T2IN and PY2IN improved the delocalization of their π-electrons due to the increased conjugation. Due to the extensive π-conjugation, all the molecules are expected to be suitable fluorophores for optoelectronic applications.48Frontier molecular orbital (FMO) analysis
To examine the optical and electronic properties of the newly synthesised fluorophores, the HOMOs (highest occupied molecular orbitals), LUMOs (lowest unoccupied molecular orbitals), and their energy gap (HOMO–LUMO gap) were investigated.49 In general, the relative ordering of the molecular orbitals qualitatively indicated the excitation properties and the ability of the molecule for electron/hole transportation in optoelectronic applications. Theoretically, the energy gap is the orbital energy difference between the HOMO and LUMO, termed as the HOMO–LUMO gap, whereas experimental band gaps are obtained from the absorption spectra.50–52 Generally, the lowest transition energy from the ground state to the first dipole allowed excited state is denoted as the optical band gap.53 Thus, a band gap is defined as the energy difference between the ground and first excited state. The HOMO–LUMO gaps and the band gaps were approximately equal when the excitation to the first excited state purely corresponded to the HOMO to LUMO transition. This is generally predicted in most organic fluorophores.43,48,54–57The frontier molecular orbital energy level diagram (from HOMO−3 to LUMO+3) is given in Fig. 4. As seen from the figure, the HOMO–LUMO gap lies over the range from 3.01 to 3.77 eV. The observed band gap is the maximum for NIN and minimum for T2IN. The HOMO values lie in the range from −5.32 to −5.85 eV, whereas the LUMO levels lie in the range from −1.87 to −2.40 eV. The band gaps of AIN, NIN, and PYRIN follow the order PYRIN (3.08 eV) < AIN (3.22 eV) < NIN (3.63 eV), and this is due to the extensive delocalization in the donor part that drastically stabilizes the HOMO level. When comparing T2IN and PY2IN, T2IN is found to have a narrow band gap (3.01 eV), and the triphenyl amine group stabilizes the HOMO, as expected.
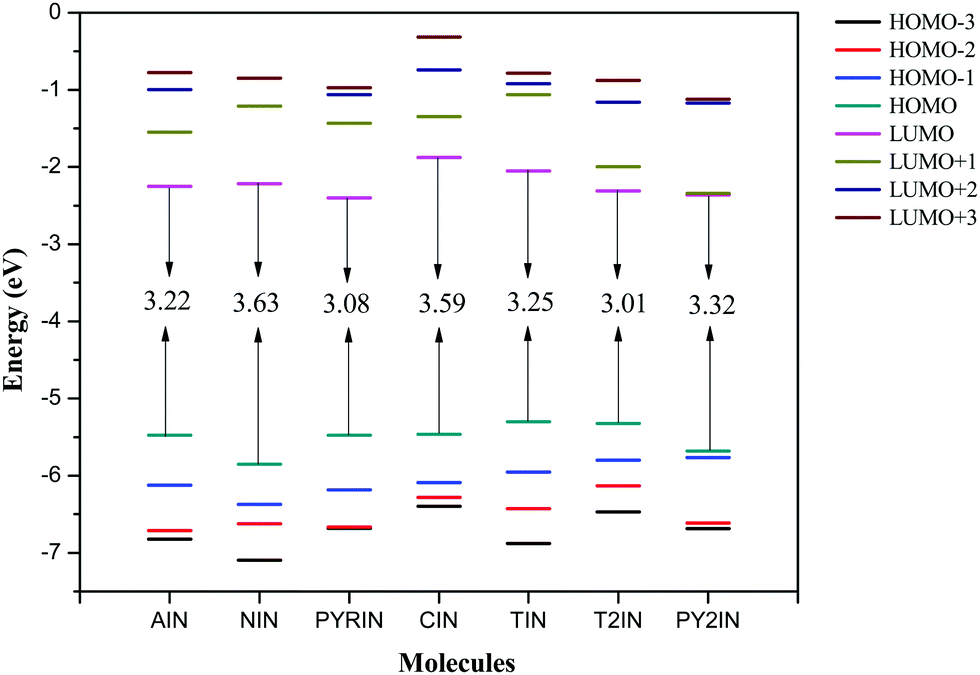 | ||
| Fig. 4 Frontier molecular orbital energy levels of fluorophores computed at the B3LYP/6-311+G(d,p) level. | ||
As is known that a hole transporting material (HTM) with a smaller negative value for the HOMO should more easily lose their electron, whereas an electron transport material (ETM) with a large negative value for the LUMO should more easily accept electrons.53 Therefore, the frontier molecular energies were compared herein. Note that the HOMO energies were found to be higher, i.e., from −5.32 to −5.85 eV, and this largely reduced the energy barrier for the hole creation process. However, the LUMO energies were in the range from −1.84 to −2.40 eV and this reduced the energy barrier for electron acceptance in optoelectronic applications. Interestingly, PYRIN and T2IN show the lowest bang gaps among the new fluorophores and are suitable candidates for improved OLED performance. It was evident that the charge injection ability improves with suitable substitutions.
The energy levels of the fluorophores matched well with the work function of the indium tin oxides (ITO; from −4.8 to −5.1 eV)58 and the LUMO levels of tris(8-hydroxyquinoine)aluminium (Alq3, −1.81 eV).59 We also compared our results with those obtained for BNPB (Mes2B[p-4,4′-biphenyl-NPh(1-napthyl)]), which is widely used in OLEDs as a hole transport material (HTM). The HOMO energy level of our fluorophores (from −5.32 to −5.85 eV) were lower than those of BNPB48,60,61 (−5.27 eV), and this suggests that the selected fluorophores can be used in OLEDs as hole transport materials. The HOMO energies of AIN, PYRIN, CIN, and T2IN were increased by the change in the substituent, and this can be used to fine tune the HOMO energy level to make them efficient hole transport materials. However, the LUMO energy of BNPB (1.88 eV) is slightly higher than that of the studied molecules (from −1.84 to −2.40 eV) and this suggests that all the fluorophores are suitable for electron transport application. Overall, all the reported fluorophores may act as trifunctional materials, such as emitters, hole and electron transporters, in OLED applications.
The frontier molecular orbitals of all the chosen fluorophores are shown in Fig. 5. Note that the HOMO and LOMO are localized throughout the molecule. The electron density distribution of the HOMO is mainly localized on the donor unit rather than on the acceptor and π–spacer. However, the LUMO is spread over the entire molecule: the donor, spacer, and acceptor units. This indicates that the lowest transition has significant π → π* character. To quantify the contributions of the donor, π–spacer, and acceptor towards the HOMO and LUMO, we performed a %MO contribution analysis using QMForge software.62 For this, we segmented the molecules into three fragments, namely, the donor, π–spacer, and acceptor. From Table 3, it is evident that 55–72% of the HOMO is contributed by the donor part and the remaining 38–45% comes from the acceptor (14–30%) and π–spacer (10–20%). On the other hand, 30–57% of the LUMO contribution is from the donor, 19–49% is from the acceptor, and 20–32% is from the π–spacer. Therefore, these findings suggest that the donating group has a significant effect on the band gap as it stabilizes the HOMO, whereas the electron-withdrawing groups stabilizes the LUMO.
 | ||
| Fig. 5 Plots of the frontier molecular orbitals of the studied fluorophores computed at the B3LYP/6-311+G(d,p) level. | ||
| Molecule | HOMO | LUMO | ||||
|---|---|---|---|---|---|---|
| Donor | π–Spacer | Acceptor | Donor | π–Spacer | Acceptor | |
| AIN | 70.25 | 9.75 | 20.00 | 49.82 | 31.57 | 18.61 |
| NIN | 57.16 | 23.86 | 18.98 | 64.9 | 12.23 | 22.87 |
| PYRIN | 61.09 | 8.26 | 30.66 | 56.86 | 18.34 | 24.81 |
| CIN | 58.72 | 24.46 | 16.83 | 44.63 | 16.96 | 38.4 |
| TIN | 67.26 | 11.84 | 20.9 | 30.29 | 20.97 | 48.73 |
| T2IN | 54.98 | 8.15 (5.00) | 18.16 (13.71) | 36.93 | 13.83 (11.14) | 19.93 (18.17) |
| PY2IN | 70.18 | 7.19 (7.19) | 7.72 (7.72) | 59.14 | 9.57 (9.57) | 10.86 (10.86) |
Electroluminescence performance
We investigated the electroluminescence property of TIN and T2IN for a non-doped green OLED. The EL performance was assessed by fabricating a device with the configuration of ITO (150 nm)/PEDOT:PSS (40 nm)/TIN or TI2N (65 nm)/LiF (0.5 nm)/Al (120 nm), where ITO acts as the anode, PEDOT:PSS as the hole-injecting layer, TIN and T2IN as the emissive layers, LiF as the electron-injecting layer, and Al as the anode. Fig. 6 shows the current density–voltage–luminescence (I–V–L) characteristics and EL spectra of the devices. The EL device performance data are summarized in Table 4. The fabricated device with compound TIN had a turn-on voltage or threshold voltage at 6.5 V. The maximum luminescence was 14 V, the maximum brightness reached 110 cd m−2, the maximum current efficiency was 0.34 cd A−1 at 9.5 V, and the power efficiency was 0.10 lm W−1 at 7.5 V for TIN. Under the same conditions, the compound containing two acceptor and indole units displayed a maximum luminescence efficiency of 133 cd m−2, a current efficiency of 0.73 cd A−1, and a power efficiency of 0.28 lm W−1 at 7.5 V. The EL spectra of TIN (532 nm) and T2IN (533 nm) remains stable at different voltages (10, 12, and 14), which suggests the stable electroluminescence behaviour of the compounds. The CIE chromaticity coordinate values for TIN (x = 0.34; y = 0.53) and T2IN (x = 0.34; y = 0.54) matched well with those of pure green light. To the best of our knowledge, this is the first report on the 2-(1H-indol-3yl)acrylonitrile-based fluorophores and these fluorophores ensure good OLED device performance and emission of pure green light.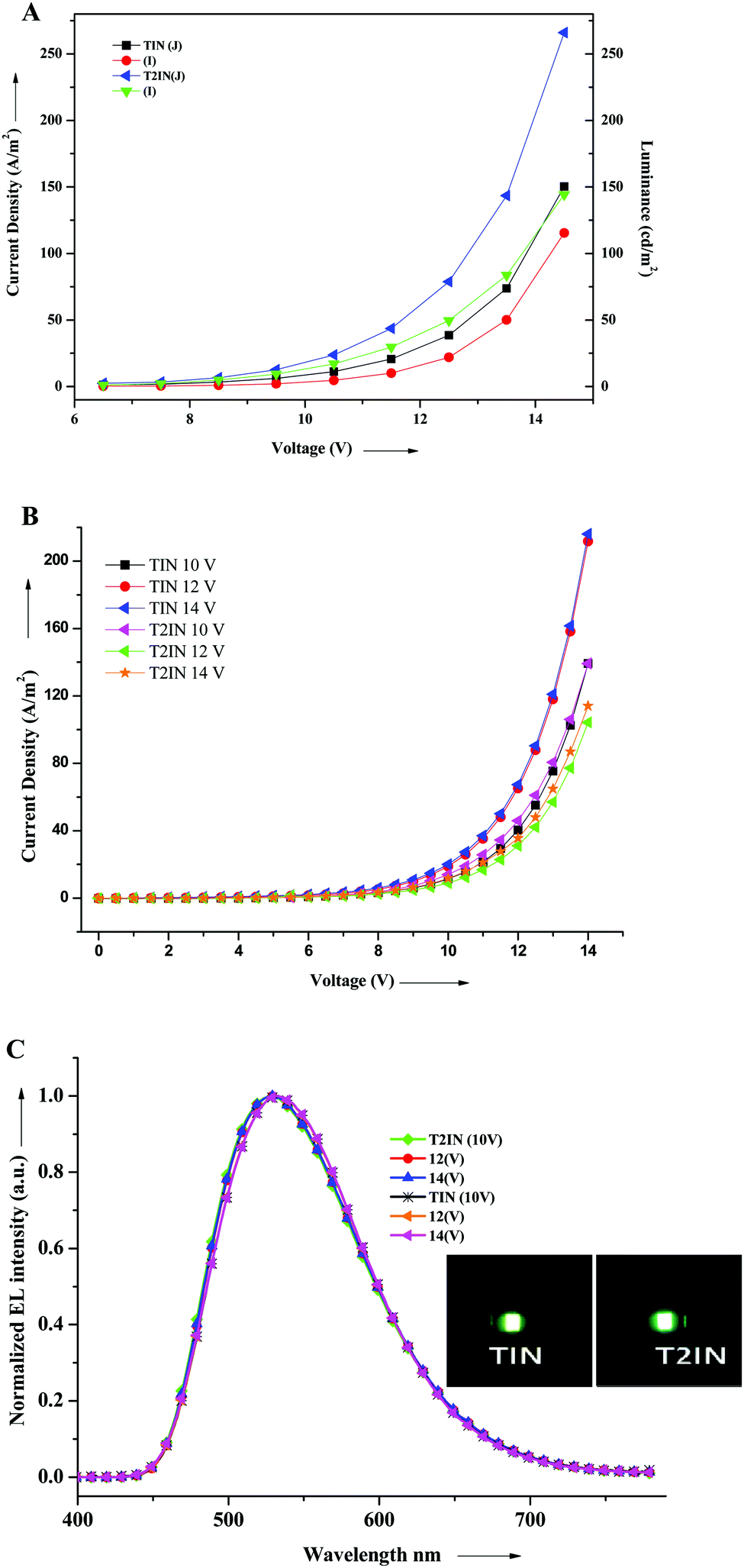 | ||
| Fig. 6 (A) Voltage–current density–luminance (V–J–L), (B) current–voltage (I–V), and (C) electroluminescence spectra of the devices TIN and T2IN. | ||
| Entry | V on | EL & Vonb | FWHMc (nm) | L max |
η
cmax
9.5 V |
η
pmax
7.5 V |
CIE [x, y]g | |
|---|---|---|---|---|---|---|---|---|
| a Turn-on voltage (V). b Maximum luminance (nm) and turn-on voltage (V). c Full width at half maximum. d Luminance efficiency (cd m−2). e Maximum current efficiency (cd A−1). f Power efficiency (lm W−1). g CIE at 14 V. | ||||||||
| TIN | 6.5 |
532
10, 12, 14 |
110 | 110 | 0.34 | 0.10 | 0.34 | 0.53 |
| T2IN | 6.5 |
533
10.5, 12.5, 14.5 |
109 | 133 | 0.73 | 0.28 | 0.34 | 0.54 |
Conclusion
We successfully designed and synthesized seven new donor–acceptor compounds and characterized their optical, thermal, and electrochemical properties. Furthermore, good emission behaviour, high fluorescence quantum yield, and excellent electrochemical and thermal stability were observed for all the compounds. Promising OLED performances were observed by fabricating non-doped single layer devices with green emission, and the maximum luminescence, current efficiency, and power efficiency were 110 cd m−2 and 133 cd m−2, 0.34 cd A−1 and 0.73 cd A−1, and 0.28 lm W−1 and 0.10 lm W−1 for TIN and T2IN, respectively. The fluorophore 2-(1H-indol-3-yl)acetonitrile emitted a stable green color with CIE for TIN (x = 0.34, y = 0.53) and T2IN (x = 0.34, y = 0.54). A modified new donor–acceptor will be rationally designed in the future to enhance the device performance and reduce the turn-on voltage of the device. DFT results showed that the higher HOMO energies of the fluorophores largely reduced the energy barrier for the hole creation process, whereas the lower LUMO energies reduced the energy barrier for electron acceptance in optoelectronic applications. As a result, all the fluorophores were found to be efficient with the lowest band gap with promising optoelectronic properties. We hope that the results of this study will be useful for creating novel OLED devices based on 2-(1H-indol-3yl)acrylonitrile fluorophores with appreciable performance. This attempt will significantly reduce the performance efficiency loss and result in economically viable OLED's because of the cheaply available component materials involved.Acknowledgements
S. M. thanks the UGC-RGNF program for the financial assistant as a JRF (Ref. No. F1-17.1/2013-14/RGNF-SC-TAM-48918). R. R. thanks DST-Nanomission (Ref. No. SR/NM/NS-1256/2013) and UGC (UGC-2013-35169/2015) for research funding and UGC (Ref. No. F.6-6/2016-17/EMERITUS-2015-17-OBC-7855/(SA-II)) for award Emeritus Fellowship. P. V. thanks CSIR, India for the award of Emeritus Scientistship (Ref. No. 21(0936)/12/EMR-II) and DST, India for Major Research Project (SB/S1/PC-52/2012).References
- S. P. Anthony, ChemPlusChem, 2012, 77, 518–531 CrossRef CAS.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., 2007, 119, 5637–5640 CrossRef.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- A. Montali, C. Bastiaansen, P. Smith and C. Weder, Nature, 1998, 392, 261–264 CrossRef CAS.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541–5544 CrossRef CAS PubMed.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- T. J. Dale and J. Rebek, J. Am. Chem. Soc., 2006, 128, 4500–4501 CrossRef CAS PubMed.
- F. Cicoira and C. Santato, Adv. Funct. Mater., 2007, 17, 3421–3434 CrossRef CAS.
- B. Geffroy, P. Le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- B. Geffroy, P. le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- Z. Ma, P. Sonar and Z.-K. Chen, Curr. Org. Chem., 2010, 14, 2034–2069 CrossRef CAS.
- J. Ding, J. Gao, Y. Cheng, Z. Xie, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 575–581 CrossRef CAS.
- F. Liu, W.-Y. Lai, C. Tang, H.-B. Wu, Q.-Q. Chen, B. Peng, W. Wei, W. Huang and Y. Cao, Macromol. Rapid Commun., 2008, 29, 659–664 CrossRef CAS.
- D. Katsis, Y. Geng, J. Ou, S. Culligan, A. Trajkovska, S. H. Chen and L. Rothberg, Chem. Mater., 2002, 14, 1332–1339 CrossRef CAS.
- L. Li, J. Liang, S.-Y. Chou, X. Zhu, X. Niu and Q. Pei, Sci. Rep., 2014, 4, 4307 Search PubMed.
- T.-Y. Hwu, T.-C. Tsai, W.-Y. Hung, S.-Y. Chang, Y. Chi, M.-H. Chen, C.-I. Wu, K.-T. Wong and L.-C. Chi, Chem. Commun., 2008, 4956–4958 RSC.
- T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed.
- G. Zhou, W. Y. Wong, B. Yao, Z. Xie and L. Wang, Angew. Chem., Int. Ed., 2007, 46, 1149–1151 CrossRef CAS PubMed.
- H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li and P. Lu, Chem. Sci., 2015, 6, 3797–3804 RSC.
- J. Sivanadanam, P. Ganesan, P. Gao, M. K. Nazeeruddin, A. Emeline, D. Bahnemann and R. Rajalingam, RSC Adv., 2016, 6, 37347–37361 RSC.
- S. H. Kim, I. Cho, M. K. Sim, S. Park and S. Y. Park, J. Mater. Chem., 2011, 21, 9139–9148 RSC.
- D. Kondakov, J. Appl. Phys., 2007, 102, 114504 CrossRef.
- J. Lee and J. Park, Org. Lett., 2015, 17, 3960–3963 CrossRef CAS PubMed.
- S. Chidirala, H. Ulla, A. Valaboju, M. R. Kiran, M. E. Mohanty, M. N. Satyanarayan, G. Umesh, K. Bhanuprakash and V. J. Rao, J. Org. Chem., 2015, 81, 603–614 CrossRef PubMed.
- K. J. Thomas, N. Kapoor, M. P. Bolisetty, J.-H. Jou, Y.-L. Chen and Y.-C. Jou, J. Org. Chem., 2012, 77, 3921–3932 CrossRef CAS PubMed.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- Y. Li, B.-X. Li, W.-Y. Tan, Y. Liu, X.-H. Zhu, F.-Y. Xie, J. Chen, D.-G. Ma, J. Peng and Y. Cao, Org. Electron., 2012, 13, 1092–1099 CrossRef CAS.
- S.-i. Kato, H. Noguchi, A. Kobayashi, T. Yoshihara, S. Tobita and Y. Nakamura, J. Org. Chem., 2012, 77, 9120–9133 CrossRef CAS PubMed.
- N. Singla, V. S. Bhadram, C. Narayana and P. Chowdhury, J. Phys. Chem. A, 2013, 117, 2738–2752 CrossRef CAS PubMed.
- H. Du, R. C. A. Fuh, J. Li, L. A. Corkan and J. S. Lindsey, Photochem. Photobiol., 1998, 68, 141–142 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, H. P. H. X. Li, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. B.01, Gaussian Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- M. P. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS PubMed.
- G. Lai, X. R. Bu, J. Santos and E. A. Mintz, Synlett, 1997, 1275–1276 CrossRef CAS.
- G. Jones, Org. React., 1967, 15, 204–599 CAS.
- N. Nagarajan, G. Velmurugan, A. Prakash, N. Shakti, M. Katiyar, P. Venuvanalingam and R. Renganathan, Chem. – Asian J, 2014, 9, 294–304 CrossRef CAS PubMed.
- J. M. Chudomel, B. Yang, M. D. Barnes, M. Achermann, J. T. Mague and P. M. Lahti, J. Phys. Chem. A, 2011, 115, 8361–8368 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science & Business Media, 2013 Search PubMed.
- T. Y. Wu and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1444–1448 CrossRef CAS.
- Q. Fang, B. Xu, B. Jiang, H. Fu, X. Chen and A. Cao, Chem. Commun., 2005, 1468–1470 RSC.
- G. Velmurugan, S. A. Vedha and P. Venuvanalingam, RSC Adv., 2014, 4, 53060–53071 RSC.
- M. A. De Oliveira, H. A. Duarte, J.-M. Pernaut and W. B. De Almeida, J. Phys. Chem. A, 2000, 104, 8256–8262 CrossRef CAS.
- A. Curioni, W. Andreoni, R. Treusch, F. Himpsel, E. Haskal, P. Seidler, C. Heske, S. Kakar, T. Van Buuren and L. J. Terminello, Appl. Phys. Lett., 1998, 72, 1575–1577 CrossRef CAS.
- P. J. Hay, J. Phys. Chem. A, 2002, 106, 1634–1641 CrossRef CAS.
- S. Y. Hong, D. Y. Kim, C. Y. Kim and R. Hoffmann, Macromolecules, 2001, 34, 6474–6481 CrossRef CAS.
- L. Y. Zou, A. M. Ren, J. K. Feng, Y. L. Liu, X. Q. Ran and C. C. Sun, J. Phys. Chem. A, 2008, 112, 12172–12178 CrossRef CAS PubMed.
- N. Nagarajan, G. Velmurugan, G. Prabhu, P. Venuvanalingam and R. Renganathan, J. Lumin., 2014, 147, 111–120 CrossRef CAS.
- N. Nagarajan, A. Prakash, G. Velmurugan, N. Shakti, M. Katiyar, P. Venuvanalingam and R. Renganathan, Dyes Pigm., 2014, 102, 180–188 CrossRef CAS.
- N. Nagarajan, G. Velmurugan, P. Venuvanalingam and R. Renganathan, J. Photochem. Photobiol., A, 2014, 284, 36–48 CrossRef CAS.
- R. Jagadeesan, G. Velmurugan and P. Venuvanalingam, RSC Adv., 2016, 6, 44569–44577 RSC.
- C. H. Huang, F. Li and W. Huang, Introduction to Organic Light-Emitting Materials and Devices, Fudan University, Shanghai, 2005 Search PubMed.
- X. Liu, A. Ren, J. Feng, L. Yang, H. Xu, M. Shi and C. Sun, Chem. Res. Chin. Univ., 2006, 27, 2156–2159 CAS.
- W. L. Jia, X. D. Feng, D. R. Bai, Z. H. Lu, S. Wang and G. Vamvounis, Chem. Mater., 2005, 17, 164–170 CrossRef CAS.
- L. Y. Zou, A. M. Ren, J. K. Feng and X. Q. Ran, J. Phys. Org. Chem., 2009, 22, 1104–1113 CrossRef CAS.
- A. Tenderholt, QMForge: A Program to Analyze Quantum Chemistry Calculations, Version 2.3.2, http://qmforge.sourceforge.net.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption and emission spectra of all the compounds in different solvents, absorption spectra of the thin films, results of the fluorescence decay in different solvents, cyclic voltammogram results, AFM images for all the compounds, DFT optimized structures, lifetime data for all the solvents, frontier molecular orbital energies, and molecular orbital composition (%) of various fragments in the ground state. See DOI: 10.1039/c6qm00326e |
| This journal is © the Partner Organisations 2017 |